

Abstract
The anti-apoptotic proteins Bcl-2 and Bcl-XL bind and inhibit Beclin-1, an essential mediator of autophagy. Here, we demonstrate that this interaction involves a BH3 domain within Beclin-1 (residues 114–123). The physical interaction between Beclin-1 and Bcl-XL is lost when the BH3 domain of Beclin-1 or the BH3 receptor domain of Bcl-XL is mutated. Mutation of the BH3 domain of Beclin-1 or of the BH3 receptor domain of Bcl-XL abolishes the Bcl-XL-mediated inhibition of autophagy triggered by Beclin-1. The pharmacological BH3 mimetic ABT737 competitively inhibits the interaction between Beclin-1 and Bcl-2/Bcl-XL, antagonizes autophagy inhibition by Bcl-2/Bcl-XL and hence stimulates autophagy. Knockout or knockdown of the BH3-only protein Bad reduces starvation-induced autophagy, whereas Bad overexpression induces autophagy in human cells. Gain-of-function mutation of the sole BH3-only protein from Caenorhabditis elegans, EGL-1, induces autophagy, while deletion of EGL-1 compromises starvation-induced autophagy. These results reveal a novel autophagy-stimulatory function of BH3-only proteins beyond their established role as apoptosis inducers. BH3-only proteins and pharmacological BH3 mimetics induce autophagy by competitively disrupting the interaction between Beclin-1 and Bcl-2 or Bcl-XL.
Keywords: apoptosis, autophagy, Bax, Bcl-2, mitochondria
Introduction
Two self-destructive processes, apoptosis (self-killing) and autophagy (self-eating), have captured the attention of cell biologists over the last decades. While apoptosis involves the activation of catabolic enzymes leading to the demolition of cellular structures and organelles, autophagy is a slow, localized phenomenon in which parts of the cytoplasm are sequestered within double-membraned autophagic vacuoles and finally digested by lysosomal hydrolases (Gozuacik and Kimchi, 2004; Kroemer and Jaattela, 2005). The relationship between apoptosis and autophagy is complex and autophagy may either contribute to cell death (Shimizu et al, 2004; Yu et al, 2004) or constitute a cellular defense against acute stress, in particular stress induced by starvation from nutrients or obligate growth factors (Boya et al, 2005; Lum et al, 2005). The crosstalk between autophagy and apoptosis is mediated at least in part by the functional and physical interaction between Beclin-1, an essential autophagy gene, and Bcl-2, one of the paradigmatic apoptosis-inhibitory proteins (Liang et al, 1999; Pattingre et al, 2005; Takacs-Vellai et al, 2005).
Bcl-2 is the prototype of a family of proteins containing at least one Bcl-2 homology (BH) region. The family is split into anti-apoptotic multidomain proteins (such as Bcl-2 and Bcl-XL), which contain four BH domains (BH1234), pro-apoptotic multidomain proteins (prototypes Bax and Bak), which contain three BH domains (BH123), and the pro-apoptotic BH3-only protein family (Letai et al, 2002). As a rule, BH1234 proteins (Bcl-2, Bcl-XL, Mcl-1) mainly reside in mitochondria, protecting these organelles against mitochondrial outer membrane permeabilization (MOMP), one of the rate-limiting events of apoptosis induction. Either of the two BH123 proteins (Bax and Bak) are required for MOMP, in a series of different models of apoptosis induction (Wei et al, 2001). BH3-only proteins are suggested to kill cells by interacting with the BH3 receptor, which is a hydrophobic groove formed by apposition of the BH1, BH2 and BH3 domains, hence activating BH123 proteins and/or by neutralizing BH1234 proteins. The so-called ‘BH3 mimetics', pharmacological compounds that bind to BH3 receptors, can induce apoptosis or facilitate apoptosis induction in cancer cells (Letai et al, 2002; Oltersdorf et al, 2005).
Beclin-1 (also called ATG6) is a phylogenetically conserved protein that is essential for the initiation of autophagy, perhaps via its interaction with the class III phosphatidylinositol-3-kinase Vps34 (Zeng et al, 2006). Originally, human Beclin-1 has been identified as an interactor of Bcl-2 (Liang et al, 1999). Caenorhabditis elegans Beclin-1 (BEC-1) interacts with the Bcl-2 homolog CED-9, and inactivation of the C. elegans bec-1 gene causes apoptosis (Takacs-Vellai et al, 2005). In mammalian cells, knockdown of beclin-1 sensitizes to apoptosis induction by starvation (Boya et al, 2005; Lum et al, 2005). However, Beclin-1 downregulation can also inhibit cell death induction by conditions in which essential pro-apoptotic MOMP or caspase activation are blocked (Shimizu et al, 2004; Yu et al, 2004), and restoration of normal Beclin-1 levels in Beclin-1 deficient tumor cells can facilitate the induction of cell death by vitamin D analogues (Hoyer-Hansen et al, 2005). Importantly, beclin-1 is a haploinsufficient tumor suppressor gene (Qu et al, 2003; Yue et al, 2003). Transfection-enforced overexpression of Beclin-1 stimulates autophagy, and this autophagy-stimulatory effect is enhanced by depletion of Bcl-2 and reduced by Bcl-2 overexpression (Pattingre et al, 2005).
Based on these premises and incognita, we explored the fine mechanisms which govern the interaction between Beclin-1 and Bcl-2/Bcl-XL. As shown here, Beclin-1 possesses a BH3-like domain, thus elucidating the structural basis for the functional Beclin-1-Bcl-2/Bcl-XL interaction. Disrupting this interaction by BH3-only proteins or BH3 mimetics increases the autophagic activity of Beclin-1, thus revealing a novel physiological role for BH3 domains.
Results
Identification of a BH3-like domain in Beclin-1
In multiple yeast two-hybrid screens, using complex human DNA libraries, a number of Beclin-1 fragments interacted with Bcl-2 as well as with Bcl-XL, allowing us to narrow down the precise interaction domain of Beclin-1 to amino acids (aa) 112 to 159 (Figure 1A). A stretch of 10 amino acids (aa 114–123) within this domain showed significant similarity with BH3 domains from the Bcl-2 family (Figure 1B). An eicosahexapeptide (aa 107–132) comprising this BH3-like domain in an α-helical conformation displaced a Bak-derived BH3-containing peptide binding to the hydrophobic groove of purified recombinant Bcl-XL measured in a fluorescence anisotropy assay. Mutation of the most conserved residue in the BH3-like domain of Beclin-1 (L116A) as well as that of another amino acid (F123A) (Pattingre et al, 2005) abolished the competition with Bak-BH3 for Bcl-XL binding (Figure 1C). The affinity of the wild-type BH3 peptide from Beclin-1 for Bcl-XL was high (Kd 203±6 nM, n=3) (Figure 1D) and of an order of magnitude similar to Bax-BH3 (145 nM) or Bak-BH3 (40 nM), although lower than Bad-BH3 (10 nM) (data not shown). In contrast Bcl-XL carrying a single mutation (G138A) in the BH3 binding groove (Ottilie et al, 1997) was unable to interact with the Beclin-derived BH3-like peptide.
Figure 1.
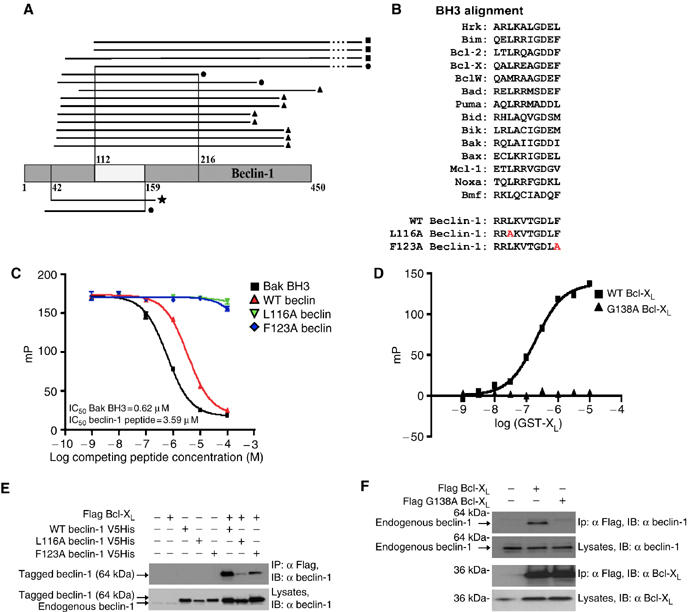
Physical interaction between Bcl-XL/Bcl-2 and the BH3-like domain of Beclin-1. (A) Selection of Beclin-1 fragments that interact with Bcl-XL/Bcl-2 in the yeast-two-hybrid system. Black lines indicate the fragments of Beclin-1 (grey) that interact with Bcl-XL (upper part of the graph) or with Bcl-2 (lower part). ▪, CEMC7 library; •, human thymocyte library; ▴, placenta library and ★, human brain library. The minimal interacting domain required for interaction with Bcl-XL/Bcl-2 is situated between aa 112 and 159. (B) Alignment of the BH3-like domain of Beclin-1 with established BH3 domains. L116A Beclin-1 and F123A Beclin-1 describe two Beclin-1 mutant peptides. (C) Fluorescence polarization assays demonstrating that a peptide containing the BH3-like domain of Beclin-1 interacts with Bcl-XL. Recombinant Bcl-XL ΔTM was incubated with carboxyfluorescein-labeled Bak BH3 peptide, in the absence or presence of wild-type (WT) Beclin-1 BH3 or either of the two mutant peptides described in B. (D) Affinity determination of the interaction between recombinant Bcl-XL ΔTM and fluorescent Beclin-1 BH3, by fluorescence polarization. A comparison between WT Bcl-XL(squares) and G138A Bcl-XL (triangles) is shown (means±s.d., n=3 separate experiments). (E) Co-immunoprecipitation of Bcl-XL and WT or mutant Beclin-1. HeLa cells were transfected with the indicated constructs, followed by immunoprecipitation of Flag-tagged Bcl-XL and immunochemical detection of Beclin-1. (F) Co-immunoprecipitation of Beclin-1 with-WT and mutant (G138A) Bcl-XL. Results are representative of at least three experiments yielding similar results.
