

Abstract
The secretion of transthyretin (TTR) variants contributes to the pathogenesis of amyloidosis because they form aggregates in the extracellular environment. However, the mechanism of how TTR variants pass the quality control system in the endoplasmic reticulum (ER) has not yet been elucidated. We investigated here the mechanism of how TTR passes ER monitoring. Monomeric mutation introduced in TTRs (M-TTRs) resulted in the ER retention of amyloidogenic M-TTRs but not non-amyloidogenic M-TTRs. Retention of amyloidogenic M-TTRs induced the unfolded protein response and upregulated the expression of ER chaperones BiP and glucose-regulated protein (GRP) 94. Additionally, we showed that the ER-retained amyloidogenic M-TTRs are subject to ER-associated degradation. On the other hand, the amyloidogenic TTR variants and non-amyloidogenic M-TTRs were secreted normally. These findings suggest that unlike for wild-type TTR, the ER quality control system may differentially regulate the fate of the TTR variants and their monomeric counterparts.
Keywords: amyloidosis, ER quality control, familial amyloid polyneuropathy (FAP), transthyretin, TTR variants
Introduction
Proteins entering the secretory pathway are first translocated into the endoplasmic reticulum (ER), where they undergo folding and oligomerization by the assistance of ER chaperones. Only correctly folded or assembled proteins are sent to their final destinations. Misfolded or misassembled proteins are selectively retained in the ER by the ER chaperones, a process termed ER quality control (Sitia and Braakman, 2003). Proteins that are irreversibly misfolded are retrotranslocated across the ER membrane and degraded by the cytosolic proteasome in a process referred to as ER-associated degradation (ERAD), which is an aspect of ER quality control (Hampton, 2002). ER quality control protects cells from harmful product such as aggregation of misfolded or misassembled proteins. However, some proteins that are associated with extracellular amyloidoses can pass through the ER but misfold and misassemble outside the cells (Cohen and Kelly, 2003).
Transthyretin (TTR) is a soluble non-glycosylated protein, which forms a 55 kDa homotetramer and functions as a transporter of thyroxine (T4) and holo-retinol binding protein in the blood and cerebral spinal fluid (Hamilton and Benson, 2001). TTR forms amorphous aggregates and amyloid fibrils, which cause senile systemic amyloidosis and familial TTR amyloid diseases, including familial amyloid polyneuropathy (FAP) and familial amyloid cardiomyopathy (Connors et al, 2003). The precise mechanism of TTR amyloidogenesis remains to be elucidated. However, it is now accepted that tetramer dissociation to a misfolded monomeric subunit is rate-limiting for amyloid formation (Lai et al, 1996; Hammarstrom et al, 2002). Familial TTR amyloid diseases, which are autosomal dominant, are associated with a point mutation in TTR gene. To date, over 100 different TTR variants have been reported, although onset of the disease, tissue selectivity and severity are different with variants (Connors et al, 2003; Hammarstrom et al, 2003; Sekijima et al, 2003). These variations result from differences in secretion efficiency and amyloidogenicity of TTR variants, which appear to be important parameters in disease etiology. Sekijima et al (2005) recently demonstrated that secretion efficiency and amyloidogenicity of TTR variants are predicted by a combination of thermodynamic and kinetic stabilities in vitro. D18G and A25T TTRs, which exhibit late-onset central nervous system-selective amyloidosis and are present at very low concentrations in the blood (Hammarstrom et al, 2003; Sekijima et al, 2003), have low secretion efficiencies that might be attributed to a combination of low thermodynamic and kinetic stabilities. These TTRs are recognized by the ER quality control system and are subject to ERAD. Therefore, ERAD leads to low secretion of the most highly destabilized TTR variants, protecting against severe early-onset systemic amyloidosis. However, the molecular mechanism underlying the escape from ERAD of TTR variants exhibiting systemic amyloidosis, remains unclear.
In this study, we compared the secretion pattern and cellular localization of wild-type TTR, seven TTR variants and their monomeric mutants (M-TTRs) using TTR secretion assay and immunofluorescence analysis. Five of these TTR variants (CNS-selective amyloidoses, D18G, A25T TTR; systemic amyloidoses, V30M, E54K and L55P TTR) are disease associated and are termed ‘amyloidogenic TTRs', whereas two of the variants we used, T119M and R104H, have no known pathogenic consequences and are termed ‘non-amyloidogenic TTRs' (Saraiva, 2001). Here, we report that D18G TTR and amyloidogenic M-TTRs were barely secreted into the cell media and that they were retained in the ER. Moreover, D18G TTR and amyloidogenic M-TTRs are degraded by ERAD. On the other hand, the amyloidogenic TTR variants and the non-amyloidogenic M-TTRs were efficiently secreted from the cells. Because most amyloidogenic TTR variants have moderate thermodynamic and kinetic stabilities, they exist as tetramer in the ER and could pass the ER quality control system but form aggregates or amyloid fibrils in the extracellular space.
Result
Secretion pattern of wild-type, non-amyloidogenic and amyloidogenic TTRs
We first determined the secretion pattern of TTRs using mammalian cell assay system by transiently transfecting Chinese hamster ovary (CHO-K1) cells, which do not express endogenous TTR, with wild-type TTR or TTR variants. After full confluence, transfected cells were incubated for 24 h in serum-free medium. Cell media and lysates were collected for analysis of secreted and intracellular TTRs, respectively. The samples were run under both SDS–PAGE (Figure 1) and native PAGE conditions (Supplementary Figure 1A). Tetramer forms of wild-type TTR and non-amyloidogenic TTRs, R104H and T119M TTRs were detected in the media by Western blotting using anti-human TTR antibody (Figure 1A and Supplementary Figure 1A, left panels). R104H TTR was secreted in a pattern similar to that of wild-type TTR, but T119M TTR was hardly detected in monomeric form, suggesting that this variant tetramer is stable, consistent with the trans-suppression effect of T119M TTR in individuals having the compound heterozygotes of V30M and T119M TTR mutation (Coelho et al, 1993; Hammarstrom et al, 2001). Among the amyloidogenic TTR variants, only D18G TTR was barely detected in the media of CHO-K1 cells. Because the level of intracellular D18G TTR was comparable with that of the wild-type TTR under reduced as well as non-reduced SDS–PAGE conditions (Figure 1B, right panel and Supplementary Figure 1D, respectively), the difference in its secretion pattern could not be attributed to its inability to be expressed in the cells but most likely to its cellular trafficking. We also observed that the levels of the tetramer form (T) of amyloidogenic TTRs in the media were slightly lower than that of wild type TTR, indicating that the amyloidogenic TTRs might be less likely to retain their tetramer form after secretion (Figure 1B and Supplementary Figure 1C).
Figure 1.
