

Abstract
Reduced levels of the cyclin-dependent kinase inhibitor p27Kip1 connote poor prognosis in cancer. In human Burkitt lymphoma and in precancerous B cells and lymphomas arising in Eμ-Myc transgenic mice, p27Kip1 expression is markedly reduced. We show that the transcription of the Cks1 component of the SCFSkp2 complex that is necessary for p27Kip1 ubiquitylation and degradation is induced by Myc. Further, Cks1 expression is elevated in precancerous Eμ-Myc B cells, and high levels of Cks1 are also a hallmark of Eμ-Myc lymphoma and of human Burkitt lymphoma. Finally, loss of Cks1 in Eμ-Myc B cells elevates p27Kip1 levels, reduces proliferation and markedly delays lymphoma development and dissemination of disease. Therefore, Myc suppresses p27Kip1 expression, accelerates cell proliferation and promotes tumorigenesis at least in part through its ability to selectively induce Cks1.
Keywords: Cks1, lymphomagenesis, Myc, p27Kip1 , proliferation
Introduction
Inappropriate and/or accelerated rates of cell proliferation are hallmarks of cancer. Frequent culprits that direct this response are the Myc family of transcription factors (c-Myc, N-Myc and L-Myc), which are overexpressed in ∼70% of all cancers (Oster et al, 2002). Dysregulation of Myc can occur either directly by chromosomal amplifications or translocations (Boxer and Dang, 2001), or indirectly through mutations in signaling pathways or tumor suppressors that control Myc expression. The widespread selection for Myc activation in malignancies in part reflects its essential roles in directing cell proliferation (Trumpp et al, 2001), cell growth (Iritani and Eisenman, 1999) and tumor angiogenesis (Pelengaris et al, 1999; Baudino et al, 2002). Indeed, overexpression of Myc prevents withdrawal from the cell cycle (Askew et al, 1991), accelerates rates of cell cycle traverse and is sufficient to drive quiescent cells into S phase (Roussel et al, 1991), properties which put the tumor cell at a considerable advantage (Evan and Vousden, 2001).
Under physiological conditions, Myc expression is tightly controlled by signaling pathways that restrict transcription and/or that control the stability of Myc transcripts or proteins (Welcker et al, 2004; Liu and Levens, 2006). These signals are superseded when Myc is overexpressed in cancer, but in normal cells, Myc's effects on the proliferative response are counterbalanced by apoptotic checkpoints. In particular, Myc overexpression activates the p53 apoptotic program through the nucleolar tumor suppressor Arf, which inactivates p53's endogenous inhibitor Mdm2 (reviewed in Nilsson and Cleveland, 2003; Sherr, 2004). This leads to the induction of p53's apoptotic targets, including the BH3-only Bcl-2 family protein Puma (Jeffers et al, 2003). Further, Myc can also suppress the expression of the antiapoptotic proteins Bcl-XL or Bcl-2 (Eischen et al, 2001; Maclean et al, 2003). Not surprisingly then, inactivation of proapoptotic Bcl-2 family members, or Bcl-2 overexpression, accelerates the course of Myc-driven malignancies (Strasser et al, 1990; Eischen et al, 1999, 2001; Egle et al, 2004; Hemann et al, 2004), and alterations that inactivate these checkpoints occur in tumors induced by Myc (Eischen et al, 1999, 2001).
Myc accelerates cell proliferation, at least in part, through its ability to downregulate the expression of the cyclin-dependent kinase (Cdk) inhibitor p27Kip1 (Vlach et al, 1996; Muller et al, 1997), which harnesses cell cycle traverse by inhibiting cyclin E–Cdk2 and cyclin A–Cdk2 complexes that are necessary for entry and progression through S phase, respectively (Sherr and Roberts, 1999). Myc suppresses p27Kip1 transcription (Yang et al, 2001), although its effects on p27Kip1 protein levels are more profound and include effects on the half-life and localization of the protein. For example, the induction of cyclin D2 by Myc augments levels of cyclin D2–Cdk4/6 complexes, which activate cyclin E–Cdk2 complexes by sequestering p27Kip1 (Bouchard et al, 1999; Perez-Roger et al, 1999). Activated cyclin E–Cdk2 then phosphorylates p27Kip1, directing its association with the F-box protein Skp2 and the SCFSkp2 complex that ubiquitylates p27Kip1 and targets it for proteasomal degradation (Pagano et al, 1995; Montagnoli et al, 1999). Finally, Myc also induces the transcription of Cullin1 (Cul1), a scaffolding component of SCFSkp2 (O'Hagan et al, 2000).
In Eμ-Myc transgenic mice (Adams et al, 1985), a mouse model of human Burkitt lymphoma that bears Myc/immunoglobulin gene translocations, the precancerous state is characterized by excessive B-cell proliferation (Baudino et al, 2003) that is initially offset by Myc's apoptotic programs (Eischen et al, 1999). Nonetheless, loss of p27Kip1 is sufficient to accelerate lymphoma development in Eμ-Myc;p27Kip1−/− transgenic mice without affecting Myc-induced apoptosis (Martins and Berns, 2002), and E2f1 heterozygosity or loss impairs Myc-induced proliferation and lymphoma development in Eμ-Myc mice, and abolishes Myc's ability to downregulate p27Kip1 expression. However, E2f1 alone is not sufficient to suppress p27Kip1 protein levels, indicating that other mediators also contribute to Myc-mediated degradation of p27Kip1 (Baudino et al, 2003).
E2f1 induces cyclin E expression (DeGregori et al, 1995) and signals that control p27Kip1 protein levels include its phosphorylation on Threonine-187 (Thr-187) by cyclin E–Cdk2 in S phase (Montagnoli et al, 1999; Malek et al, 2001), and on Serine-10 (Ser-10) in G0 (Ishida et al, 2000; Kotake et al, 2005). While Ser-10 phosphorylation stabilizes p27Kip1 in G0, Thr-187 phosphorylation directs p27Kip1 to the SCFSkp2 ubiquitin ligase complex that contains Skp1, Cks1, Cul1, Rbx1 and the F-box protein Skp2 (Bartek and Lukas, 2001). Skp2 binds to phosphorylated p27Kip1 (Carrano et al, 1999), but its binding is drastically reduced in the absence of Cks1 (Ganoth et al, 2001; Spruck et al, 2001), which forms part of the p27Kip1 phosphodegron binding site (Hao et al, 2005) that is necessary for efficient docking of p27Kip1 to the SCFSkp2 holoenzyme and for subsequent ubiquitylation of p27Kip1 (Ganoth et al, 2001). Consequently, the targeted deletion of Cks1 (Spruck et al, 2001) or Skp2 (Nakayama et al, 2000) leads to marked increases of p27Kip1 levels in mice, reduces cell proliferation and results in small body size, phenotypes that are the opposite of those manifest by p27Kip1 deletion (Fero et al, 1996; Kiyokawa et al, 1996; Nakayama et al, 1996).