To corroborate the role of the BH3-like domain for the Beclin-1 interaction with Bcl-XL, cells were transfected with wild-type or mutant Beclin-1 and/or with Bcl-XL, followed by co-immunoprecipitation assays. The BH3-disrupting mutation L116A almost abolished the interaction of Beclin-1 with Bcl-XL (Figure 1E), as well as that of Beclin-1 with Bcl-2 and Mcl-1 (not shown). Moreover, F123A mutation reduced the Beclin-1-Bcl-XL interaction (Figure 1E). The G138A mutation within the BH3-binding cleft of Bcl-XL abrogated the binding of Beclin-1 in the cellular context (Figure 1F). These results suggest that a novel BH3-like domain in Beclin-1 is critical for the interaction of Beclin-1 with anti-apoptotic members of the Bcl-2 family. The BH3-like domain of Beclin-1 is phylogenetically conserved, because the Beclin-1 orthologs from fugu (Takifugu rubripes), latipes (Oryzia latipes) and zebrafish (Danio rerio) exhibit sequence homology within their BH3-like domain with human Beclin-1 (Supplementary Figure 1A). Peptides corresponding to these BH3-like domains induced apoptosis in a Bax-dependent manner when they were microinjected into human HCT116 cells (Supplementary Figure 1B and C), and the peptides from fugu and latipes (but not the ones from zebrafish) are able to displace the Bak BH3 peptide from recombinant Bcl-XL or Bcl-2 protein in vitro (Supplementary Figure 1D and E). The fact that the zebrafish-derived Beclin-1 peptide did not bind to human Bcl-XL yet induced Bax-dependent apoptosis, suggests that this particular peptide induces cell death by acting on a multidomain Bcl-2 family protein other than Bcl-XL.
Inhibition of the Beclin-1-Bcl-XL/Bcl-2 interaction by a BH3 mimetic
The BH3-mimetic compound ABT737 (Oltersdorf et al, 2005), inhibits the binding of Beclin-1 BH3 peptide to Bcl-XL in a competitive manner, with an IC50 in the micromolar range, as determined by fluorescence polarization of synthetic peptide binding to purified recombinant Bcl-XL in vitro (Figure 2A). Pretreatment of cells with ABT737 inhibited the co-immunoprecipitation of Flag-tagged Bcl-XL and His-tagged (Figure 2B) or endogenous Beclin-1 (Figure 2C). ABT737 also reduced the interaction between Bcl-2 and Beclin-1 (Figure 2D), yet had no effect on the interaction between Mcl-1 and Beclin-1 (Figure 2E). This can be explained by the selectivity of ABT737, which binds to Bcl-2 and Bcl-XL but not to Mcl-1, and hence has a Bad-like profile (Oltersdorf et al, 2005, Van Delft et al, 2006). It is important to note that ABT737 also abolished the interaction between endogenous Bcl-2 and Beclin-1 (see below), meaning that these effects cannot be attributed to overexpression-associated artifacts. Altogether, these data indicate that the BH3-like domain of Beclin-1 binds to the BH3 receptor region of Bcl-XL/Bcl-2 and that ABT737 competitively disrupts this interaction.
Figure 2.
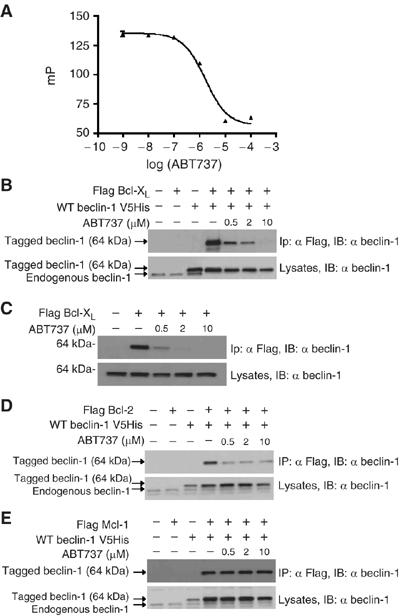
Inhibition of the interaction between Beclin-1 and anti-apoptotic Bcl-2 proteins by ABT737. (A) Competition between Beclin-1 BH3 and ABT737 for Bcl-XL binding. A fluorescent 25-mer peptide containing the BH3-like domain of Beclin-1 (Figure 1D) was docked to recombinant Bcl-XL ΔTM protein in the absence or presence of ABT737. The IC50 of ABT737, as measured in the presence of 15 nM of peptide and 100 nM of Bcl-XL ΔTM, was 1.7 μM. (B, C) Abolition of the interaction between Bcl-XL and Beclin-1 by ABT737 in intact cells. Co-immunoprecipitation assays were performed on HeLa cells transfected 48 h earlier with the indicated constructs as in Figure 1E. Sixteen hours before the immunoprecipitation, cells were exposed to ABT737. Similar results were obtained for co-transfected Bcl-XL and Beclin-1 (B) and for endogenous Beclin-1 interacting with Flag-tagged Bcl-XL (C). (D, E) Differential effect of ABT737 on the interaction between Bcl-2 (D) or Mcl-1 (E) and Beclin-1. This experiment was designed as (B). All experiments have been performed at least three times, with similar results.
The BH3 mimetic ABT737 stimulates Beclin-1-dependent autophagy
If the physiological function of the physical Beclin-1-Bcl-XL/Bcl-2 interaction were to control Beclin-1-initiated autophagy (Pattingre et al, 2005), then inhibition of this interaction would be expected to stimulate autophagy. Accordingly, ABT737 increased the frequency of cells that manifested cytoplasmic (non-nuclear) aggregation of the marker of autophagic vacuoles, LC3-GFP. This aggregation of LC3-GFP (which, in non-autophagic cells, is diffuse in the cytosol as well as in the nucleus) is an established sign of autophagy (Mizushima et al, 2004), and was induced both in complete medium as well as in conditions of nutrient depletion (Figure 3A). Knockdown of Beclin-1 by two distinct small interfering RNA (siRNA) heteroduplexes (insert in Figure 3B) inhibited the ABT737-stimulated LC3-GFP aggregation (Figure 3A and B). Similarly, knockdown of other essential ATG proteins (ATG5, 10, 12) reduced ABT737-induced LC3-GFP aggregation, confirming that ABT737 engages the classical autophagic pathway (Supplementary Figure 2S). Since LC3-GFP might interfere with the normal regulation of autophagy, cytoplasmic vacuoles (which are bona fide autophagic vacuoles) were detected by staining with CMFDA, without prior transfection with LC3-GFP. Again, ABT737 induced signs of autophagy that could be completely suppressed by the knockdown of Beclin-1 (Figure 3C and D). These results could be confirmed by transmission electron microscopy showing double-membraned autophagic vacuoles that were elicited by ABT737 and suppressed by Beclin-1-specific siRNAs (Figures 3E and 4). The ABT737-stimulated autophagy was organelle-specific in the sense that LC3-GFP colocalized with the mitochondrial marker HSP60 (Supplementary Figure 3A and C) but not with the endoplasmic reticulum (ER) marker calreticulin (Supplementary Figure 3B and C). It is noteworthy that these results were obtained by observing viable, adherent cells (Figure 3A–E) and that the cytotoxic pro-apoptotic effects of ABT737 were minor. Thus, ABT737 failed to induce a major loss of the mitochondrial transmembrane potential (which may be associated with apoptosis or necrosis) (Figure 3F) or phosphatidylserine exposure (which is associated with apoptosis) (Supplementary Figure 4), unless Beclin-1 was depleted simultaneously. This is in accord with the notion that ABT737 as a single agent only kills a limited set of transformed cell lines (Oltersdorf et al, 2005). ABT737 can stimulate autophagy without inducing cell death.