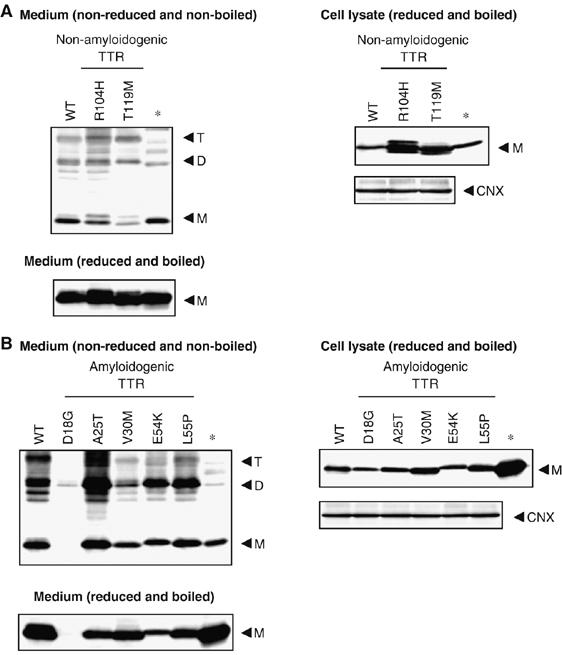
Secretion pattern of wild type and variant TTRs. (A, B) Wild-type TTR, non-amyloidogenic TTRs (A) or amyloidogenic TTRs (B) were transiently transfected in CHO-K1 cells. Medium samples were non-reduced and non-boiled (left panels), whereas the cell lysate samples were reduced and boiled (right panels) before loading. Samples were analyzed by Western blotting using anti-human TTR or anti-calnexin (CNX) antibody. The arrowheads indicate the TTR monomer (M), dimer (D) and tetramer (T). * indicates wild-type TTR protein purified from human serum.
Amyloidogenic monomeric TTR variants (M-TTRs) are not secreted from the cells
It was recently reported that wild-type TTR could be secreted as monomer (Sekijima et al, 2005). On the other hand, largely monomeric D18G TTR is inefficiently secreted from the cells. These findings suggest that amyloidogenic monomeric TTR variants might not pass the ER quality control, thus inhibiting their secretion. To examine this possibility, we first investigated the secretion of amyloidogenic monomeric TTR variants. We introduced monomeric mutation (F87M/L110M) into non-amyloidogenic and amyloidogenic TTRs to inhibit tetramerization (Jiang et al, 2001). The states of the M-TTRs after recombinant overexpression in Escherichia coli are tabulated and shown in Supplementary Table 1. Urea denaturation assay and far-UV circular dichroism (CD) analysis revealed that the thermodynamic unfolding and the secondary structure of E54K M-TTR, which we used here as representative of the other M-TTRs, were highly similar to that of E54K TTR, which indicates that the monomeric mutation have no profound effects on the stability and secondary structure of the M-TTRs (Supplementary Figure 2A and B and Supplementary Table 2). Additionally, we confirmed by near-UV CD that the tertiary structures of these proteins were not the same at neutral pH wherein TTRs retain their tetrameric form (Supplementary Figure 2C). These data on the characterization of E54K M-TTR are similar to the wild-type M-TTR performed previously (Jiang et al, 2001). Subsequently, we used these engineered M-TTRs to determine their secretion pattern in the cell system. We confirmed previous findings that wild-type M-TTR could be secreted (Sekijima et al, 2005) (Figure 2A and Supplementary Figure 3A, left panels). Moreover, non-amyloidogenic (R104H and T119M) M-TTRs were also detected in the media under reducing conditions in SDS–PAGE (Figure 2A, left panel), suggesting that non-amyloidogenic M-TTR variants could pass ER monitoring. In non-denaturing conditions, however, T119M M-TTR was detected, surprisingly, not in its monomeric form but as a tetramer, suggesting that T119M mutation confers a strong inhibitory effect against monomer formation (Supplementary Figure 3A and B). In contrast to the wild-type and non-amyloidogenic M-TTRs, all of the amyloidogenic M-TTRs were not secreted from the cells (Figure 2B and Supplementary Figure 3C and D, left panels). This was not due to the failure of the proteins to be expressed within the cells, as proven by Western blotting analysis of cell lysates (Figure 2B, right panel). A similar secretion pattern was observed in human hepatic cell line HepG2 (Supplementary Figure 4). These results imply that amyloidogenic M-TTRs might be restricted by the ER quality control.
Figure 2.
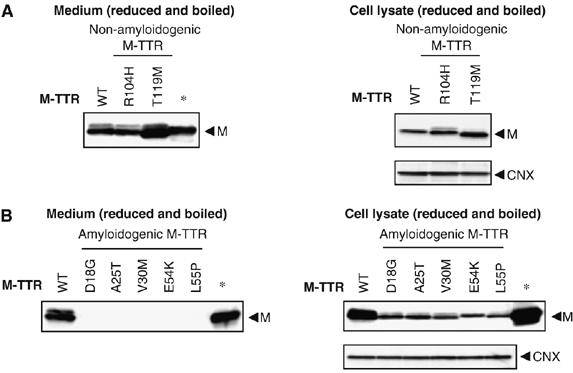
Amyloidogenic monomeric TTRs (M-TTRs) are not secreted efficiently from the cells. (A, B) Wild-type M-TTR, non-amyloidogenic M-TTRs (A) or amyloidogenic M-TTRs (B) were transiently transfected in CHO-K1 cells. Medium samples and the cell lysate samples were reduced and boiled before loading. Samples were analyzed by Western blotting using anti-human TTR or anti-calnexin (CNX) antibody. The arrowheads indicate the TTR monomer (M). *Indicates wild-type TTR protein purified from human serum.
Amyloidogenic M-TTRs are retained in the ER
To establish a relationship between intracellular trafficking and secretion of wild-type and TTR variants, we first looked at their cellular distribution. TTR constructs were transfected into HeLa cells and 48 h after, cells were fixed, permeabilized and stained with TTR and organelle marker (KDEL or GM130) antibodies. All naturally occurring TTR variants were present in the ER, as revealed by their overlap with proteins having KDEL signal (Figure 3A). Furthermore, these TTRs, except for D18G TTR, were also found in a punctate distribution pattern that overlapped with the Golgi marker GM130 (Figure 3B). The data suggest that these TTRs are efficiently trafficked through the normal protein secretory pathway. However, D18G TTR is not localized in the Golgi compartment but is retained in the ER, which would explain its low level of secretion.
Figure 3ab.