p27Kip1 is haploinsufficient for tumor suppression (Fero et al, 1998), and low levels of p27Kip1 connote poor prognosis in human cancer (reviewed in Sgambato et al, 2000; Slingerland and Pagano, 2000), although how reductions in p27Kip1 expression occur in malignancies are largely unknown. Here, we report that Myc induces Cks1 transcription and that elevated levels of Cks1 are a hallmark of precancerous Eμ-Myc transgenic B cells, and of Myc-driven lymphomas of mice and man. Further, Cks1 is shown to be necessary for Myc to repress p27Kip1 and to contribute to Myc-induced proliferation and lymphoma development. Therefore, the Myc-to-Cks1 pathway directs p27Kip1 downregulation and promotes proliferation and cancer.
Results
Myc induces Cks1 expression
Myc suppresses p27Kip1 expression by transcriptional repression (Yang et al, 2001), by provoking ubiquitin-mediated degradation of p27Kip1 (Muller et al, 1997) and through sequestering p27Kip1 via the induction of cyclin D2 (Bouchard et al, 1999). The expression of p27Kip1 (Cdkn1b) transcripts (Figure 1A and B), and especially p27Kip1 protein (Figure 1C), was markedly reduced in the precancerous (4-week old) B cells of Eμ-Myc transgenic mice relative to levels expressed in the B cells of wild-type littermates. Further, suppression of p27Kip1 protein was evident in both mature (IgM+) and especially immature (IgM−) precancerous Eμ-Myc B cells from either spleen or bone marrow (Supplementary Figure S1A).
Figure 1.
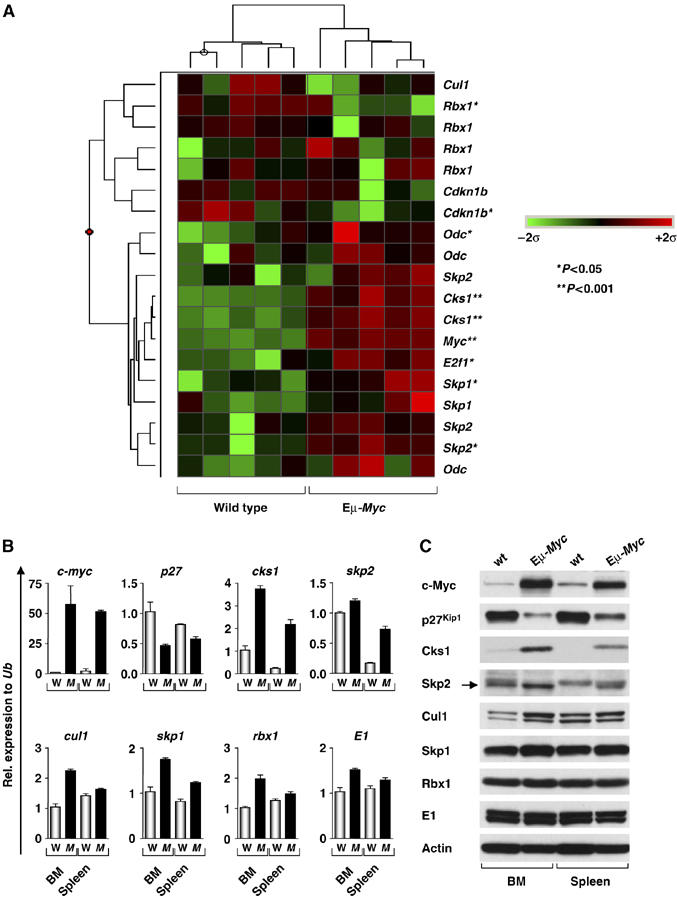
The expression of the Cks1 component of the SCFSkp2 complex is highly elevated in Eμ-Myc transgenic B cells. (A) Hierarchical clustering of components of the SCFSkp2 complex, p27 (Cdkn1b) and the selected Myc target genes (Odc, E2f1) in B220+ splenic B cells of five weanling age wild-type and five Eμ-Myc transgenic mice. Probe set signals were normalized to the mean across mice and values of each individual case are represented by a color, with green corresponding to s.d. (σ) below and red corresponding to s.d. (σ) above the mean, according to the scale shown. * Indicates a P-value of <0.05 and **P-value of <0.01. (B) SYBR-green real-time PCR analysis of B220+ bone marrow (BM) and sIgM+ splenic (spleen) B cells. W indicates wild-type mice and M denotes Eμ-Myc transgenic mice. Levels of mRNAs are standardized to the expression of ubiquitin (Ub), which is not regulated by Myc. (C) Immunoblot analysis of the indicated proteins in B220+ B cells from wild-type (wt) versus Eμ-Myc mice. Arrow indicates the Skp2-specific (lower) band on the gels.
E2f1 is necessary for Myc-induced suppression of p27Kip1, although E2f1 alone does not suppress p27Kip1 (Baudino et al, 2003); therefore, Myc regulates other components that contribute to this response. We reasoned that logical mediators could include cyclin D2 and/or components of the SCFSkp2 complex that directs p27Kip1 ubiquitylation. We therefore assessed the expression of cyclin D2 and SCFSkp2 components in splenic and bone marrow B220+ B cells isolated from several individual precancerous (4-week old) Eμ-Myc transgenic mice and their wild-type littermates. Expression profiling and quantitative real-time PCR (qRT-PCR) analyses of RNA isolated from Eμ-Myc B220+ B cells showed expected increases in the Myc target genes Odc and E2f1 (Figure 1A) but revealed that cyclin D2 (Ccnd2) levels were, surprisingly, reduced (Supplementary Figure S1B). By contrast, Eμ-Myc B cells expressed elevated levels of Cks1 mRNA, and there were also modest increases in the levels of transcripts encoding other SCFSkp2 components, including Skp2, Rbx1, Skp1 and Cul1; for Rbx1, Skp1 and Cul1, these effects were usually most evident in immature (bone marrow) versus mature (spleen) B cells (Figure 1B). However, when canvassed for changes in protein levels, it was evident that Myc's effects on SCFSkp2 components were selective, as there was little change in the levels of Cullin1, Skp1, or Rbx1, modest increases in Skp2, but profound increases in the levels of Cks1 (Figure 1C). Therefore, Myc markedly induces the expression of Cks1 and also induces Skp2, and this is associated with a dramatic reduction in p27Kip1 levels in Myc-expressing B cells.