Figure 3abcd.
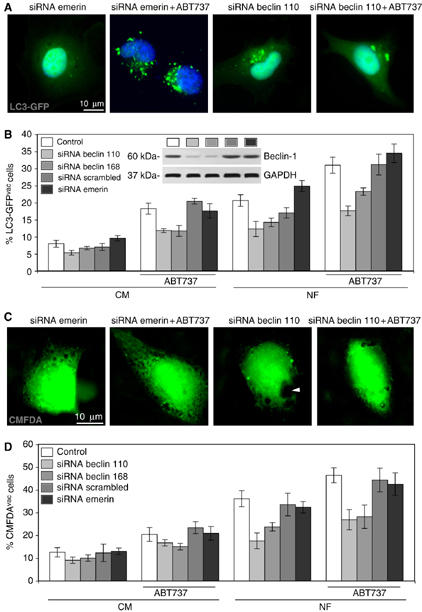
Beclin-1-dependent autophagy stimulated by ABT737. (A, B) Detection of autophagic vacuoles by LC3-GFP and their modulation by ABT737 and by Beclin-1-specific siRNAs. HeLa cells were transfected with control or Beclin-1-specific siRNAs, 24 h later re-transfected with LC3-GFP, cultured in complete medium (CM) for 24 h, and finally kept 12 h either in CM or in nutrient-free (NF) conditions, in the presence or absence of 1 μM ABT737. Representative microphotographs of cells cultured in NF medium are shown in (A) and the percentage (means±s.d., n=3 separate experiments) of LC3-GFP-transfected cells bearing LC3-GFP aggregates in the cytoplasm (LC3-GFPvac) are quantified in (B). The insert in (B) demonstrates the efficiency of the Beclin-1-specific siRNAs, as quantified by immunoblot. (C, D) Detection of cytoplasmic vacuoles using chloromethylfluorescein diacetate (CMFDA). Cells were transfected with control or Beclin-1-specific siRNAs, cultured for 48 h in CM, washed, cultured in CM (D) or NF (C, D) for 12 h, stained with CMFDA, and either photographed (C) or subjected to the quantification of the cells that bear at least one discernible cytoplasmic vacuole (arrow head) (means±s.d., n=3 separate experiments).
Figure 3ef.
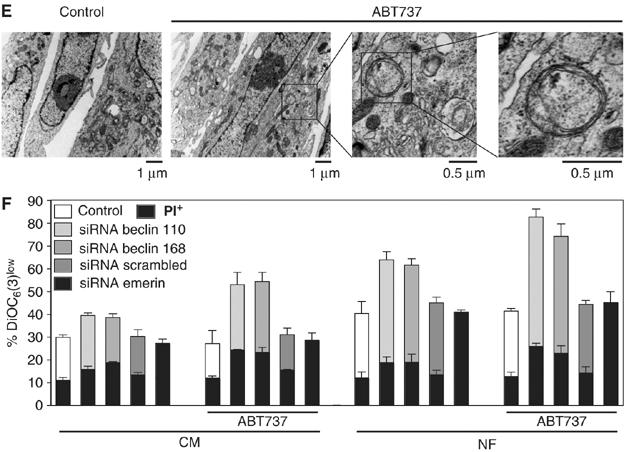
(E) Ultrastructure of autophagic vacuoles induced by ABT737. Transmission electron microphotographs are shown. (F) Detection of dead and dying cells in the cultures. Cells treated as in (A) were stained with the ΔΨm-sensitive dye DiOC6(3) and the vital dye propidium iodide (PI). The black portions of the columns refer to the DiOC6(3)low PI+ population (dead) and the remaining part of the column corresponds to the DiOC6(3)low PI− (dying) population. Results are means±s.d. of three independent experiments.
Figure 4.
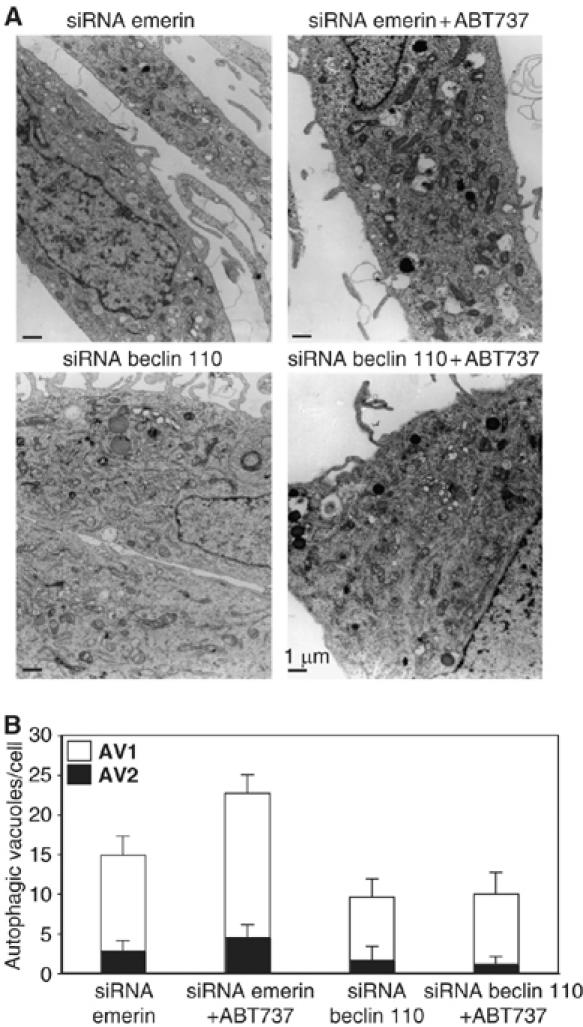
Quantification of autophagic vacuoles induced by ABT737. HeLa cells transfected with the indicated siRNAs (specific for Emerin or for Beclin-1 at 0 h) were re-transfected with LC3-GFP finally cultured in nutrient-free (NF) conditions (60–72 h), in the presence or absence 1 μM ABT737 and then subjected to electron microscopic detection of immature (AV1) or mature (AV2) autophagic vacuoles. Representative pictures are shown in (A). The number of AV1 and AV2 was determined for a minimum of 50 cells (means±s.e.m.) (B).
BH3 dependency of the functional interaction between Beclin-1 and Bcl-XL/Bcl-2
Overexpression of Beclin-1 stimulates autophagy (Liang et al, 1999; Hoyer-Hansen et al, 2005; Pattingre et al, 2005), and this effect was increased by ABT737 (Figure 5A and B). The Beclin-1 mutants L116A and F123A were more efficient than wild-type Beclin-1 in stimulating autophagy, in line with the fact that these mutations disrupt the interaction with the Beclin-1 inhibitors Bcl-2/Bcl-XL. This result was obtained when using two different read-outs, namely LC3-GFP aggregation (Figure 5A and B) and CMFDA-quantifiable vacuolization (Figure 5C and D). In the presence of ABT737, the differential capacity of wild-type Beclin-1 and of its mutants L116A and F123A to induce autophagy was matched, suggesting that it was indeed the interaction between the BH3-like domain of Beclin-1 and a BH3 receptor (inhibitable by ABT737) that regulated the pro-autophagy activity of Beclin-1. These data also suggests that endogenous Mcl-1 (which is not inhibitable by ABT737) (van Delft et al, 2006) does not play a crucial role in controlling autophagy induced by overexpressed Beclin-1. Moreover, Bcl-XL (but not Bcl-XL G138A), Bcl-2 and Mcl-1 all inhibited the induction of autophagy by Beclin-1 (Figure 5E). The autophagy-inhibitory effect of Bcl-XL and Bcl-2 was abrogated by ABT737. However, ABT737 did not affect the suppression of autophagy by Mcl-1 (Figure 5E), in accord with its incapacity to block the Beclin-1–Mcl-1 interaction (Figure 2E). Altogether, these data suggest that ABT737 stimulates (or de-inhibits) autophagy by competitively inhibiting the interaction between the BH3 domain of Beclin-1 and the BH3 receptor regions of Bcl-2/Bcl-XL.
Figure 5.