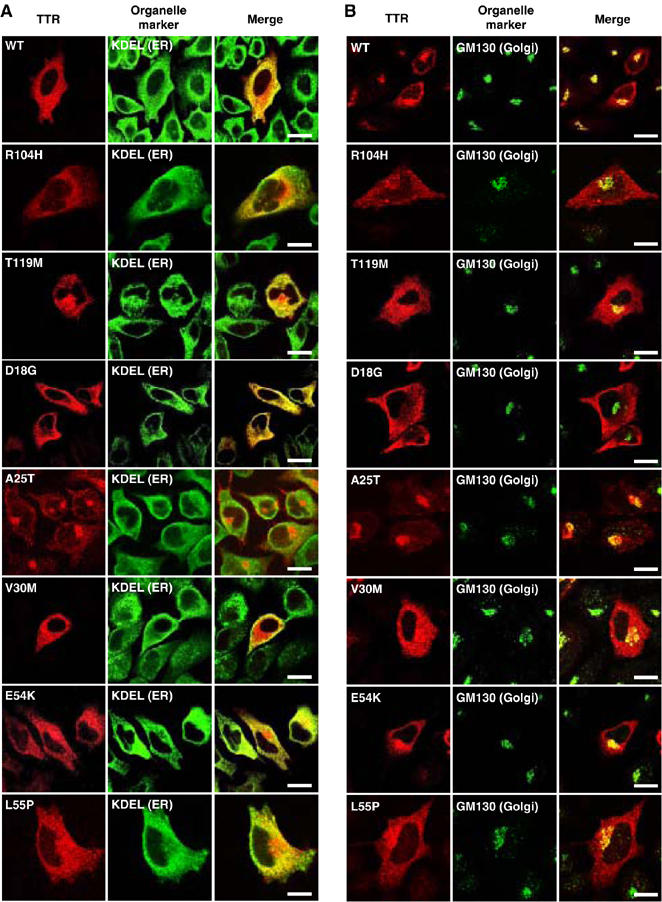
D18G TTR and amyloidogenic monomeric TTRs (M-TTRs) are retained in the ER. HeLa cells were transfected with TTRs (A, B) or M-TTRs (C, D). After 48 h, cells were fixed, permeabilized and immunostained with anti-human TTR and anti-KDEL (A, C) or anti-GM130 (B, D) antibodies and visualized with Alexa Fluor546-conjugated and Alexa Fluor488-conjugated secondary antibodies, respectively. Red fluorescence in the left subpanels represents TTRs, green fluorescence in the middle subpanels are KDEL (A, C) or GM130 (B, D), and right panels represent the overlay (merge). TTR overlap with organelle markers is indicated by the yellow fluorescence. Scale bars, 20 μm.
We next determined the cellular distribution of monomeric TTR variants. Wild-type and non-amyloidogenic (R104H and T119M) M-TTRs were present in the ER and Golgi, as revealed by the overlap with KDEL signal and GM130 (Figure 3C and D). In contrast, the amyloidogenic M-TTRs were found to overlap with the ER but not with the Golgi marker (Figure 3C and D), suggesting that these M-TTRs were mostly restricted in the ER compartment and could not be trafficked through the Golgi. These results are consistent with the inefficient secretion of amyloidogenic M-TTRs (Figure 2B and Supplementary Figure 3C and D).
Figure 3cd.
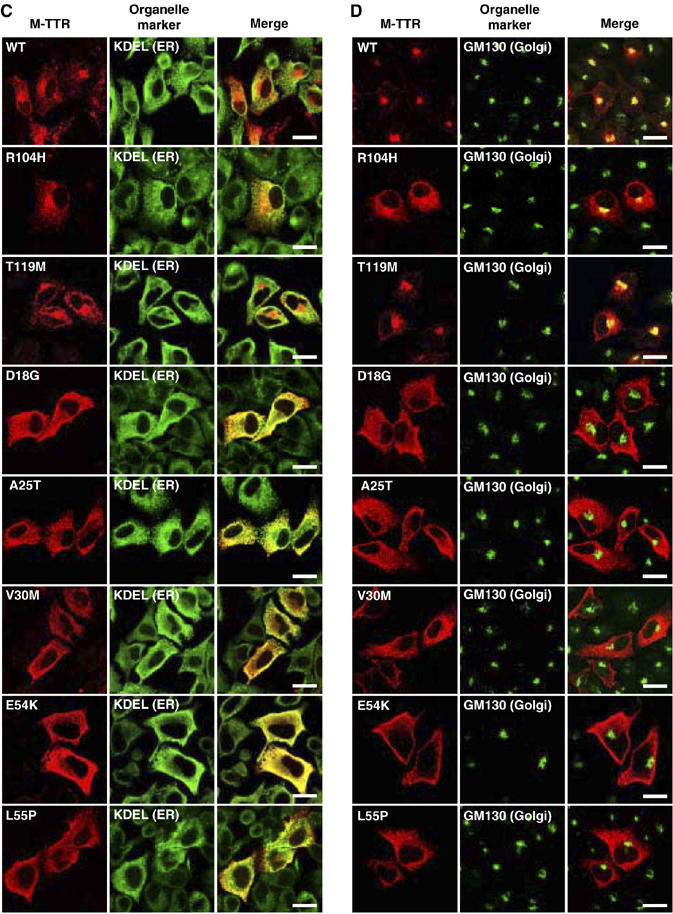
Continued.
Retention of D18G TTR and amyloidogenic M-TTRs upregulates ER chaperones, BiP and GRP94
Misfolded or misassembled proteins, which are retained in the ER, activate the unfolded protein response (UPR), which upregulates ER chaperones (Gething, 1999). We next examined whether ER retention of D18G and amyloidogenic M-TTRs induces UPR. TTR constructs were transfected into HeLa cells and cells were recovered 48 h post-transfection. As positive control for ER chaperone induction, we treated cells for 16 h with 2 μg/ml tunicamycin, a protein glycosylation inhibitor, 32 h post-transfection. We first compared the expression levels of ER chaperone in cells transfected with non-amyloidogenic and amyloidogenic M-TTRs. Densitometric quantification of blots revealed that expressions of ER chaperones, immunoglobulin heavy chain-binding protein (BiP) and glucose-regulated protein (GRP) 94 in amyloidogenic M-TTR-transfected cells were two-fold higher than that in non-amyloidogenic M-TTR-transfected cells (Figure 4A–C). We observed significant difference in the expression levels of BiP and GRP94 between cells transfected with wild-type and D18G TTR (Figure 4E and Supplementary Figure 5). Cells expressing V30M M-TTR also exhibited significant upregulation of BiP and GRP94 compared with cells transfected with wild-type TTR or wild-type M-TTR (Figure 4E). BiP and GRP94 expression levels also differed significantly between cells expressing V30M TTR and its monomeric counterpart V30M M-TTR (Supplementary Figure 5). The protein expressions were quantified (Figure 4F and G). Another molecular chaperone, protein disulfide isomerase (PDI), was not upregulated in D18G TTR and amyloidogenic M-TTR-transfected cells (Figure 4D and H). Expression of BiP, GRP94 and PDI did not change between non-amyloidogenic and amyloidogenic TTR-expressing cells, except for those in D18G TTR-transfected cells (Supplementary Figure 5). Collectively, these results indicate that ER retention of D18G and amyloidogenic M-TTRs significantly induced UPR, which upregulated BiP and GRP94 but not PDI.
Figure 4.