Myc's ability to induce Cks1 largely occurred through increases in Cks1 mRNA. The Cks1 promoter-regulatory region lacks consensus Myc responsive elements, but nonetheless could be activated through non-consensus elements. To address this issue, primary early-passage mouse embryo fibroblasts (MEFs) were infected with the MSCV-Myc-ERTAM-IRES-GFP retrovirus, which harbors the Myc-ERTAM transgene, a chimeric fusion of c-Myc that can be selectively activated by the ER agonist 4-hydroxy tamoxifen (4-HT; Eischen et al, 1999), and the gene for green fluorescence protein (GFP), expressed in cis from an internal ribosome entry site (IRES). Myc-ERTAM- and GFP-only-expressing MEFs were left untreated or were pretreated with 1 μg/ml cycloheximide (Chx) for 30 min (which blocked protein synthesis by >95%) and then were treated with 4-HT and the levels of Cks1 transcript were assessed by real-time PCR (Figure 2A). Myc activation led to the induction of Cks1 RNA (Figure 2A) and protein (Figure 2B), although unlike the bona fide Myc transcription targets Odc (Bello-Fernandez et al, 1993) and Rcl (Lewis et al, 1997), the induction of Cks1 transcripts was blocked by Chx (Figure 2A); therefore, the induction of Cks1 by Myc is indirect. Similar results applied to the induction of Skp2 by Myc (Figure 2A), although Skp2 induction was more delayed and less robust than that of Cks1, especially regarding changes in Skp2 protein (Figure 2B).
Figure 2.

Myc regulation of Cks1 is indirect and independent of E2f1. (A) Primary early-passage MEFs were infected with MSCV-Myc-ERTAM-IRES-GFP (Myc-ER) or MSCV-IRES-GFP (GFP) virus. GFP+ cells were then left untreated (−) or were treated (+) with 2 μm 4-HT±Chx pretreatment (30 min) for 24 h and assessed for their expression of the indicated mRNAs by SYBR-green real-time PCR analysis. Levels of mRNA were standardized to Ub. (B) Immunoblot analysis of Cks1 and Skp2 expression upon Myc activation by 4-HT in primary MEFs. * Denotes nonspecific band detected by the Myc antiserum. (C) qRT-PCR analysis of the expression of the indicated mRNAs in splenic B220+ B cells of 4-week-old wild-type (wt) and Eμ-Myc mice. The E2f1 genotypes are indicated. Levels of mRNA are standardized to Ub. (D) Immunoblot analysis of Cks1 expression upon Myc-ER activation by 4-HT in primary, paired E2f1+/+ and E2f1−/− MEFs. (E) Immunoblot analysis of Cks1 expression in 4-week-old splenic B220+ B cells of wt and Eμ-Myc mice of the indicated E2f1 genotypes.
Myc did not activate a Cks1–promoter–luciferase reporter that harbors the 5′-regulatory region and exon 1 of mouse Cks1 and, as might be expected for an indirect target, Myc also did not bind to the Cks1 promoter-regulatory region in chromatin immunoprecipitation assays (data not shown). However, we reasoned that Myc might induce Cks1 through the agency of other transcription factors. One obvious candidate was E2f1, which is induced by Myc and which is required for Myc-mediated suppression of p27Kip1 protein (Baudino et al, 2003). We therefore assessed the effects of E2f1 loss on the expression of Cks1 in vivo in splenic B220+ B cells from (precancerous) 4-week old E2f1+/+-, E2f1+/−- and E2f1−/−-Eμ-Myc transgenics and compared these to non-transgenic E2f1+/+ and E2f1−/− littermates. E2f1 loss had no effect on the markedly increased levels of Cks1 mRNA in Eμ-Myc B cells and also had no effect on the induction of Rcl (Figure 2C). As expected, the suppression of p27Kip1 protein following activation of Myc-ERTAM was impaired in E2f1−/− MEFs compared with that of paired E2f1+/+ MEFs. Nonetheless, there was no effect of E2f1 loss on the magnitude of the induction of Cks1 protein by Myc in either E2f1-deficient MEFs (Figure 2D) or in precancerous E2f1−/−-Eμ-Myc B cells (Figure 2E). Furthermore, E2f1 activation alone was not sufficient to induce Cks1 and/or suppress p27Kip1 in early-passage MEFs engineered to express ERTAM-E2f1, a chimeric form of E2f1 that activates E2f1 targets (Vigo et al, 1999), although E2f1 activation did, as expected, induce cyclin E1 and E2 (Supplementary Figure S2A). Finally, induction of Cks1 was not associated with significant differences in the expression of E2f2 or E2f3, which belong to the same E2f activator subclass and have some overlapping functions (Black et al, 2005), in wild-type versus splenic Eμ-Myc B cells (Supplementary Figure S2B). Therefore, Myc activates Cks1 expression indirectly and in an E2f-independent manner.
At face value, the effects of Myc on Cks1 protein levels appeared more robust than effects on Cks1 transcripts. We therefore tested whether turnover of Cks1 protein might be regulated by c-Myc, especially given that the anaphase promoting complex/cyclosome-Cdh1 (APC/CCdh1) E3 ubiquitin ligase directs the turnover of Cks1 and Skp2 (Bashir et al, 2004). However, the expression of Cdh1 transcripts and protein, and of other components of the APC/CCdh1 complex, was similar in Eμ-Myc versus wild-type B cells (Supplementary Figure S3, and data not shown). Furthermore, measurements of the rate of Cks1 turnover in Myc-ERTAM- versus puromycin-only-expressing MEFs by pulse–chase immunoprecipitation experiments demonstrated no significant differences in Cks1 half-life (Supplementary Figure S4A). By contrast, the rates of Cks1 biosynthesis were two- to three-fold higher in Myc-ERTAM-expressing MEFs than in puromycin-only-expressing MEFs (Supplementary Figure S4B), a finding similar to the increased levels of Cks1 transcripts observed in Myc-expressing MEFs.