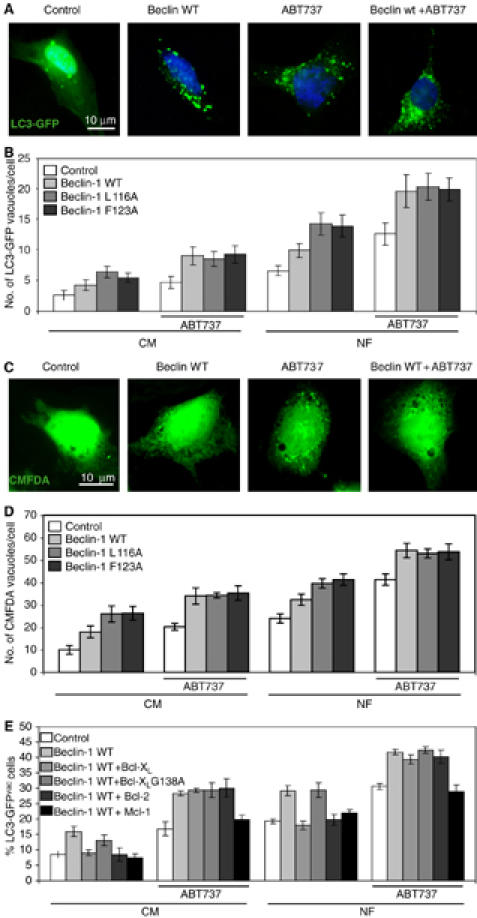
Impact of the BH3-like domain on Beclin-1-induced autophagy. (A–D) Modulation of Beclin-1-induced autophagy by BH3 mutants and ABT737. Cells were transfected with GFP-LC3 together with an empty control vector or Beclin-1 (WT, L116A, F123A) and cultured for 48 h, followed by overnight culture in CM (B) or NF (A, B) in the absence or presence of 1 μM ABT737. Representative microphotographs are shown in (A) and the LC3-GFP-positive vacuoles per cell are quantified in (B) (means±s.d.; n=3 separate experiments). Alternatively, cells were not transfected and stained with CMFDA to detect vacuoles, as shown in (C) and (D), yielding similar results as for the LC3-GFP method. (E) BH3-dependent modulation of Beclin-1-induced autophagy by anti-apoptotic Bcl-2 proteins. Cells were transfected simultaneously with Beclin-1 (or control vector) and equivalent amounts of plasmids coding for WT Bcl-XL, Bcl-XL G138A, Bcl-2 or Mcl-1 (or control vector), as well as GFP-LC3. The culture in CM or NF in the absence or presence of ABT737 was performed during the last 12 h of the 60 h-long experiment. The percentage of cells exhibiting the accumulation of LC3-GFP in vacuoles (LC3-GFPvac) is quantified as means±s.d. (n=3 separate experiments).
Spatially restricted, regulated interactions between Bcl-2 and Beclin-1 are inhibited by ABT737
ABT737 as well as nutrient depletion reduced the amount of endogenous Beclin-1 that immunoprecipitated with endogenous Bcl-2, both in HeLa and in MV4.11 cells (Figure 6A), confirming that physiological levels of these proteins can interact in a fashion that is inhibited by nutrient depletion or ABT737. Bcl-2 associates both with ER and mitochondrial membranes (Germain and Shore, 2003). Wild-type and ER-targeted Bcl-2 (Bcl-2-Cb5) but not mitochondrion-targeted Bcl-2 (Bcl-2-Acta) inhibits starvation-induced autophagy (Pattingre et al, 2005), suggesting that the autophagy-regulatory pool of Bcl-2 is localized on ER. To investigate this, we determined the inhibitory effect of ABT737 and nutrient depletion on the Bcl-2-Beclin-1 interaction in cell lines that stably express wild-type Bcl-2, Bcl-2-Cb5, or Bcl-2-Acta. The amount of Beclin-1 that co-immunoprecipitated with wild type and Bcl-2-Cb5 diminished after treatment with ABT737 or starvation, whereas the amount of Beclin-1 that co-immunoprecipiated with Bcl-2-Acta remained constant (Figure 6B). These results could be further substantiated by subcellular fractionation. The interaction between wild-type Bcl-2 and Beclin-1 measurable in microsomes (ER) was reduced by ABT737 or starvation, but remained constant within the heavy membrane fraction (mitochondria) (Figure 6C and D). Hence, only the ER-targeted pool of Bcl-2 is relevant to the inhibition of autophagy.
Figure 6.
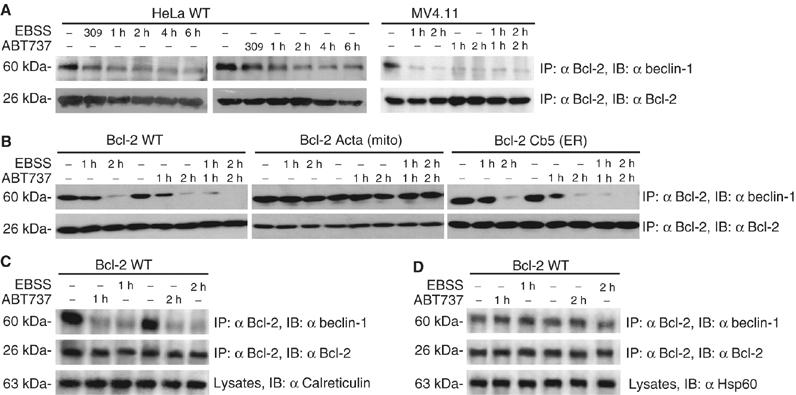
Interaction between endogenous and spatially restricted Bcl-2 and Beclin-1 in starvation and after addition of ABT737. (A) Immunoprecipitation of endogenous Bcl-2 and Beclin-1. Untransfected HeLa or MV4.11 cells were treated by culture in nutrient-free EBSS or with ABT737 (1 μM). (B) Immunoprecipitation of wild-type and ER-targeted Bcl-2 with Beclin-1. Cells stably transfected with wild type, ER- and mitochondrion-targeted Bcl-2 were subjected to starvation or treated with ABT737, followed by immunoprecipitation of Bcl-2 and immunodetection of Beclin-1. (C, D) Immunoprecipitation of Bcl-2 and Beclin-1 in subcellular fractions. Cells treated as in (B) were lysed and subjected to subcellular fractionation to enrich either ER vesicles (microsomes, (C)) or mitochondria (heavy membrane fraction, (D)), followed by immunoprecipitation of Bcl-2 and immunodetection of Beclin-1. Calreticulin and Hsp60 were used as internal controls. Results are representative of three independent determinations.
Starvation-induced induction of autophagy via the BH3-only protein Bad
The results above imply that, to the very least in certain instances, induction of Beclin-1-mediated autophagy should correlate with its release from inhibitory complexes. Upon starvation, the amount of endogenous Beclin-1 that co-immunoprecipitated with Bcl-XL declined within the first hour of serum and nutrient withdrawal, whereas the amount of the BH3 protein Bad (whose activation is known to be triggered by serum withdrawal) (Danial and Korsmeyer, 2004) that co-immunoprecipitated with Bcl-XL increased (Figure 7A). In contrast, addition of rapamycin (which induces autophagy by inhibition of mTOR) (Sarbassovdos et al, 2005) or other autophagy inducers that affect the level of phosphatidyl inositol-3-phoshate (Sarkar et al, 2005) had less marked effects on the interactions between Beclin-1, Bcl-XL and Bad (Figure 7B). Upon starvation (but not upon ABT737 addition), the amount of endogenous Bad that immunoprecipitated with endogenous Bcl-2 also increased (Figure 7C), at the same time as the Bcl-2–Beclin-1 interaction was reduced (Figure 6A). The siRNA-mediated depletion of Bad (Figure 7D) reduced the starvation-induced activation of autophagy, yet had no or little effect on rapamycin-induced autophagy, whereas depletion of Vps34 inhibited both starvation and rapamycin-induced autophagy (Figure 7E). Bad−/− mouse embryonic fibroblasts (MEF) (Ranger et al, 2003) also exhibited a decreased starvation-induced autophagy as compared with WT MEF, although this difference disappeared in the presence of ABT737 (Figure 7F). The absence of Bad compromised the disruption of the Beclin-1/Bcl-2 interaction induced by starvation, yet had no effect on the disruption of this interaction by ABT737 (Figure 7G). The effect of Bad depletion/deletion on starvation-induced autophagy (Figure 7D and F) and on the changing Beclin-1/Bcl-2 interaction (Figure 7G) was partial, suggesting that Bad is not the sole BH3 protein linking starvation to autophagy induction or that BH3-independent mechanisms may be involved. Transfection-enforced overexpression of Bad was sufficient to induce autophagy both in normal conditions and conditions of caspase inhibition (Figure 7H). These results indicate that BH3-only proteins including Bad may participate in the activation of autophagy by starvation.
Figure 7.