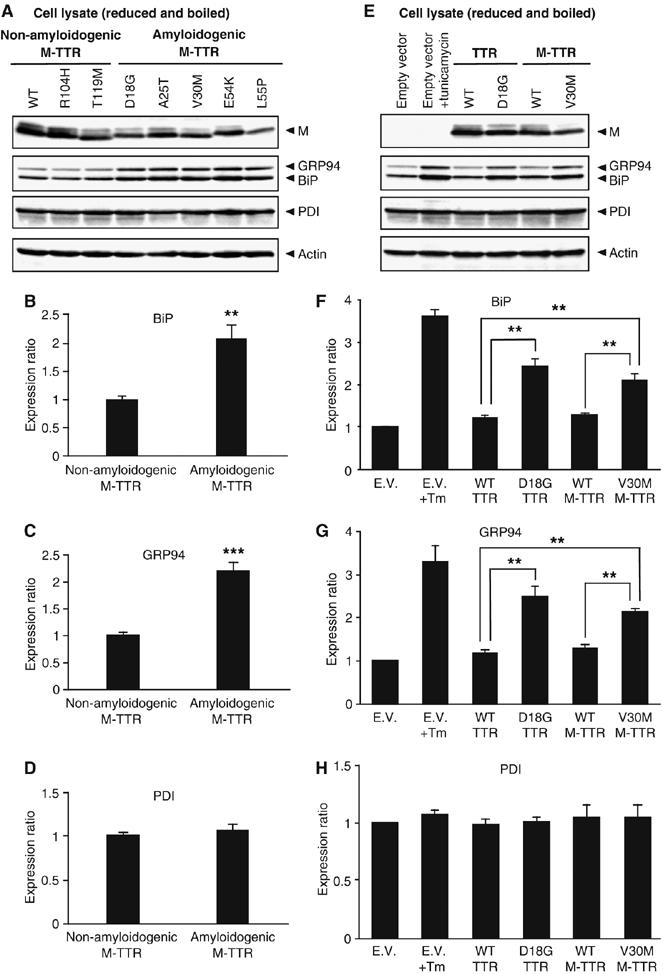
Retention of D18G TTR and amyloidogenic monomeric TTRs (M-TTRs) upregulates ER chaperone, BiP and GRP94. (A, E) HeLa cells were transfected with empty vector (E.V.) or the indicated M-TTR constructs. Cell lysates were recovered 48 h post-transfection, boiled and subjected to SDS–PAGE under reduced condition. Proteins were probed with anti-human TTR, anti-KDEL (BiP and GRP94), anti-PDI and anti-actin antibodies. As positive control for ER chaperones induction, cells transfected with empty vector were treated for 16 h with 2 μg/ml tunicamycin (Tm) 32 h post-transfection (Figure 4E, lane 2). (B–D and F–H) Densitometric quantifications were performed on BiP, GRP94 and PDI Western blots of HeLa cells. (B–D) Bars shown are the average expression of ER chaperones in cells transfected with non-amyloidogenic and amyloidogenic M-TTRs. (F–H) Bars shown are the average of three independent experiments for the expression ratio of ER chaperones in cells transfected with the indicated constructs. Values represent the mean±s.e. (**P<0.01, ***P<0.001)
D18G TTR and amyloidogenic M-TTRs are degraded by ERAD
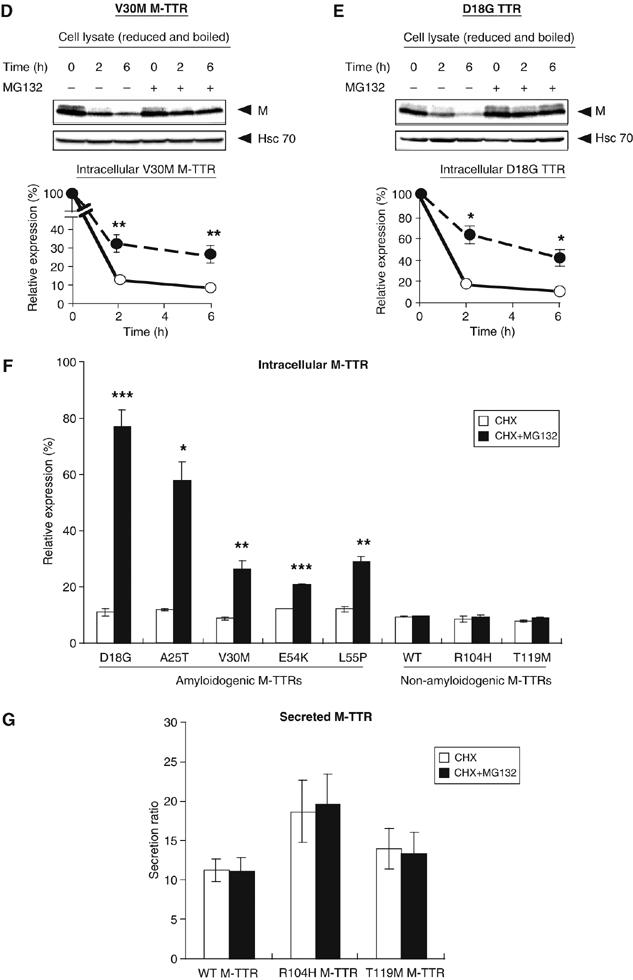
Prolonged retention of misfolded or misassembled protein in the ER leads to their degradation by the proteasome, and ERAD of highly destabilized proteins leads to low secretion (Ellgaard and Helenius, 2003). The substantially reduced secretion of amyloidogenic M-TTRs and their retention within the ER suggests that amyloidogenic M-TTRs may be targeted for ERAD. To investigate this possibility, degradation experiments were performed in the presence of 500 μM cycloheximide, a protein synthesis inhibitor, and a proteasome inhibitor, MG132, or its solvent, dimethyl sulfoxide (DMSO), in the fresh medium added to cells. Cell media and lysates from HeLa cells transfected with each TTR constructs were collected at 0, 2 or 6 h after treatment. For 0 time point, samples were taken immediately after adding cycloheximide and MG132 or DMSO. Western blots of intracellular TTRs were quantified (see Materials and methods) to determine the extent of intracellular TTR degradation relative to the quantity of TTR synthesized. We did not observe a significant difference in the expression levels of intracellular wild-type TTR between MG132-treated and untreated cells at different time points (Figure 5A, left panel, graph). Densitometric quantification of secreted TTRs indicated that similar amounts of wild-type TTR were secreted from cells with or without MG132 2 h after cycloheximide treatment (Figure 5A, right panel). The proteasome inhibitor did not also significantly affect the intracellular wild-type M-TTR level and the protein was secreted from both treated and untreated cells with a similar efficiency (Figure 5B). Moreover, we determined by quantification that under the same conditions, the levels of secreted wild-type TTR and wild-type M-TTR were comparable (Figure 5A and B, graph of secreted TTRs). These data indicate that wild-type TTR and wild-type M-TTR are not destined for degradation by the proteasome. On the other hand, the intracellular V30M TTR level was slightly increased by MG132 treatment (Figure 5C, left panel), indicating that this amyloidogenic variant might be partially targeted to ERAD. Corresponding to the intracellular levels, higher amount of V30M TTR was secreted into the medium of treated cells (Figure 5C, right panel). Compared with the effect of MG132 on V30M TTR, however, the effect of the proteasome inhibitor on intracellular V30M M-TTR was stronger. In MG132-treated cells, the relative amount of intracellular V30M M-TTR was three-fold higher than in untreated cells (Figure 5D; Table I). Since this monomeric variant TTR was incapable of being secreted (Figure 2B, left panel), it is plausible to assume that it was degraded in the absence of a proteasome inhibitor. We confirmed previous findings (Sekijima et al, 2005) that the highly unstable D18G TTR is targeted to ERAD (Figure 5E). We also determined the intracellular relative expression (%) of other non-amyloidogenic and amyloidogenic M-TTRs in MG132-treated and untreated cells at 6 h after cycloheximide treatment (Figure 5F; Table I). From the densitometric quantification of TTR expression, we found that all amyloidogenic M-TTRs were significantly stabilized by MG132, indicating that they are subject to proteasomal degradation (Figure 5F). On the other hand, neither the intracellular nor secreted wild-type and non-amyloidogenic M-TTRs were affected by MG132 treatment (Figure 5F and G; Table I). Taken together, we conclude that the ER retention of D18G TTR and amyloidogenic M-TTR variants leads to the ERAD of these proteins, thus inhibiting their secretion from mammalian cells.