Collectively, these findings suggested that the predominant mode of upregulation of Cks1 by Myc was transcriptional. To confirm this, we performed quantitative primary transcript real-time PCR (qPT-PCR) analyses, which measure the relative abundance of primary, unspliced transcripts relative to those of mature mRNA. Using Cks1 exon 1–intron 1, intron 2–exon 2, exon 1–exon 2 and exon 2–exon 3 primer pairs for qPT-PCR, the relative abundance of immature and mature Cks1 transcripts were similarly elevated in Eμ-Myc B cells (Figure 3A). qPT-PCR can also be utilized to determine the relative rates of synthesis of unspliced versus mature, spliced mRNA. We therefore assessed the rates of synthesis of immature versus mature Cks1 transcripts in puromycin-only versus Myc-ERTAM-expressing MEFs following the addition of 4-HT. As expected, the induction of unspliced Cks1 transcripts in Myc-ERTAM-expressing MEFs preceded the appearance of mature Cks1 mRNA, which, with time, increased to levels that were similar to unspliced Cks1 transcripts, indicating no overt effects of Myc on the splicing of Cks1 transcripts (Figure 3B). Finally, Myc did not affect the half-life of Cks1 transcripts in experiments assessing the effects of actinomycin-D on the turnover of Cks1 mRNA (data not shown). Therefore, Myc provokes increases in Cks1 at the level of transcription.
Figure 3.
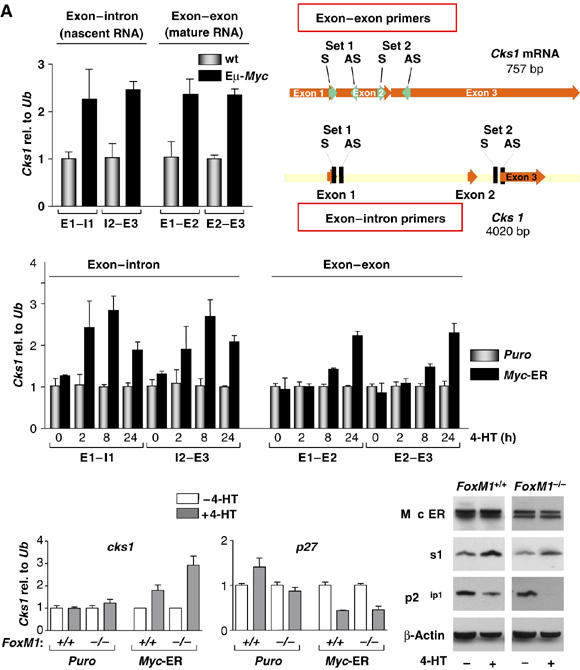
Myc regulates Cks1 transcription independent of FoxM1. (A) Total RNA was isolated from B220+ B cells from 4-week-old wild-type (wt) and Eμ-Myc transgenic mice and qPT-PCR analysis was performed with primers designed to detect unspliced, nascent RNA (intron–exon–PCR product) or spliced, mature mRNA only (exon–exon–PCR product). The location of the two sets of primers used to detect nascent versus mature Cks1 mRNA is illustrated at right. Levels of Cks1 were standardized to Ub. (B) The induction of nascent and mature RNA was analyzed in MEFs expressing Puro alone or Myc-ERTAM+Puro. MEFs were treated for the indicated times with 4-HT and levels of Cks1 were standardized to Ub. (C) Left, FoxM1+/+ or FoxM1−/− MEFs expressing Puro alone or Myc-ERTAM+Puro were treated with 4-HT for 24 h and RNA levels were determined by qRT-PCR. Right, extracts from the same cells were assessed for the indicated proteins by immunoblot analyses.
Logical mediators of the Myc-to-Cks1 response would include transcription factors that are essential for Cks1 transcription. One potential candidate was the Forkhead box M1 transcription factor (FoxM1), as targeted deletion of Foxm1 has revealed its role in regulating Cks1 transcription (Wang et al, 2005). To address a potential role for FoxM1, paired Foxm1−/− and Foxm1+/+ MEFs were transduced with the MSCV-Myc-ERTAM-IRES-puro or MSCV-IRES-puro retroviruses and puromycin-resistant cultures were treated with 4-HT. Activation of Myc-ERTAM led to a marked induction of Cks1 transcripts in Foxm1−/− MEFs and also to increases in Cks1 protein, and to corresponding reductions in p27Kip1 protein (Figure 3C). Therefore, Myc-mediated induction of Cks1 is also independent of FoxM1.
Alterations of the Cks1-p27Kip1 pathway are a hallmark of Myc-induced lymphoma
Given the effects of Myc on the expression of p27Kip1, Skp2 and Cks1, we reasoned that alterations in their expression might be evident in the Myc-driven lymphomas that arise in Eμ-Myc transgenic mice, and in human Burkitt lymphoma. Expression of p27Kip1, Skp2 and Cks1 was determined by qRT-PCR (Figure 4A) and by Western blotting of 49 lymphomas from Eμ-Myc transgenics, and of 17 primary human Burkitt lymphoma, and was compared with their expression in normal B cells (B220+ cells from wild-type littermates, or CD19+ peripheral blood human B cells), and to precancerous Eμ-Myc splenic B220+ B cells (Figure 4B). Most Eμ-Myc lymphomas expressed very low levels of p27Kip1 mRNA and protein and elevated levels of Skp2, and all tumors expressed much higher levels of Cks1 (Figure 4). Specifically, in Eμ-Myc mice, 94% of lymphomas had lower p27Kip1 levels, and Skp2 and Cks1 were greatly elevated in 85 and 95% of lymphomas, respectively (Figure 4B, representative tumors shown). Furthermore, dramatic alterations in p27Kip1 and CKS1 expression were also evident in all Burkitt lymphoma analyzed, which expressed reduced levels of p27Kip1 and much higher levels of CKS1, when compared with levels expressed in normal human B cells (Figure 4B). The marked increases in Cks1 transcripts (Figure 4A) were not due to Cks1 amplification, as assessed by Southern blotting (data not shown). Furthermore, the high levels of Cks1 in Eμ-Myc lymphomas were not associated with corresponding changes in FoxM1 expression, as FoxM1 transcripts were reduced in Eμ-Myc lymphomas compared with levels expressed in wild-type or precancerous Eμ-Myc B220+ bone marrow cells, and markedly increased levels of Cks1 protein were also evident in lymphomas that arise in E2f1+/−-, and E2f1−/−-Eμ-Myc transgenics (Supplementary Figure S5). Therefore, the induction of Cks1 and Skp2, and the suppression of p27Kip1 expression, are hallmarks of Myc-induced lymphoma.