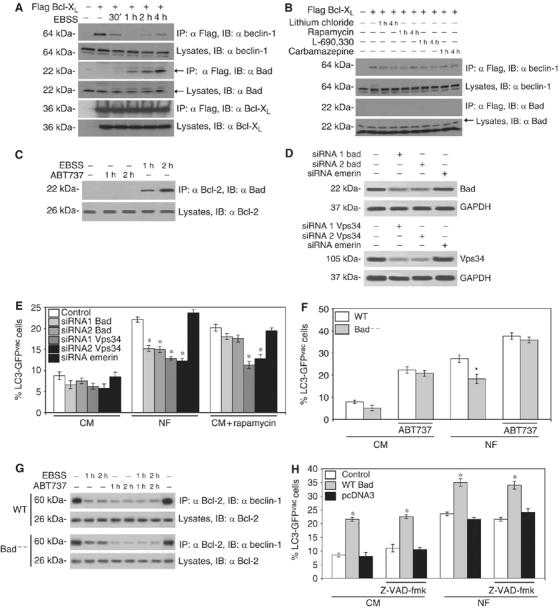
Impact of the BH3-only protein Bad on autophagy. (A, B) Interactions between Bcl-XL, Beclin-1 and Bad in conditions of autophagy induction. Cells were either treated by nutrient depletion (A) or addition of 1 μM rapamycin, 1 mM lithium chloride, 100 μM L-690,330 or 50 μM carbamazepine (B), followed by immunprecipitation of Bcl-XL (as in Figure 2C) and revealing the immunoblots by antibodies specific for Bcl-XL, Beclin-1 or Bad. (C) Interaction between endogenous Bad and Bcl-2. HeLa cells were treated with ABT737 (1 μM) or nutrient-depleted, followed by immunoprecipitation of Bcl-2 and immunodetection of Bad. (D, E) Impact of Bad depletion on autophagy. Cells were transfected with LC3-GFP together with siRNAs specific for Bad, siRNAs specific for emerin or the Beclin-1-associated PI3 kinase Vps34. Forty-eight hours later, when the siRNAs had down-regulated the proteins of interest (D), cells were subjected by nutrient depletion (NF) or addition of rapamycin (E) and the frequency of cells exhibiting LC3-GFP aggregation as a marker of autophagy was assessed after 18 h (means±s.d., *P<0.05, n=3 separate experiments). (F) Effect of the Bad knock-out on autophagy. Wild Type (WT) or Bad−/− MEF were transfected with LC3-GFP and then subjected to nutrient depletion (NF) and/or treatment with ABT737 (18h; means±s.d., n=3). The asterisks denotes a significant (P<0.05) effect of Bad deficiency. (G) Effect of the Bad knockout on the Beclin-1/Bcl-2 interaction. WT or Bad−/− MEF were subjected to the indicated treatments, followed by immunoprecipitation of Bcl-2 and immunobloting of Beclin-1. (H) Overexpression of Bad triggers autophagy. Cells were transfected with LC3-GFP alone (Control), together with vector-only or with a vector encoding Bad, and subjected 48 h later to nutrient depletion (NF). Cells were cultured in the continuous presence or absence of the pan-caspase inhibitor Z-VAD-fmk (50 μM) for 16 h, followed by assessment of autophagy as in (E) (means±s.d., n=3 separate experiments, *P<0.01). Immunoblots are representative of at least three independent experiments.
Regulation of autophagy by the BH3-only protein EGL-1 in C. elegans
EGL-1 is the sole pro-apoptotic BH3-only protein in C. elegans and is required for developmental cell death in this nematode (Conradt and Horvitz, 1998, 1999). Hence, this model organism is uniquely suitable to determine the phylogenetically conserved regulation of autophagy by BH3-only proteins. In C. elegans, lgg-1 (the nematode orthologue of Atg8/LC3) is ubiquitously expressed throughout development and induced in conditions of autophagy (Melendez et al, 2003). Autophagy was monitored using an LGG-1∷DsRED reporter in embryos that carry a wild-type, gain-of-function or deletion allele of egl-1, in normal conditions or after starvation. Starvation strongly induced autophagy, and this induction was blunted in egl-1-deficient nematode embryos. In contrast, the gain-of-function mutation of egl-1 caused an increase in constitutive autophagy that was not enhanced further by starvation (Figure 8). These results underscore the phylogenetic conservation of autophagy control by BH3-only proteins.
Figure 8.
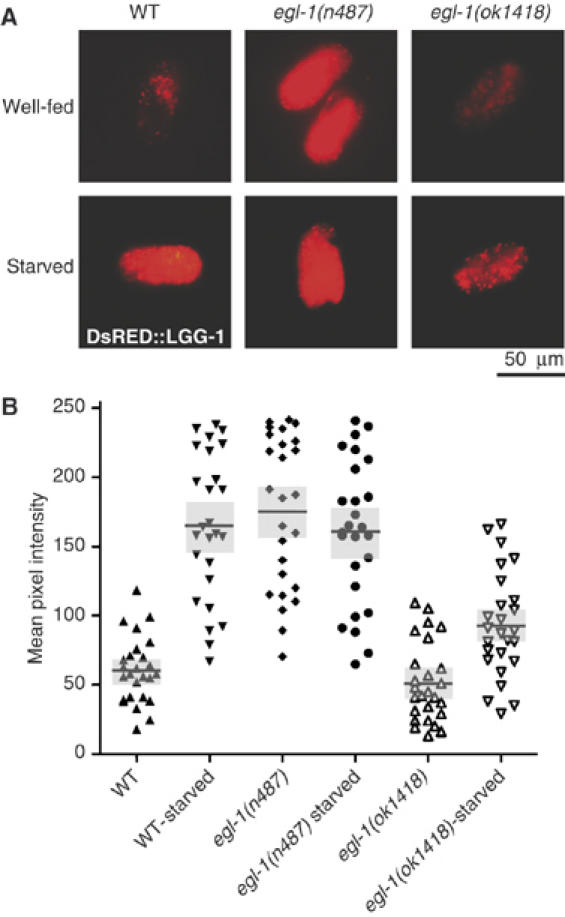
The BH3-domain protein EGL-1 modulates starvation-induced autophagy in C. elegans. L4 Larvae expressing an LGG-1∷DsRED reporter (whose expression level provides an indication of autophagy) as well as a gain-of-function mutation of egl-1 (n487) or a deletion allele of egl-1 (ok1418) were maintained in rich medium or starved overnight. (A) Representative images of wt and egl-1 mutant embryos, bearing the plgg−1DsRED∷LGG-1 reporter transgene are shown under conditions of normal growth and under starvation. Scale bars denote approximately 50 μm. (B) Quantification of LGG-1∷DsRED fluorescence as determined in (A). Each point represents the measurement for one individual embryo. Horizontal bars denote means and gray areas denote standard errors.
Discussion
As shown here, Beclin-1 possesses a BH3 domain that dictates its interaction with the BH3 receptor domain of anti-apoptotic proteins of the Bcl-2 family including Bcl-2, Bcl-XL and Mcl-1. A recent report, which was published when this paper was under review, revealed the crystallographic structure of human Bcl-XL interacting with a peptide (aa 107–135) derived from human Beclin-1 and corroborated the hypothesis that Beclin-1 possesses a BH3-like amphipathic α-helix that can bind to the conserved hydrophobic groove of Bcl-XL (Oberstein et al, 2007). As shown here, this interaction is physiologically important because its inhibition by the BH3-only protein Bad and by a Bad-like BH3 mimetic can stimulate autophagy.
Beclin-1 possesses a BH3 domain that, in the form of a synthetic peptide excised from the context of the whole protein, induces Bax-dependent apoptosis. Upon overexpression, however, it promotes Bcl-XL-inhibitable autophagy rather than apoptosis, Beclin appears therefore as a new type of BH3 protein, in which this domain plays a regulatory, rather than an effector function.
The prevalence of the autophagic activity of BH3-containing Beclin-1 and its failure to induce apoptosis in an in vivo context raises important mechanistic questions. Regulatory sequences in the Beclin-1 full-length protein, or the structure of the Beclin-1 BH3 domain itself, may normally prevent the BH3 motif in Beclin-1 from activating apoptosis, perhaps because of different affinities between BH3-only proteins for anti-apoptotic Bcl-2 family members in a cellular context. Indeed, in the recent report of the structure of Beclin-1 BH3 peptide binding to Bcl-XL, interaction features suggest a distinct binding specificity (Oberstein et al, 2007). Alternatively, there could be different pools of anti-apoptotic multidomain Bcl-2 proteins, some sequestering pro-apoptotic Bcl-2 family members, others Beclin-1, and that these may be selectively displaced by other BH3 proteins, again perhaps reflecting their different affinities and/or subcellular compartmentalization.