Figure 5abc.
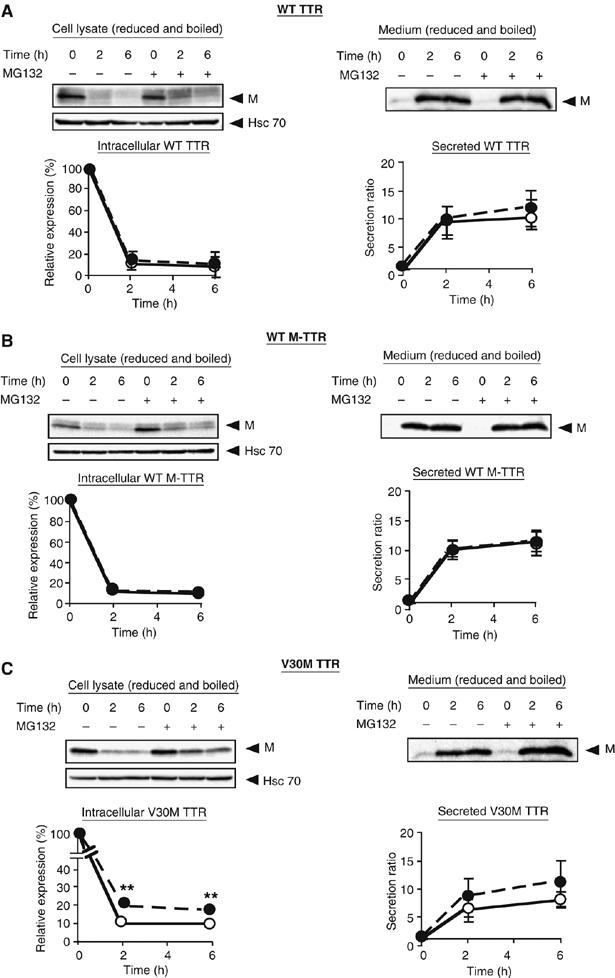
Amyloidogenic M-TTRs are degraded by proteasome. (A–E) HeLa cells were transfected with the indicated TTR constructs. After 42 h, cells were treated with 500 μM cycloheximide and the proteasome inhibitor, MG132 (30 μM) or DMSO. Cell lysate and media were collected immediately (0 h) or 2, 6 h after the addition of cycloheximide. Densitometric quantifications were performed on the intracellular TTR and secreted TTR from HeLa cells. Intracellular TTR expression at different time points was calculated as relative expression: relative expression (%)=100 × (intracellular TTR at given time t/intracellular TTR at 0 h). Secreted TTR was calculated as follows: (secretion level at given time t/secretion level at 0 h). White circles, CHX and black circles, CHX+MG132. (F) The % relative expression level of intracellular M-TTRs at 6 h after cycloheximide treatment in HeLa cells untreated or treated with 30 μM MG132. (G) The secretion ratio of wild-type or non-amyloidogenic M-TTRs at 6 h after cycloheximide treatment in HeLa cells untreated or treated with 30 μM MG132. Conditions in (F) and (G) are identical to those described above. All experiments were performed in triplicate. Values represent the mean±s.e. (*P<0.05, **P<0.01, ***P<0.001).
Figure 5defg.
Continued.
Table 1.
Summary of TTR variants stability, cellular distribution and ERAD efficiency
| TTR variant | Phenotype | Character | Monomer TTR (%) T4 (1 μM) | Monomer TTR (%) diflunisal (1 μM) | Cellular localization | Intracellular protein (%) (6 h treatment) | |
|---|---|---|---|---|---|---|---|
| CHX | CHX+MG132 | ||||||
| WT | SSA | 12.2±0.8 | 20.3±3.4 | ER and Golgi | 9.1±1.3 | 11.6±0.7 | |
| R104Ha | Non-amyloid | Low suppressive effect | 6.1±0.9 | 1.0±2.6 | ER and Golgi | ||
| T119Ma | Non-amyloid | Suppresive effect | ND | ND | ER and Golgi | ||
| D18G | CNS, LM | Largely monomeric form, late onset | ND | ND | ER | 11.2±1.0 | 42.0±6.5* |
| A25T | CNS, PN | Unstable tetramer, late onset | 83.1±9.5 | 92.2±4.3 | ER and Golgi | ||
| V30M | AN, E, LM, PN | Prevalent, mild progression | 43.4±3.3 | 58.3±3.4 | ER and Golgi | 10.0±0.6 | 15.1±0.3** |
| E54K | AN, H, PN | Aggressive progression | 92.2±4.9 | 96.6±1.4 | ER and Golgi | ||
| L55P | AN, E, H, PN | Aggressive progression | 61.5±2.6 | 85.3±2.6 | ER and Golgi | ||
| Monomeric mutant | |||||||
| Wild type | ER and Golgi | 9.5±0.2 | 9.7±0.1 | ||||
| R104H | ER and Golgi | 8.4±1.1 | 9.4±0.6 | ||||
| T119M | ER and Golgi | 7.8±0.5 | 8.8±0.4 | ||||
| D18G | ER | 11.1±1.3 | 77.2±6.1*** | ||||
| A25T | ER | 8±0.3 | 58.0±6.8* | ||||
| V30M | ER | 8.8±0.6 | 26.2±3.3** | ||||
| E54K | ER | 12.3±0.0 | 20.9±0.3*** | ||||
| L55P | ER | 12.1±1.0 | 29.1±1.9** | ||||
| Abbreviations: AN, autonomic neuropathy; CNS, central nervous system; CTS, carpal tunnel syndrome; E, eye; H, heart; LM, leptomeningeal; PN, polyneuropathy; S, skin. | |||||||
| aNon-amyloidogenic variant. | |||||||
| Monomer TTR (%)=100 × (non-reduced and non-boiled monomer at 1 μM T4 or diflunisal/reduced and boiled monomer at 0 μM T4 or diflunisal) | |||||||
| ND: not detected. *P<0.05, **P<0.01 and ***P<0.001. | |||||||
Effects of thyroxine (T4) and diflunisal (DF) on the level of TTR monomers in the cell media
It has been demonstrated that T4 and DF inhibit fibril formation in vitro (Baures et al, 1999; Adamski-Werner et al, 2004). We investigated here whether these small molecules have comparable effects on TTRs in mammalian cell system. CHO-K1 cells overexpressing the TTR constructs were treated with 1 μM T4 or DF for 24 h and the cell media were collected for analysis of the expression of secreted TTRs. The addition of T4 or DF suppressed the monomer levels of wild-type and non-amyloidogenic TTR, R104H, in the media, as determined by quantification of TTR monomer ratio under non-boiled, non-reduced condition (Figure 6A and B; Table I). The effect of T4 on wild-type TTR was similar in various cell lines tested (Supplementary Figure 6). T4 and DF had a dose-dependent effect on wild-type and R104H TTRs (Supplementary Figure 7A and B). The monomer levels of the amyloidogenic TTR variants V30M and L55P were slightly decreased by T4 and DF treatment (Figure 6A and B; Table I), suggesting that the small molecules weakly stabilize these TTR variants. But this effect was not so apparent and a higher concentration of the small molecules may be needed to stabilize V30M and L55P TTRs (Supplementary Figure 7C and D). T4 or DF did not decrease the monomer levels of amyloidogenic TTRs E54K and A25T (Figure 6A and B; Table I). The E54K and A25T TTR monomers in the media were unaffected by the presence of these small molecules at various concentrations (Supplementary Figure 7E and F, non-boiled, non-reduced, blot a). The total amount of secreted amyloidogenic TTRs did not change upon addition of T4 or DF (Supplementary Figure 7, blot b), except for A25T in which T4 dose dependently increased its secretion, an observation consistent with the report by Sekijima et al (2005). As expected, the highly unstable D18G variant was refractory to T4 and DF treatment and its secretion was not improved by the addition of the small molecules (Supplementary Figure 7G). These data collectively suggest that in mammalian cells, the sensitivity to T4 and DF effect on TTR stabilization is higher for wild-type and non-amyloidogenic TTRs compared with amyloidogenic TTR variants.