Figure 4.
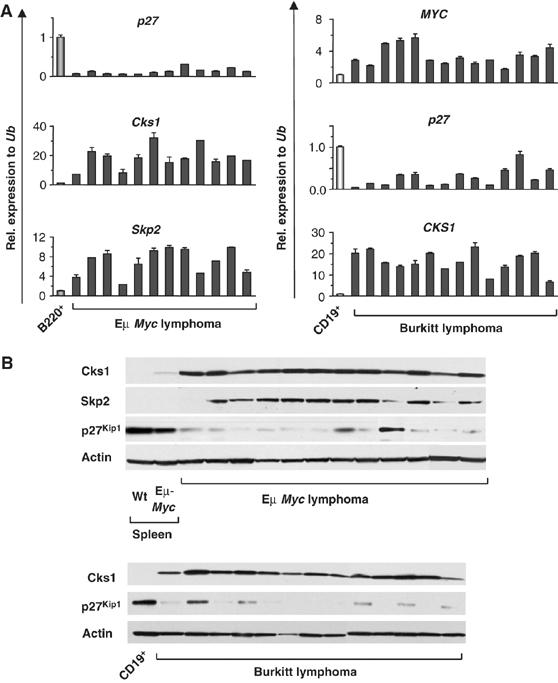
Alterations in the Cks1–p27Kip1 axis are hallmarks of Myc-induced lymphomas in mice and man. (A) qRT-PCR analysis of c-Myc (only in human samples), p27, Skp2 and Cks1 in Eμ-Myc lymphomas compared with wild-type B220+ splenic B cells (B220+, left panel), and of 14 human Burkitt lymphoma samples compared with CD19+ control human B cells (right panel). Levels of mRNAs were standardized to Ub. (B) Lymphomas arising in Eμ-Myc transgenic mice and B220+ splenic B cells from precancerous mice (Eμ-Myc) and wild-type controls (wt) were analyzed for expression of the indicated proteins by immunoblotting (top panel). Thirteen human Burkitt lymphoma samples were analyzed by immunoblots for expression of the indicated proteins (bottom panel). CD19+ peripheral B cells from healthy donors were used as control.
Loss of Cks1 impairs Myc-induced lymphomagenesis
The dramatic changes in p27Kip1 and Cks1 expression in Myc-driven lymphoma suggested that this pathway plays a critical role in tumorigenesis. Indeed, loss of p27Kip1 accelerates lymphoma development in Eμ-Myc mice (Martins and Berns, 2002), although the role of Cks1 in tumorigenesis is unknown. If Cks1 induction was important for Myc to suppress p27Kip1, we predicted that Cks1 loss would delay lymphoma development. To test this hypothesis, Eμ-Myc transgenic mice were mated to Cks1−/− mice (Spruck et al, 2001) and F1 offspring were bred to obtain Cks1+/+-, Cks1+/−- and Cks1−/−-Eμ-Myc transgenics. These littermates were then followed for the onset of disease, which is first manifest by a marked lymphocytosis and splenomegaly. To evaluate the precancerous phase, 4-week old littermates were analyzed for these parameters. Non-transgenic Cks1−/− mice had normal white blood cell and lymphocyte counts and no splenomegaly (data not shown). Notably, the spleens of Eμ-Myc;Cks1−/− transgenics were significantly smaller than those from Eμ-Myc;Cks1+/+ littermates (spleen sizes 157±7 versus 83±11 mg for wild-type versus Cks1-null cohorts; Figure 5A, right panel). There were also obvious reductions in white blood cell numbers in Eμ-Myc;Cks1−/− transgenics (Cks1-null, 7.7±1.1 × 106/μl versus Cks1+/+, 11.1±0.7 × 106/μl; Figure 5A, left panel) and corresponding reductions in lymphocyte counts (Cks1-null, 5.4±0.4 × 106/μl versus Cks1+/+, 6.9±0.4 × 106/μl; Figure 5A, middle panel). Heterozygosity effects in Eμ-Myc;Cks1+/− littermates were not observed (data not shown). Thus, Cks1 loss effectively cancels the precancerous phase of disease in Eμ-Myc transgenic mice.
Figure 5.
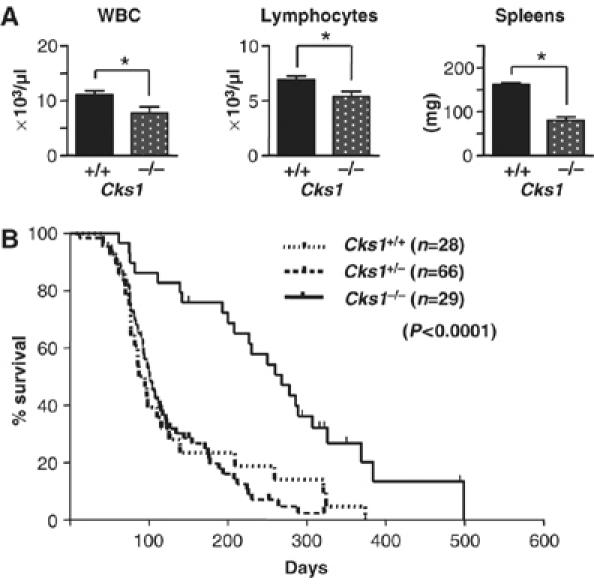
Cks1 loss impairs Myc-induced lymphomagenesis. (A) Precancerous Eμ-Myc transgenic mice of different Cks1 genotypes were analyzed for white blood counts (WBC), lymphocyte number in the peripheral blood and spleen weights. * Indicates a P-value of <0.05. (B) Kaplan–Meier survival curves of Eμ-Myc transgenic animals of different Cks1 genotypes. The differences in the rates of tumor incidence between the Cks1+/+ and the Cks1−/− group are highly statistically significant (P<0.0001).
Eμ-Myc transgenic mice succumb to aggressive lymphoma within 3–6 months of birth (Adams et al, 1985). Non-transgenic Cks1−/− littermates showed no signs of tumor development throughout their lifespan. Importantly, Eμ-Myc;Cks1−/− transgenic mice had significant delays in lymphoma development, with a median survival of 268 days compared with 91 and 101 days median survival of their Eμ-Myc;Cks1+/+ and Eμ-Myc;Cks1+/− littermates, respectively (Figure 5B, P<0.0001). Thus, Cks1 is a critical mediator of Myc-driven tumorigenesis in B cells.