In favour of this latter possibility, we found that ABT737 as well as nutrient withdrawal only affected the interaction between Beclin-1 and ER-localized Bcl-2. The mechanisms through which ABT737 may effect the interaction between ER-localized Bcl-2-Beclin–1 complexes (but not those found on mitochondria) are elusive. On the one hand, the affinity of ABT737 for Bcl-2 may be influenced by post-transcriptional alterations (such as Bcl-2 phosphorylation; Konopleva et al, 2006) that may correlate with its subcellular localization (Ruvolo et al, 1998). On the other hand, the nature of the physicochemical interaction between Bcl-2 and Beclin-1 might be conditioned by the presence of additional organelle-specific proteins, such as IP3R (1,4,5-inositol trisphosphate receptor) in ER (Chen et al, 2004; Criollo et al, 2007) and VDAC (voltage-dependent Anion Channel) in mitochondria (Shimizu et al, 1999). While mitochondrial Bcl-2 and Bcl-XL could be the preferential target for the pro-apoptotic function of BH3-only proteins, ER-resident Bcl-2-Beclin-1 complexes could be selectively disrupted by competing BH3 domains (such as that of Bad) or BH3 mimetics for the induction of autophagy, which is agreement with the findings of Pattingre et al (2005) showing that only ER-targeted Bcl-2 inhibits autophagy.
It is noteworthy that the regulation of autophagy by BH3-only proteins is phylogenetically old. This does not only apply to the conservation of the BH3-like domain of Beclin-1 in non-mammalian species but extends to the observation that EGL-1, the sole BH3-only protein of C. elegans, suffices to trigger autophagy and is a mediator of starvation-induced autophagy. It should be noted that loss-of-function mutants of egl-1 (in C. elegans) and depletion/deletion of Bad (in human and mouse cells) did not completely abolish starvation-induced autophagy, in line with the presence of redundant regulatory pathways (Shintani and Klionsky, 2004) in which BH3-only proteins play a prominent but not exclusive role.
By virtue of its BH3-mimetic action, ABT737 exerts two distinct cellular effects. On the one hand, it inhibits the anti-apoptotic action of Bcl-2 or Bcl-XL, as established previously (Oltersdorf et al, 2005), thus triggering apoptosis in cells in which neutralization of Bcl-2 or Bcl-XL is sufficient to de-inhibit the mitochondrial cell death pathway. On the other hand, ABT737 stimulates autophagy, which can be viewed as a cytoprotective mechanism (Boya et al, 2005; Lum et al, 2005). Although at present no clinically applicable autophagy inhibitors are available, these results point to the possibility of using autophagy inhibitors for sensitizing tumor cells to BH3 mimetics or indirect activators of BH3-dependent apoptotic pathways.
In conclusion, our data support a novel function of the BH3-binding site of Bcl-2 homolog, namely regulation of autophagy. Beyond their established function as cell death inducers, BH3-only proteins or BH3 mimetics can induce autophagy by competitively disrupting the interaction of Beclin-1 with Bcl-2/Bcl-XL. Our findings indicate a hitherto unexpected crosstalk between self-killing and self-eating. Both phenomena are controlled by BH3-only proteins.
Materials and methods
Cell lines and culture conditions
HeLa cells were cultured in DMEM containing 10% fetal calf serum (FCS), 1 mM pyruvate and 10 mM Hepes at 37°C under 5% CO2. MV4.11 cells were cultured in RPMI 1640 medium containing 10% FCS, 1 mM pyruvate, 10 mM Hepes, 100 U/ml penicillin G sodium, 100 μg/ml streptomycin sulfate and 5 ng/ml Granulocyte/Macrophage Colony-Stimulating Factor (GM-CSF) at 37°C under 5% CO2. wild type and Bad−/− MEF (Ranger et al, 2003), as well as cells expressing Bcl-2 Acta or Bcl-2 Cb5 (Zhu et al, 1996) were maintained in DMEM supplemented with 10% FCS, 1 mM pyruvate, 10 mM Hepes and 1% non-essential amino acids (NEAA, SIGMA) at 37°C under 5% CO2. All media and supplements were purchased from Gibco-Invitrogen (Carlsbad, USA). For serum and amino-acid starvation, cells were cultured in serum-free Earle's Balanced Salt Solution medium (Sigma) (Vahsen et al, 2004), optionally supplemented with ABT737 (1 μM; synthesized by Servier).
Plasmids, transfection and RNA interference
Cells were cultured in six-well plates and transfected at 80% confluence with Oligofectamine (Invitrogen), in the presence of 100 nM of siRNAs specific for human Beclin-1 and other atg genes (Boya et al, 2005; Gonzalez-Polo et al, 2005), Bad (siRNA1 from Jin et al, 2004; siRNA2: CUGGGCAGCCAUCUUGAAU dTdT), Vps (siRNA1 from Nobukuni et al, 2005 and siRNA2 from Byfield et al, 2005), a scrambled siRNA or an siRNA targeting the unrelated protein emerin (Harborth et al, 2001). siRNA effects were controlled by immunoblots with suitable antibodies specific for Beclin-1 (SantaCruz), Bad (SantaCruz) or Vps34 (Zymed). Transient transfections with cDNAs were performed with Lipofectamine 2000 reagent (Invitrogen), and cells were used 24 h after transfection unless specified differently. Cells were transfected with empty vector alone or together with LC3-GFP (Kabeya et al, 2000), in the presence or absence of Beclin-1 wild type, Beclin-1 L116A, Beclin-1 F123A, Bcl-XL wild type, Bcl-XL G138A, Bcl-2 wild type, or Mcl-1.
Yeast two-hybrid system
Bcl-XL and Bcl-2 without their C-terminal transmembrane domains (Bcl-XL 1–624and Bcl-2 1–630) were PCR amplified (Pfu, Stratagene) and cloned N-terminal to the lexA DNA binding domain in the pB24 vector. cDNA libraries constructed in pP6 vector (Fromont-Racine et al, 2002) were transformed into the Y187 yeast strain. Ten million independent yeast colonies were collected, pooled and stored at −80°C as equivalent aliquot fractions of the same library. The mating protocol has been described (Fromont-Racine et al, 2002). Each screen was performed to ensure a minimum of 50 million interactions. The prey fragments of the positive clones were amplified by PCR and sequenced at their 5′ and 3′ junctions on a PE3700 Sequencer. The resulting sequences were used to identify the corresponding gene in the GenBank database (NCBI).
Peptides and recombinant protein
Recombinant GST fused to Bcl-XL wild type or Bcl-XL G138A (pGEX 5X vector, Amersham) deleted of the C-terminal 21 aa were produced in Escherichia Coli (BL21) and purified on glutathione–Sepharose resin (Amersham). HPLC-purified Beclin wild-type (GTMENLSRRLKVTGDLFDIMSGQTDV), Beclin L116A (GTMENLSRRAKVTGDLFDIMSGQTDV), Beclin F123A (GTMENLSRRLKVTGDLADIMSGQTDV) and Bak BH3 (GQVGRQLAIIGDDINR) peptides were obtained from Neosystem.
Fluorescence polarization assays
Fluorescence polarization (FP) assays (measured with a Fusion™- Packard equipment) were performed essentially as described (Letai et al., 2002). Briefly, for competition experiments, fluorescent Bak BH3 peptide (15 nM) and GST- Bcl-XL (100 nM) were mixed in binding buffer (20 mM Na2HPO4 pH 7.4, 50 mM NaCl, 1 mM EDTA and 0.05% pluronic acid) with titrations of competing peptides (Beclin wild type, Beclin L116A, Beclin F123A or Bak BH3, from 10−9 to 10−4 M). For affinity constant (Kd) calculations, fluorescent Beclin peptide (15 nM) was mixed with increasing amounts of GST- Bcl-XL wild type or GST- Bcl-XL G138A (10−9–10−5 M).
Co-immunoprecipitation experiments
HeLa cells were transiently transfected using Effecten reagent (Qiagen) with pcDNA3topoD vectors (Invitrogen) expressing V5-His-tagged Beclin constructs (Wild type, L116A or F123A) alone or together with Flag-tagged Bcl-XL wild type, Bcl-XL G138A, Bcl-2 wild type or Mcl-1 wild type (p3Xflag vectors, Sigma). Twenty-four hours later, cells were collected and lysed in RIPA buffer. The cleared lysates were then subjected to immunoprecipitation with anti-Flag beads (Sigma). The immunoprecipitates were analysed by immunoblot with anti-Beclin antibody (Santa Cruz, H300), anti-Bcl-XL (Transduction laboratory) or anti-Mcl-1 (SantaCruz, S19). HeLa cells, MV4.11, WT and Bad−/− MEF and cells expressing Bcl-2 Acta or Bcl-2 Cb5 (10 × 106 cells) were collected, lysed and/fractionated (FOCUS™ SubCell, G Biosciences, USA), and the immunoprecipitation was performed (Chen et al, 2004) using anti-Bcl-2 (Santa Cruz) antibody and protein G-Sepharose (GE Healthcare), followed by immunoblot detection of Beclin-1 and Bad (antibodies from SantaCruz).