Figure 6.
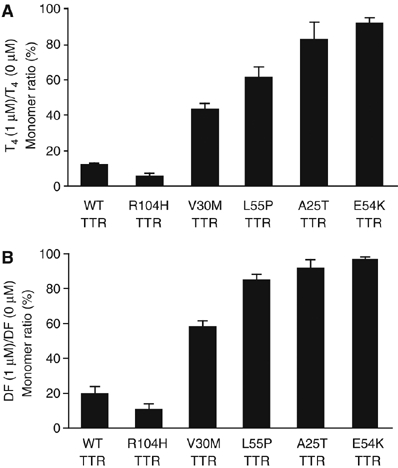
Effect of thyroxine (T4) and DF on the level of TTR monomers in the cell media. (A, B) Wild-type TTR or variant TTRs were transiently transfected in CHO-K1 cells. Cells were maintained for 24 h in serum-free DMEM with or without 1 μM T4 or 1 μM DF. Medium samples were analyzed by Western blotting using anti-human TTR antibody. Western blots of secreted TTR monomer were quantified by densitometric scanning. Ratio of 1 μM T4 or DF monomer to 0 μM T4 or DF monomer in the media was calculated as follows: T4 (DF) (1 μM)/T4 (DF) (0 μM) monomer ratio (%)=100 × (non-reduced, non-boiled monomer at 1 μM T4 or DF)/(non-reduced, non-boiled monomer at 0 μM T4 or DF). Bars shown are representative of three independent experiments. Values represent the mean±s.e.
Discussion
It is widely known that mutations, which cause aberrant folding, mostly lead to the degradation of the protein by ERAD. However, TTR variants having disease-associated mutations are efficiently secreted by mammalian cells (Sekijima et al, 2005). How these amyloidogenic variants pass through ER quality control is yet to be elucidated. Here, we first assessed the cellular secretion pattern of the wild-type, non-amyloidogenic and amyloidogenic TTRs by examining the TTRs expression in the media using SDS–PAGE and native PAGE. By taking a cue on the observation that the mostly monomeric D18G TTR is barely detectable in the media (Figure 1B and Supplementary Figure 1C), we next investigated whether the monomeric forms of amyloidogenic variants (M-TTRs) are also not secreted from the cells. The results of our assay under denaturing and non-denaturing conditions (Figure 2 and Supplementary Figure 3) demonstrated that this is indeed the case, wild type and non-amyloidogenic M-TTRs were detected in the media but amyloidogenic M-TTR variants were not.
To exclude the possibility that the non-secretion of the amyloidogenic M-TTRs might be due to the compromised stability of the protein arising from the introduced monomeric mutation, we determined the structure and stability of E54K M-TTR. Because the unfolding curve of E54K M-TTR was similar to the naturally occurring E54K TTR, and that the secondary structures of these proteins were almost identical (Supplementary Figure 2A and B), we could conclude that the monomeric mutation did not compound the misfolding propensity of E54K M-TTR. The inefficient secretion of amyloidogenic M-TTRs and of D18G TTR was not likely due to their lack of expression within the cells because these proteins were detected in the cell lysates, albeit, mostly at reducing conditions and faintly at non-denaturing conditions (Figure 2B and Supplementary Figure 3C and D, respectively). It is interesting to note here that in non-reduced, non-boiled SDS–PAGE, ‘dimer' bands were detected in the cell lysates of M-TTRs (Supplementary Figure 3B and D). This was not without precedence, however, because in a previous report, the SDS–PAGE of wild-type and V30M M-TTRs showed distinct ‘dimer' bands, which could be monomers that have dimerized in solution under mildly denaturing conditions during sample preparation (Reixach et al, 2004).
Our findings that: (1) the naturally occurring amyloidogenic TTRs, except D18G, are secreted with comparable efficiency as the wild type TTR; (2) the wild type and non-amyloidogenic M-TTRs are secreted from the cells; (3) the amyloidogenic M-TTRs are not detected in the cell media and (4) D18G TTR and the amyloidogenic M-TTRs were retained in the ER and degraded by ERAD provide proof of principle that at the monomeric level, the TTR variants are remarkably differentially regulated by the ER quality control system in comparison with the wild type TTR. The retention of the amyloidogenic M-TTRs in the ER (Figure 3) led to activation of UPR and the ERAD of these proteins (Figures 4 and 5). On the other hand, amyloidogenic TTR variants, except D18G TTR, pass through the ER quality control because the tetramer–monomer equilibrium would favor the tetrameric state due to the fast rate of folding and assembly of the TTR tetramer. Since tetrameric variant TTRs do not engage the ERAD machinery, export of these variants from the ER could be facilitated (Sekijima et al, 2005). However, we detected that intracellular V30M TTR level was slightly increased by MG132 treatment (Figure 5C), which indicated that a small portion of TTR variants that exists as monomer may be subjected to proteasomal degradation. This result further supports our observations that the monomeric forms of amyloidogenic TTR variants are targeted to ERAD and implies that this may be an endogenously occurring process.