Cks1 contributes to Myc's proliferative response
Loss of inhibitors of the Arf-p53 apoptotic program such as Mdm2 (Alt et al, 2003), or of regulators of the Myc-to-p27Kip1 pathway such as E2f1 (Baudino et al, 2003), impair lymphoma development. Therefore, the profound effects of Cks1 loss on lymphoma development in Eμ-Myc transgenics might reflect an amplification of Myc's apoptotic response and/or the inhibition of its proliferative response. To address the effect of Cks1 loss on Myc-induced apoptosis, the apoptotic indices of precancerous B cells from both immature (sIgM−) and mature (sIgM+) B220+ cells from wild-type versus Cks1-null Eμ-Myc transgenic littermates were assessed by staining with Annexin-V–FITC and propidium iodide (PI). There were no significant differences in the apoptotic indices of Eμ-Myc;Cks1−/− precancerous B cells when compared with those of Eμ-Myc;Cks1+/+ littermates (Figure 6A). Further, the frequency of alterations in the Arf-p53 pathway, which occur through bi-allelic deletions of Arf or mutations in p53 (Eischen et al, 1999), were similar in wild-type, Cks1+/− and Cks1−/− Eμ-Myc lymphomas (Supplementary Figure S6). Therefore, the delay in lymphoma development evident in Eμ-Myc;Cks1−/− transgenic mice was not due to overt effects on Myc's apoptotic program.
Figure 6.
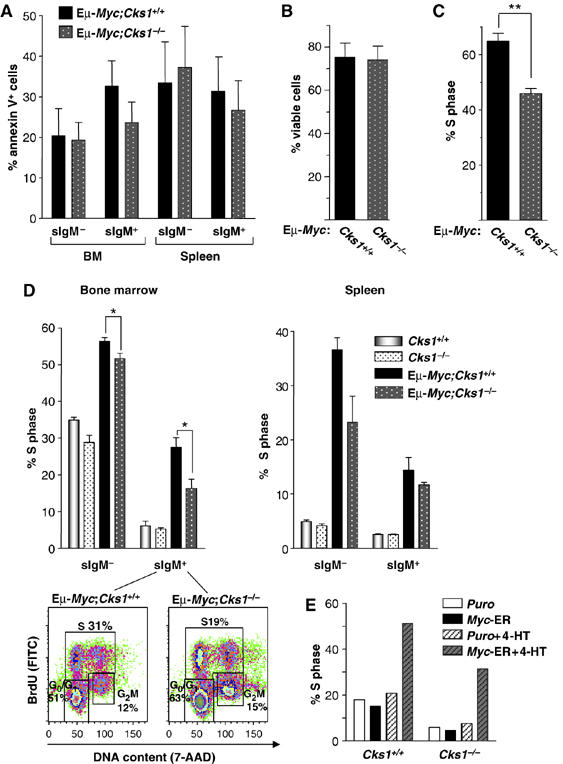
Loss of Cks1 affects proliferation, but not apoptosis, of Eμ-Myc transgenic B cells. (A) Apoptosis in B220+ sIgM− or sIgM+ Eμ-Myc B cells of the indicated Cks1 genotypes was analyzed using Annexin-V+ assays. The bars represent the mean±s.e.m. (n=3 independent experiments). (B, C) B cells from ex vivo cultured bone marrow of littermate Eμ-Myc transgenic mice of different Cks1 genotypes were analyzed for spontaneous rates of apoptosis by Annexin-V staining (B) or BrdU incorporation (C) into DNA (S phase) to assess proliferation (n=3; bars indicate mean±s.e.m.). ** Indicates P<0.01. (D) Wild-type and Eμ-Myc littermates of different Cks1 genotypes received intraperitoneal injections of BrdU and cells from bone marrow and spleen were harvested 12 h later. BrdU incorporation was analyzed by an antibody-dependent fluorescence assay. The bars are the mean±s.e.m. of three independent experiments. * Indicates P<0.05. A representative FACS analysis of BrdU- and 7-AAD-stained B220+ B cells from Eμ-Myc;Cks1+/+ and Eμ-Myc;Cks1−/− littermates is shown below, along with the percentages of these cells in G0/G1, S and G2/M phases. (E) Paired wild-type and Cks1−/− MEFs expressing Myc-ER were treated with 4-HT for 24 h and the percent of cells in S phase were determined by PI/FACS.
To evaluate potential effects of Cks1 loss on Myc's proliferative response, we initially assessed the rates of growth of B cells derived from transgenic littermates by ex vivo culture of bone marrow on S17 stromal cells in medium containing interleukin-7 (Eischen et al, 1999). Again, there were no differences in the apoptotic indices of Eμ-Myc;Cks1+/+ versus Eμ-Myc;Cks1−/− B cells (Figure 6B). To quantify their rates of proliferation, these B cells were labeled with BrdU and analyzed by flow cytometry. Eμ-Myc;Cks1−/− B cells had much slower growth rates than B cells derived from Eμ-Myc;Cks1+/+ littermates (Figure 6C). Therefore, at least ex vivo, the Cks1 deficiency reduces the proliferative rates of Myc-expressing B cells.
To address whether Cks1 loss might inhibit Myc's ability to augment cell growth (mass) (Iritani and Eisenman, 1999) and/or to accelerate rates of cell cycle traverse in vivo, BrdU was injected intraperitoneally into 4-week-old Eμ-Myc transgenic littermates of different Cks1 genotypes. After 12 h, B220+sIgM+ and B220+sIgM− cells were assessed for their size by forward versus side scatter, and for their S phase indices by FACS analysis. There were no significant changes in the sizes of B cells from wild-type versus Cks1-null transgenics, although all Myc transgenic cells were, as reported (Iritani and Eisenman, 1999), larger in size than non-transgenic B cells. Further, as expected (Baudino et al, 2003), both bone marrow- and spleen-derived Eμ-Myc B cells had higher proliferative rates than wild-type B cells. Loss of Cks1 alone had only minor effects on B-cell proliferation as compared with wild-type littermate controls (Figure 6D). Importantly, Cks1 deficiency impaired Myc's proliferative response, especially in Eμ-Myc;Cks1−/− B cells from bone marrow and pro-/pre- (IgM−) splenic B cells, and FACS analyses demonstrated that this was associated with decreased numbers of cells in S phase and concomitant increases in the percentage of cells in G0/G1 (Figure 6D). Therefore, the delay in Myc-induced lymphomagenesis caused by Cks1 deficiency is associated with defects in Myc's proliferative response. Finally, to confirm the effects of Cks1 deficiency on Myc's ability to drive cells into S phase in other cell contexts, we also transduced paired early-passage Cks1+/+ and Cks1−/− MEFs with puro- or Myc-ERTAM-encoding retroviruses. Puromycin-resistant cells were then treated with 4-HT and evaluated by PI/FACS analyses. Again the percentage of Myc-expressing cells in S phase was markedly reduced by the Cks1 deficiency (Figure 6E). Therefore, the Cks1 deficiency impairs Myc's proliferative response.