Western blot analysis
Cells were washed with cold PBS at 4°C and lysed. Forty μg of protein were loaded on a 10% SDS–PAGE and transferred to nitrocellulose. The membrane was incubated for 1 h in PBS-Tween 20 (0.05%) containing 5% nonfat milk. Primary antibody anti-Beclin1 (SantaCruz), Bad (SantaCruz), hVps (Zymed) was incubated for 15 h at 4°C and revealed with the appropriate horseradish peroxidase-labeled secondary antibodies (Southern Biotechnologies Associates) plus the SuperSignal West Pico chemoluminiscent substrate (Pierce). Anti-GAPDH (Chemicon), anti-Hsp60 (Sigma) or anti-calreticulin (Stressgen) antibody was used to ensure equal loading.
Flow cytometry
The following fluorochromes were employed to determine apoptosis-associated changes by cytofluorometry: 3,3′-dihexyloxacarbocyanine iodide (DiOC6(3) (40 nM) for quantification of the mitochondrial transmembrane potential (ΔΨm) and propidium iodide (PI; 1 μg/ml) for determination of cell viability (Molecular probes) (Gonzalez-Polo et al, 2005). Trypsinized cells were labeled with the fluorochromes at 37°C, followed by cytofluorometric analysis with a fluorescence-activated cell sorter (FACS) scan (Becton Dickinson).
Light microscopy, immunofluorescence, and electron microscopy
Cells cultured on coverslips were stained with Cell Tracker Green 5-chloromethylfluorescein diacetate (CMFDA (1 μM); Molecular Probes) and Hoechst 33342 (2 μM; Sigma). Alternatively, cells were fixed with paraformaldehyde (4% w:v) for LC3-GFP and immunofluorescence assays (Obeid et al, 2007). Cells presenting a mostly diffuse distribution of LC3-GFP in the cytoplasm and nucleus were considered as non-autophagic, whereas cells representing several intense punctuate LC3-GFP aggregates with no nuclear LC3-GFP were classified as autophagic. Each LC3-GFP staining was read by two independent investigators (MC Maiuri and A Criollo or E Tasdemir). Transmission electron microscopy was performed as described (Gonzalez-Polo et al, 2005).
Nematode experiments
To generate the plgg−1DsRED∷LGG-1 reporter construct, we fused DsRED to the N-terminus of C. elegans LGG-1. A 750 bp fragment containing the lgg-1 gene was amplified using the following oligos: 5′-GGAATTCGAAGTGGGCTTACAAGGAGGA-3′ and 5′-GGAATTCGTCTTCTTCGTTTATTCATG-3′, and inserted at the C-terminus of DsRED. The reporter fusion was placed under the lgg-1 promoter. To obtain the promoter, we amplified a 1934 bp fragment upstream of the lgg-1 gene using the following oligos: 5′-ACATGCATGCGCACTTTCAAGGCGACAGTA-3′ and 5′-GGGGTACCTCCTCCTTGTAAGCCCACTT-3′. The reporter construct was injected into the gonads of N2 animals together with a plasmid that carries a pmyo−2GFP reporter fusion, expressing GFP in the pharyngeal muscle cells, as a transformation marker. Transgenic males were crossed with egl-1 mutant hermaphrodites to obtain mutants carrying the autophagosomal marker (Brenner, 1974). The C. elegans strains N2: wild-type Bristol isolate, MT1082: egl-1(n487), RB1305: egl-1(ok1418), N2;Ex[plgg−1DsRED∷LGG-1], egl-1(n487);Ex[plgg−1DsRED∷LGG-1] and egl-1(ok1418);Ex[plgg−1DsRED∷LGG-1] were obtained from the C. elegans Knockout Consortium (Robert Barstead, Oklahoma Medical Research Foundation, USA) or the Caenorhabditis Genetics Center (Theresa Stiernagle, University of Minnesota, Minneapolis, USA). To monitor DsRED∷LGG-1 levels in embryos, eggs were obtained from transgenic gravid hermaphrodite adults, expressing plgg−1DsRED∷LGG-1. For starvation experiments, late L4 larval stage animals were grown in the absence of food and eggs were collected between 12–24 h into adulthood.
Transgenic embryos expressing DsRED were photographed on an Axioskop 2 Plus, epifluorescence microscope (Carl Zeiss, Jena, Germany). Images were acquired using a 480±10 nm band-pass excitation filter and a 515±15 nm band-pass emission filter. Experiments were performed at 20°C, with photography exposure time kept identical for each experiment. Emission intensity was measured on greyscale images with a pixel depth of 8 bit (256 shades of gray). The mean and maximum pixel intensity for each embryo in these images (minimum 25 over three independent trials) was determined with ImageJ software (http://rsb.info.nih.gov/ij/).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We thank Bert Vogelstein (Johns Hopkins University, Bethesda, MA) for the gift of Bax-deficient HCT116 cells, Andreas Villunger (University of Innsbruck, Austria) for Bad-negative MEF, David Andrews for Bcl-2 Acta cb5 expressing cells, Abdelali Jalil for confocal microscopy, and Thierry Le Diguarher for synthesis of ABT737. Nematode strains were provided by the C. elegans Gene Knockout Project at OMRF (http://www.mutantfactory.ouhsc.edu/), which is part of the International C. elegans Gene Knockout Consortium, and the Caenorhabditis Genetics Center, which is funded by the NIH National Center for Research Resources (NCRR). GK is supported by Ligue contre le Cancer, European Commission (Active p53, Chemores, TransDeath, Right, Death-Train) and Institut de Recherche Servier. PJ is supported by Institut de Recherche Servier.
References
- Boya P, Gonzalez-Polo R-A, Casares N, Perfettini J-L, Dessen P, Larochette N, Metivier D, Meley D, Souquere S, Pierron G, Ohsumi Y, Codogno P, Kroemer G (2005) Inhibition of macroautophagy triggers apoptosis. Mol Cell Biol 25: 1025–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S (1974) The genetics of Caenorhabditis elegans. Genetics 77: 71–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byfield MP, Murray JT, Backer JM (2005) hVps34 is a nutrient-regulated lipid kinase required for activation of p70 S6 kinase. J Biol Chem 280: 33076–33082 [DOI] [PubMed] [Google Scholar]
- Chen R, Valencia I, Zhong F, McColl KS, Roderick HL, Bootman MD, Berridge MJ, Conway SJ, Holmes AB, Mignery GA, Velez P, Distelhorst CW (2004) Bcl-2 functionally interacts with inositol 1,4,5-trisphosphate receptors to regulate calcium release from the ER in response to inositol 1,4,5-trisphosphate. J Cell Biol 166: 193–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR (1998) The C. elegans protein EGL-1 is required for programmed cell death and interacts with the Bcl-2-like protein CED-9. Cell 93: 519–529 [DOI] [PubMed] [Google Scholar]
- Conradt B, Horvitz HR (1999) The TRA-1A sex determination protein of C. elegans regulates sexually dimorphic cell deaths by repressing the egl-1 cell death activator gene. Cell 98: 317–327 [DOI] [PubMed] [Google Scholar]
- Criollo A, Maiuri MC, Tasdemir E, Vitale I, Fiebig AA, Andrews D, Molgo J, Diaz J, Lavandero S, Harper F, Pierron G, Di Stefano D, Rizzuto R, Szabadkai G, Kroemer G (2007) Regulation of autophagy by the inositol trisphosphate receptor. Cell Death Diff (in press) [DOI] [PubMed] [Google Scholar]
- Danial NN, Korsmeyer S (2004) Cell death: critical control points. Cell 116: 205–219 [DOI] [PubMed] [Google Scholar]
- Fromont-Racine M, Rain JC, Legrain P (2002) Building protein-protein networks by two-hybrid mating strategy. Methods Enzymol 350: 513–524 [DOI] [PubMed] [Google Scholar]
- Germain M, Shore GC (2003) Cellular distribution of Bcl-2 family proteins. Sci STKE 2003: pe10. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Polo RA, Boya P, Paulau A-L, Jalil A-A, Larochette N, Souquere S, Eskelinen EL, Pierron G, Saftig P, Kroemer G (2005) The apoptosis/autophagy paradox. Accumulation of autophagic vacuoles triggers apoptosis. J Cell Sci 118: 3091–3102 [DOI] [PubMed] [Google Scholar]
- Gozuacik D, Kimchi A (2004) Autophagy as a cell death and tumor suppressor mechanism. Oncogene 23: 2891–2906 [DOI] [PubMed] [Google Scholar]
- Harborth J, Elbashir SM, Bechert K, Tuschl T, Weber K (2001) Identification of essential genes in cultured mammalian cells using small interfering RNAs. J Cell Science 114: 4557–4565 [DOI] [PubMed] [Google Scholar]