A recent study by Hammarström's group using E. coli system revealed that D18G TTR and partly, A25T TTR, interact with BiP (Sorgjerd et al, 2006), which suggests a likely role for this ER chaperone in the ERAD of these highly unstable proteins. Consistent with this report, we determined here that BiP and GRP94 expressions were significantly higher in cells overexpressing D18G TTR and amyloidogenic M-TTRs (Figure 4B and F), and that all amyloidogenic M-TTRs were retained in the ER by the ER quality control system (Figure 3). Secretory proteins unable to fold correctly are retained in the ER and form stable complexes with BiP and other molecular chaperones (Ellgaard and Helenius, 2003).
Because T4 and DF have been shown to stabilize amyloidogenic TTRs in vitro, we assessed here their effects in mammalian cells. We showed that T4 and DF had stronger stabilizing effect on wild-type and non-amyloidogenic TTR, R104H, and weaker effect on amyloidogenic TTR variants, V30M and L55P, but no perceptible effect on E54K, A25T and D18G TTRs (Figure 6 and Supplementary Figure 7), suggesting that T4 and DF have selective functions on different TTRs. Considering that we observed only the level of monomers, we cannot fully determine whether these small molecules could inhibit amyloid fibril formation, even if they decreased the level of monomeric TTRs as in the case of wild-type TTR. These current findings argue against the notion of a ‘general effectiveness' of the small molecules to prevent amyloid formation and in fact, for certain variants, these treatment might even compound the extracellular monomer formation due to the increased secretion efficiency of the variants at first afforded by the treatment. We then propose that since the small molecules have selective effects on the amyloidogenic variants, each variant must be carefully analyzed for its response to these treatments.
In conclusion, we have shown here that ER quality control differentially regulates the fate of amyloidogenic TTRs and their monomers in comparison with wild-type and non-amyloidogenic TTRs/M-TTRs, and that similar to D18G TTR, the amyloidogenic M-TTRs may be more likely targeted to ERAD, leading to their non-secretion. These findings support the hypothesis that enhancing the clearance of misfolded TTRs through increased interaction with molecular chaperones may be a possible therapeutic strategy for amyloidosis. Future studies need to address the retention mechanism of D18G TTR and amyloidogenic M-TTRs in mammalian cells.
Materials and methods
Cell culture and transfection
CHO-K1 cells, HEK293 cells and HepG2 cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% fetal bovine serum. Baby hamster kidney (BHK) cells and HeLa cells were cultured in minimal essential medium supplemented with 10% fetal bovine serum. Cells were maintained at 37°C in a humidified atmosphere of 5% CO2 and 95% air. Transient transfections of TTR constructs were performed with TransIT-LT-1 (Mirus Corp., Madison, WI) according to the manufacturer's recommendations. Specifically, TransIT-LT-1 reagent diluted with reduced serum Opti-MEM (Invitrogen, Carlsbard, CA) was mixed with total DNA in a ratio of 1:4 (DNA/LT1) and applied to subconfluent cells.
Plasmid constructs and antibodies
Human wild-type TTR cDNA sequence was generated and inserted into the XhoI and BamHI sites of the pEF-BOS vector for expression from the EF-1α promoter. TTR variants were generated utilizing the QuikChange II site-directed mutagenesis kit procedure from Stratagene (La Jolla, California) using WT TTR DNA as template.
Antibodies used are as follows: the polyclonal anti-human TTR (prealbumin; FL-147) and goat polyclonal anti-actin (I-19; sc-1616) antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, CA); polyclonal anti-human TTR (prealbumin; A00027) was from Dakocytomation (Glostrup, Denmark); rabbit polyclonal anti-calnexin (anti-CNX; SPA-860), rat monoclonal anti-Hsc 70 (SPA-815), mouse monoclonal anti-KDEL (SPA-827) and rabbit polyclonal anti-protein disulfide isomerase (PDI; SPA-890) were from Stressgen Biotechnologies (Victoria BC, Canada) and HRP-conjugated anti-rabbit, anti-goat, anti-rat and anti-mouse antibodies were from Jackson ImmunoResearch Laboratories (Westgrove, PA).
Immunofluorescence microscopy
HeLa cells were grown on glass-bottomed culture dishes and transfected with 2 μg of pEF-BOS plasmid DNA encoding the wild type or variant TTR. Forty-eight hours after transfection, cells were fixed and stained following the procedure described previously (Okiyoneda et al, 2004). Cells were incubated with mouse anti-KDEL (Stressgen), mouse anti-GM130 (BD Biosciences) or rabbit anti-human TTR (Santa Cruz). For visualization, the following secondary antibodies were used: anti-mouse Alexa Fluor488- or anti-rabbit Alexa Fluor546-conjugated antibodies (Molecular Probes, Eugene, OR). Stained cells were observed using a Fluoview FV300 confocal laser scanning microscope (Olympus, Japan).
SDS–PAGE and Western blotting
Serum-free media were centrifuged at 13 000 g for 15 min at 4°C to remove the debris and prepared as medium samples (extracellular forms of TTR). Cells were washed twice with ice-cold phosphate-buffer saline (PBS) and lysed at 4°C in radio-immunoprecipitation assay (RIPA) buffer (50 mM Tris–HCl, 150 mM NaCl, 1 mg/ml sodium deoxycholate and 1% NP-40) containing 1% protease inhibitor cocktail (Sigma-Aldrich) and centrifuged at 13 000 g for 15 min at 4°C. The supernatant was prepared as cell lysate samples (intracellular forms of TTR). Protein samples were either suspended in 2% SDS sample buffer (0.05 M Tris–HCl (pH 6.8), 2% SDS, 0.82 M β-mercaptoethanol and 10% glycerol) and boiled or were suspended in native sample buffer (0.05 M Tris–HCl (pH 6.8), 10% glycerol) and left unboiled before loading onto gels. Samples were analyzed by SDS–PAGE on 14% gels. Proteins were transferred onto polyvinylidene difluoride membranes (Millipore Corp., Bedford, MA) and then probed with the indicated antibodies. Immunological bands were identified with HRP-conjugated secondary antibody followed by reaction with chemiluminescence reagent SuperSignal (PIERCE, Rockford, IL).
ER chaperone induction
HeLa cells were transfected with 2 μg of pEF-BOS plasmid DNA encoding the TTRs or M-TTRs. Thirty-two hours after transfection, the medium was changed to fresh complete medium and cells were incubated for another 16 h. As positive control for ER chaperone induction, cells transfected with empty vector were treated for 16 h with 2 μg/ml tunicamycin 32 h post-transfection. Proteins from cell lysates and media were analyzed by SDS–PAGE and Western blotting. Densitometric quantification of blots was performed using Image Gauge software (Fujifilm). All experiments were performed in triplicate. Statistical significance was calculated using Student's t-test.