Cks1 loss abolishes Myc's ability to suppress p27Kip1
The targeted deletion of Cks1 (Spruck et al, 2001) revealed its essential role in the SCFSkp2 complex that directs p27Kip1 degradation. Given Myc's ability to induce Cks1 while repressing p27Kip1, and the effects of Cks1 loss on Myc-induced proliferation and lymphomagenesis, we evaluated p27Kip1 expression in Eμ-Myc;Cks1−/− transgenic B cells cultured ex vivo. Importantly, there was a dramatic increase in p27Kip1 protein levels in B cells cultured from Eμ-Myc;Cks1−/− mice (Figure 7A). Similar changes in p27Kip1 expression were also evident in vivo, where Myc's ability to suppress p27Kip1 protein levels was impaired in Eμ-Myc;Cks1−/− B cells (Figure 7B). Interestingly, the effects of Cks1 loss on p27Kip1 levels in Eμ-Myc B cells only occurred at the level of the protein, as levels of p27Kip1 transcripts were equally suppressed in all Eμ-Myc B cells, regardless of their Cks1 status (Figure 7C). Finally, we also assessed whether the proliferative defects of Eμ-Myc;Cks1−/− B cells (e.g. Figure 6C) could indeed be attributed to elevated levels of p27Kip1 levels in these cells. To test this issue, we knocked down p27Kip1 expression using a p27Kip1-specific shRNA by transducing Eμ-Myc;Cks1−/− B cells with MSCV-p27shRNA-IRES-GFP or with a control MSCV-pBluescript-shRNA-IRES-GFP virus (Nilsson et al, 2004). p27shRNA-expressing Eμ-Myc;Cks1−/− B cells had approximately 50% reduced levels of p27Kip1 protein compared with pBluescript-shRNA-expressing controls (Supplementary Figure S7A), and p27shRNA-expressing cells demonstrated a ∼20% increase in their uptake of 3H-thymidine, and a 40% overall increase in their growth rate, relative to control Eμ-Myc;Cks1−/− B cells (Supplementary Figure S7B and C). Therefore, Cks1 is specifically required for Myc-mediated suppression of p27Kip1 protein levels, and this pathway contributes to Myc's proliferative response.
Figure 7.
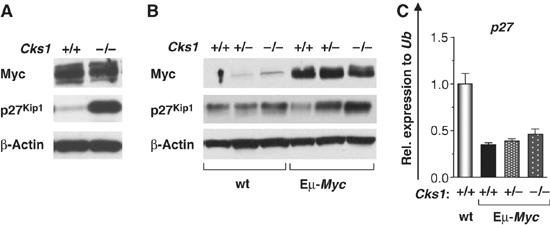
Cks1 is required for Myc to suppress p27Kip1. (A) Immunoblot analysis of B cells from ex vivo cultured bone marrow of paired Eμ-Myc littermates of the indicated Cks1 genotypes. (B) Immunoblot analysis of Myc and p27Kip1 expression was performed on splenic B220+ B cells from precancerous mice of the indicated Cks1 genotypes. (C) SYBR-green real-time PCR analysis of p27 mRNA expression in splenic B cells from precancerous Eμ-Myc transgenics of different Cks1 genotypes compared with non-transgenic wild-type littermates. Levels of mRNAs were standardized to Ub.
The Skp2 component of the SCFSkp2 E3 ubiquitin ligase has been suggested to regulate Myc protein stability and its transcriptional activity (Kim et al, 2003; von der Lehr et al, 2003), although Fbw7, a component of the SCFFbw7 ubiquitin ligase, has been more recently been demonstrated to regulate Myc turnover (Welcker et al, 2004). Nonetheless, it was possible that Cks1 status affected Myc turnover and/or function. However, Cks1 loss had no effect on the steady-state levels of Myc protein in transgenic B cells (Figure 7A and B). Furthermore, Cks1 loss did not affect Myc's ability to suppress p27Kip1 mRNA levels in Eμ-Myc transgenic B cells (Figure 7C) and also had no effect on Myc's ability to regulate a number of other well-established targets, including rcl, cad and many others (Supplementary Figure S8B), and there were no dramatic effects of Cks1 loss on the expression of Myc target genes in the ‘Myc Target gene database', http://www.myc-cancer-gene.org (Supplementary Figure S8A). Therefore, the effects of Cks1 loss on Myc-induced proliferation and tumorigenesis appear independent of effects on Myc stability or function, and rather are specifically associated with blocking Myc's ability to downregulate p27Kip1 protein expression.
Loss of Cks1 impairs the metastasis of Myc-induced lymphoma
The immunophenotypes of lymphomas arising in Cks1-null Eμ-Myc transgenics were identical (pre-B and mature B-cell lymphomas) to those arising in wild-type Eμ-Myc transgenic littermates (data not shown). However, unlike lymphomas that arose in Eμ-Myc;Cks1+/+ or Eμ-Myc;Cks1+/− littermates (Figure 8A and B), nearly all Eμ-Myc;Cks1−/− lymphomas maintained high levels of p27Kip1 protein expression (Figure 8A and C). Elevated levels of p27Kip1 were also evident in the late-onset lymphomas of Eμ-Myc;E2f1−/− mice (data not shown). Therefore, these pathways are not bypassed during lymphoma development.
Figure 8.