- Hoyer-Hansen M, Bastholm L, Mathiasen IS, Elling F, Jaattela M (2005) Vitamin D analog EB1089 triggers dramatic lysosomal changes and Beclin 1-mediated autophagic cell death. Cell Death Differ 12: 1297–1309 [DOI] [PubMed] [Google Scholar]
- Jin Z, Gao F, Flagg T, Deng X (2004) Nicotine induces multi-site phosphorylation of Bad in association with suppression of apoptosis. J Biol Chem 279: 23837–23844 [DOI] [PubMed] [Google Scholar]
- Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T (2000) LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J 19: 5720–5728 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konopleva M, Contractor R, Tsao T, Samudio I, Ruvolo PP, Kitada S, Deng X, Zhai D, Shi Y-X, Sneed T, Verhaegen M, Soengas M, Ruvolo VR, McQueen T, Schober WD, Watt JC, Jiffar T, Ling X, Marini FC, Harris D, Dietrich M, Estrov Z, McCubrey J, Stratford May W, Reed JC, Andreeff M (2006) Mechanisms of apoptosis sensitivity and resistance to the BH3 mimetic ABT-737 in acute myeloid leukemia. Cancer Cell 10: 375–388 [DOI] [PubMed] [Google Scholar]
- Kroemer G, Jaattela M (2005) Lysosomes and autophagy in cell death control. Nat Rev Cancer 5: 886–897 [DOI] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ (2002) Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell 2: 183–192 [DOI] [PubMed] [Google Scholar]
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, Levine B (1999) Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 402: 672–676 [DOI] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris HM, Li C, Lindsten T, Thompson CB (2005) Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120: 237–248 [DOI] [PubMed] [Google Scholar]
- Melendez A, Talloczy Z, Seaman M, Eskelinen EL, Hall DH, Levine B (2003) Autophagy genes are essential for dauer development and life-span extension in C. elegans. Science 301: 1387–1391 [DOI] [PubMed] [Google Scholar]
- Mizushima N, Yamamoto A, Matsui M, Yoshimori T, Ohsumi Y (2004) In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15: 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobukuni T, Joaquin M, Roccio M, Dann SG, Kim SY, Gulati P, Byfield MP, Backer JM, Natt F, Bos JL, Zwartkruis FJ, Thomas G (2005) Amino acids mediate mTOR/raptor signaling through activation of class 3 phosphatidylinositol 3OH-kinase. Proc Natl Acad Sci USA 102: 14238–14243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obeid M, Tesniere A, Ghiringhelli F, Fimia GM, Apetoh L, Perfettini JL, Castedo M, Mignot G, Panaretakis T, Casares N, Metivier D, Larochette N, van Endert P, Ciccosanti F, Piacentini M, Zitvogel L, Kroemer G (2007) Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med 13: 54–61 [DOI] [PubMed] [Google Scholar]
- Oberstein A, Jeffrey P, Shi Y (2007) Crystal structure of the BCL-XL–beclin 1 peptide complex: Beclin 1 is a novel BH3-only protein. J Biol Chem [E-pub ahead of print] [DOI] [PubMed] [Google Scholar]
- Oltersdorf T, Elmore SW, Shoemaker AR, Armstrong RC, Augeri DJ, Belli BA, Bruncko M, Deckwerth TL, Dinges J, Hajduk PJ, Joseph MK, Kitada S, Korsmeyer SJ, Kunzer AR, Letai A, Li C, Mitten MJ, Nettesheim DG, Ng S, Nimmer PM, O'Connor JM, Oleksijew A, Petros AM, Reed JC, Shen W, Tahir SK, Thompson CB, Tomaselli KJ, Wang B, Wendt MD, Zhang H, Fesik SW, Rosenberg SH (2005) An inhibitor of Bcl-2 family proteins induces regression of solid tumours. Nature 435: 677–681 [DOI] [PubMed] [Google Scholar]
- Ottilie S, Diaz JL, Chang J, Wilson G, Tuffo KM, Weeks S, McConnell M, Wang Y, Oltersdorf T, Fritz LC (1997) Structural and functional complementation of an inactive Bcl-2 mutant by Bax truncation. J Biol Chem 272: 16955–16961 [DOI] [PubMed] [Google Scholar]
- Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, Levine B (2005) Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122: 927–939 [DOI] [PubMed] [Google Scholar]
- Qu X, Yu J, Bhagat G, Furuya N, Hibshoosh H, Troxel A, Rosen J, Eskelinen EL, Mizushima N, Ohsumi Y, Cattoretti G, Levine B (2003) Promotion of tumorigenesis by heterozygous disruption of the beclin 1 autophagy gene. J Clin Invest 112: 1809–1820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranger AM, Zha J, Harada H, Datta SR, Danial NN, Gilmore AP, Kutok JL, Le Beau MM, Greenberg ME, Korsmeyer SJ (2003) Bad-deficient mice develop diffuse large B cell lymphoma. Proc Natl Acad Sci USA 100: 9324–9329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruvolo PP, Deng X, Carr BK, Stratford May W (1998) A functional role for mitochondrial protein kinase Cα in Bcl-2 phosphorylation and suppression of apoptosis. J Biol Chem 278: 25436–25442 [DOI] [PubMed] [Google Scholar]
- Sarbassovdos D, Ali SM, Sabatini DM (2005) Growing roles for the mTOR pathway. Curr Opin Cell Biol 17: 596–603 [DOI] [PubMed] [Google Scholar]
- Sarkar S, Floto RA, Berger Z, Imarisio S, Cordenier A, Pasco M, Cook LJ, Rubinsztein DC (2005) Lithium induces autophagy by inhibiting inositol monophosphatase. J Cell Biol 170: 1101–1111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu S, Kanaseki T, Mizushima N, Mizuta T, Arakawa-Kobayashi S, Thompson CB, Korsmeyer SJ, Tsujimoto Y (2004) A role of Bcl-2 family of proteins in non-apoptotic programmed cell death dependent on autophagy genes. Nature Cell Biology 6: 1221–1228 [DOI] [PubMed] [Google Scholar]
- Shimizu S, Narita M, Tsujimoto Y (1999) Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC. Nature 399: 411–412 [DOI] [PubMed] [Google Scholar]
- Shintani T, Klionsky DJ (2004) Autophagy in health and disease: a double-edged sword. Science 306: 990–995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takacs-Vellai K, Vellai T, Puoti A, Passannante M, Wicky C, Streit A, Kovacs AL, Muller F (2005) Inactivation of the autophagy gene bec-1 triggers apoptotic cell death in C. elegans. Curr Biol 15: 1513–1517 [DOI] [PubMed] [Google Scholar]
- Vahsen N, Cande C, Briere J-J, Benit P, Joza N, Mastroberardini PG, Pequignot M, Casares N, Larochette N, Metivier D, Feraud O, Debili N, Piacentini M, Penninger JM, Rustin P, Kroemer G (2004) AIF deficiency compromises oxidative phosphorylation. EMBO J 23: 4679–4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Delft MF, Wei AH, Mason KD, Vandenberg CJ, Chen L, Czabotar PE, Willis SN, Scott CL, Day CL, Cory S, Adams JM, Roberts AW, Huang DCS (2006) The BH3 mimetic ABT-737 targets selective Bcl-2 proteins and efficiently induces apoptosis via Bak/Bax if Mcl-1 is neutralised. Cancer Cell 10: 389–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei MC, Zong W-X, Cheng EH-Y, Lindsten T, Panoutsakopoulou V, Ross AJ, Roth KA, MacGregor GR, Thompson CB, Korsmeyer SJ (2001) Proapoptotic BAX and BAK: A requisite gateway to mitochondrial dysfunction and death. Science 292: 727–730 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu L, Alva A, Su H, Dutt P, Freundt E, Welsh S, Baehrecke EH, Lenardo MJ (2004) Regulation of an ATG7-beclin 1 program of autophagic cell death by caspase-8. Science 304: 1500–1502 [DOI] [PubMed] [Google Scholar]
- Yue Z, Jin S, Yang C, Levine AJ, Heintz N (2003) Beclin 1, an autophagy gene essential for early embryonic development, is a haploinsufficient tumor suppressor. Proc Natl Acad Sci USA 100: 15077–15082 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng X, Overmeyer JH, Maltese WA (2006) Functional specificity of the mammalian Beclin-Vps34 PI 3-kinase complex in macroautophagy versus endocytosis and lysosomal enzyme trafficking. J Cell Sci 119: 259–270 [DOI] [PubMed] [Google Scholar]
- Zhu W, Cowie A, Wasfy GW, Penn LZ, Leber B, Andrews DW (1996) Bcl-2 mutants with restricted subcellular location reveal spatially distinct pathways for apoptosis in different cell types. EMBO J 15: 4130–4141 [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4