Proteasome inhibitor treatment
HeLa cells were transfected with 2 μg of pEF-BOS plasmid DNA encoding the TTRs (wild-type, D18G, V30M) or M-TTRs. Forty-two hours after transfection, the media were replaced with fresh media containing 500 μM cycloheximide (Sigma-Aldrich, St Louis, MO) and 30 μM MG132 (Calbiochem) or DMSO alone. Cells were reincubated at 37°C for additional 2 or 6 h. For 0 time point, lysates and media were recovered immediately after medium change and treatment. Proteins from cell lysate and media were analyzed by SDS–PAGE and Western blotting. Densitometric quantification of blots was performed using Image Gauge software (Fujifilm). To eliminate the possible distortion of the quantification of intracellular TTRs due to the high background in the Western blots, we carried out a dilution series and used these AU values to correct for the values obtained for intracellular TTR protein expression. All experiments were performed in triplicate. Statistical significance was calculated using Student's t-test.
TTR stabilization assay
CHO-K1, BHK, HeLa, HEK293 and HepG2 cells grown on six-well plates were transfected with 2 μg of pEF-BOS DNA encoding the wild-type or mutated TTR according to the above procedure. After cells reached 90–100% confluence, the medium was changed to serum-free medium and cells were incubated for another 24 h, then cell lysates and medium were collected for analysis. To examine the effect of thyroxine (T4) and DF on TTR stabilization, serum-free medium containing T4 or DF was added to the cells. Stock solutions of 15 mM T4 and 15 mM DF were prepared in 0.3 M NaOH and DMSO, respectively. TTR was assayed by Western blotting. Quantification of TTR expression was accomplished using Image Gauge software (version 3.4; Fujifilm, Japan). One micromolar of T4 and DF treatment experiments were performed in triplicate.
Supplementary Material
Supplemental Information
Acknowledgments
This work was supported by grants from the Amyloidosis Research Committee, the Pathogenesis and Therapy of Hereditary Neuropathy Research Committee, Surveys and Research on Specific Diseases, the Ministry of Health and Welfare of Japan, the Charitable Trust Clinical Pathology Research Foundation of Japan and Grants-in-Aid for Scientific Research from the Ministry of Education, Science, Sports and Culture of Japan.
References
- Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW (2004) Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem 47: 355–374 [DOI] [PubMed] [Google Scholar]
- Baures PW, Oza VB, Peterson SA, Kelly JW (1999) Synthesis and evaluation of inhibitors of transthyretin amyloid formation based on the non-steroidal anti-inflammatory drug, flufenamic acid. Bioorg Medicin Chem 7: 1339–1347 [DOI] [PubMed] [Google Scholar]
- Coelho T, Carvalho M, Saraiva MJ, Alves I, Almeida MR, Costa PP (1993) A strikingly benign evolution of FAP in an individual compound heterozygote for two TTR mutations: TTR Met30 and TTR Met119. J Rheumatol 20: 179 [Google Scholar]
- Cohen FE, Kelly JW (2003) Therapeutic approaches to protein-misfolding diseases. Nature 426: 905–909 [DOI] [PubMed] [Google Scholar]
- Connors LH, Lim A, Prokaeva T, Roskens VA, Costello CE (2003) Tabulation of human transthyretin (TTR) variants, 2003. Amyloid 10: 160–184 [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Helenius A (2003) Quality control in the endoplasmic reticulum. Nat Rev Mol Cell Biol 4: 181–191 [DOI] [PubMed] [Google Scholar]
- Gething MJ (1999) Role and regulation of the ER chaperone BiP. Semin Cell Dev Biol 10: 465–472 [DOI] [PubMed] [Google Scholar]
- Hamilton JA, Benson MD (2001) Transthyretin: a review from a structural perspective. Cell Mol Life Sci 58: 1491–1521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom P, Jiang X, Hurshman AR, Powers ET, Kelly JW (2002) Sequence-dependent denaturation energetics: a major determinant in amyloid disease diversity. Proc Natl Acad Sci USA 99 (Suppl 4): 16427–16432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hammarstrom P, Schneider F, Kelly JW (2001) Trans-suppression of misfolding in an amyloid disease. Science 293: 2459–2462 [DOI] [PubMed] [Google Scholar]
- Hammarstrom P, Sekijima Y, White JT, Wiseman RL, Lim A, Costello CE, Altland K, Garzuly F, Budka H, Kelly JW (2003) D18G transthyretin is monomeric, aggregation prone, and not detectable in plasma and cerebrospinal fluid: a prescription for central nervous system amyloidosis? Biochemistry 42: 6656–6663 [DOI] [PubMed] [Google Scholar]
- Hampton RY (2002) ER-associated degradation in protein quality control and cellular regulation. Curr Opin Cell Biol 14: 476–482 [DOI] [PubMed] [Google Scholar]
- Jiang X, Smith CS, Petrassi HM, Hammarstrom P, White JT, Sacchettini JC, Kelly JW (2001) An engineered transthyretin monomer that is nonamyloidogenic, unless it is partially denatured. Biochemistry 40: 11442–11452 [DOI] [PubMed] [Google Scholar]
- Lai Z, Colon W, Kelly JW (1996) The acid-mediated denaturation pathway of transthyretin yields a conformational intermediate that can self-assemble into amyloid. Biochemistry 35: 6470–6482 [DOI] [PubMed] [Google Scholar]
- Okiyoneda T, Harada K, Takeya M, Yamahira K, Wada I, Shuto T, Suico MA, Hashimoto Y, Kai H (2004) Delta F508 CFTR pool in the endoplasmic reticulum is increased by calnexin overexpression. Mol Biol Cell 15: 563–574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reixach N, Deechongkit S, Jiang X, Kelly JW, Buxbaum JN (2004) Tissue damage in the amyloidoses: transthyretin monomers and nonnative oligomers are the major cytotoxic species in tissue culture. Proc Natl Acad Sci USA 101: 2817–2822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saraiva MJ (2001) Transthyretin mutations in hyperthyroxinemia and amyloid diseases. Hum Mutat 17: 493–503 [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Hammarstrom P, Matsumura M, Shimizu Y, Iwata M, Tokuda T, Ikeda S, Kelly JW (2003) Energetic characteristics of the new transthyretin variant A25T may explain its atypical central nervous system pathology. Lab Invest 83: 409–417 [DOI] [PubMed] [Google Scholar]
- Sekijima Y, Wiseman RL, Matteson J, Hammarstrom P, Miller SR, Sawkar AR, Balch WE, Kelly JW (2005) The biological and chemical basis for tissue-selective amyloid disease. Cell 121: 73–85 [DOI] [PubMed] [Google Scholar]
- Sitia R, Braakman I (2003) Quality control in the endoplasmic reticulum protein factory. Nature 426: 891–894 [DOI] [PubMed] [Google Scholar]
- Sorgjerd K, Ghafouri B, Jonsson BH, Kelly JW, Blond SY, Hammarstrom P (2006) Retention of misfolded mutant transthyretin by the chaperone BiP/GRP78 mitigates amyloidogenesis. J Mol Biol 356: 469–482 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Information