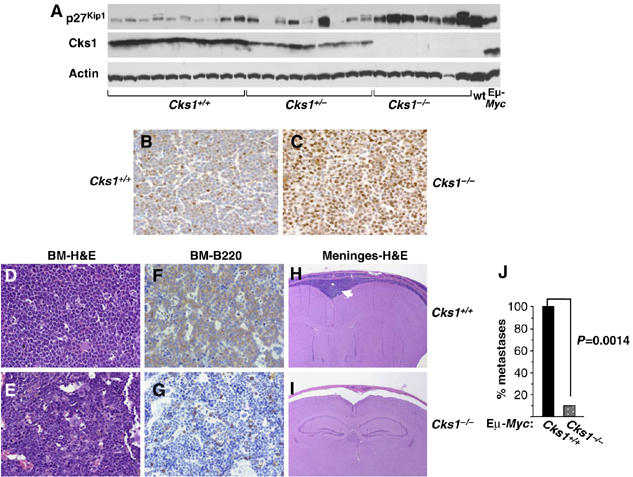
Lymphomas arising in Cks1−/− Eμ-Myc transgenic mice show a dramatic reduction of extranodal dissemination and retain elevated p27Kip1 protein levels. (A) Immunoblot analysis of lymphomas arising in mice of the indicated Cks1 genotypes were compared with precancerous B220+ B cells from wild-type (wt) and Eμ-Myc transgenic mice (Eμ-Myc). Immunohistochemical detection of p27Kip1 in representative lymphomas from Eμ-Myc;Cks1+/+ (B) and Eμ-Myc;Cks1−/− (C) transgenic mice (× 40 magnification). H&E staining of bone marrow sections from sick Cks1+/+ (D) and Cks1−/− (E) Eμ-Myc transgenic mice (× 40 magnification). B220—immunohistochemistry in bone marrow from sick Cks1+/+ (F) and Cks1−/− (G) Eμ-Myc transgenic mice (× 40 magnification). H&E staining of brain sections from Cks1+/+ (H) and Cks1−/− (I) Eμ-Myc transgenic mice (× 10 magnification). The white arrow in (H) points to the meningeal lymphoma infiltration. BM denotes bone marrow. (J) Nine Eμ-Myc;Cks1−/− mice and six Eμ-Myc;Cks1+/+ mice were histologically evaluated for lymphoma dissemination. Shown are the percent of mice with bone marrow involvement (P=0.0014).
Lymphomas that arise in Eμ-Myc transgenics are aggressive and typically display disseminated disease with invasion of all of the major organ systems (Eischen et al, 1999). Surprisingly, necropsy revealed marked differences in the lymphomas that arose in Eμ-Myc;Cks1−/− mice versus those that arose in Eμ-Myc;Cks1+/+ littermates. Histological examination of six Eμ-Myc;Cks1+/+ mice revealed that all suffered from disseminated disease, with diffuse bone marrow infiltrates of B220+ lymphocytes (Figure 8D and F) and infiltration of the spleen, liver and the meninges (Figure 8H, and data not shown). In contrast, only one of nine diseased Eμ-Myc;Cks1−/− mice showed a moderate bone marrow infiltrate (Figure 8E and G show unaffected Eμ-Myc;Cks1−/− bone marrows), and none of these diseased mice presented liver or meningeal infiltrates (Figure 8I and J, and data not shown). Therefore, Cks1 loss also compromises the aggressive nature of Myc-driven lymphoma.
Discussion
In normal cells, Myc activation accelerates cell cycle traverse, although this is counterbalanced by apoptotic checkpoints such as the Arf-p53 pathway, that guard against transformation (Nilsson and Cleveland, 2003). Thus, events which bypass apoptotic regulators are rate-limiting for tumor development (Eischen et al, 1999; Schmitt et al, 1999). However, these findings do not discount an important role for Myc's proliferative response in tumorigenesis. Indeed, as shown here, one key mediator of this response is Cks1, which is induced by Myc and is overexpressed in Myc-driven lymphomas, and which contributes to Myc-induced proliferation and lymphomagenesis. Strikingly, the effects of Cks1 in promoting Myc's proliferative response underscores the importance of the Myc-to-p27Kip1 pathway in regulating cell proliferation and tumorigenesis, where loss of p27Kip1 accelerates lymphomagenesis (Martins and Berns, 2002), and where loss of E2f1 also compromises Myc's ability to suppress p27Kip1 and impairs Myc-induced proliferation and lymphoma development (Baudino et al, 2003). Further, these studies suggest that Myc's ability to induce both E2f1 and Cks1, and perhaps also Skp2, is required in concert to suppress p27Kip1 expression and to accelerate cell proliferation and tumor development, and that in the absence of Cks1, the elevation of p27Kip1 levels is not bypassed during tumor development (Figure 8).
Cks1 is necessary for recognition of Thr-187-phosphorylated p27Kip1 by Skp2 (Ganoth et al, 2001; Spruck et al, 2001; Hao et al, 2005), and is thus essential to downregulate p27Kip1 during cell cycle traverse. Recognition of Thr-187-phosphorylated p27Kip1 by the SCFSkp2 complex occurs during S and G2 phases (Malek et al, 2001), and other levels of control, for example, Ser-10 phosphorylation of p27Kip1 and the cytoplasmic ubiquitin ligase complex KPC, regulate p27Kip1 proteolysis at the G0 and G1 phases of the cell cycle, respectively (Ishida et al, 2000; Kamura et al, 2004; Kotake et al, 2005). Despite these multiple levels of control, the targeted deletion of Cks1 revealed that it is a critical arbiter of p27Kip1 levels, as its loss leads to profound increases in p27Kip protein (Ganoth et al, 2001; Spruck et al, 2001). Further, Cks1 has other targets that may contribute to the proliferative response, as Cks1 is also required for SCFSkp2-mediated degradation of the pRb-related protein p130 (Tedesco et al, 2002). In turn, this leads to reduced rates of proliferation in Cks1-deficient cells and ultimately, in the animal, to a reduced body size (Spruck et al, 2001). Underscoring the importance of Cks1, recent studies have shown that Cks1 expression is highly elevated in a number of malignancies, where it is associated with reduced p27Kip1 levels, high proliferative rates and poor outcome (Masuda et al, 2003; Shapira et al, 2004; Slotky et al, 2005).
The underlying cause(s) for Cks1 overexpression in cancer are unknown, but our findings suggest that they are linked to Myc oncoproteins, which are overexpressed in a large number of malignancies. Whether tested ex vivo or when examined in vivo, Myc was revealed to upregulate Cks1 RNA and protein levels and, further, Cks1 was shown to be an essential mediator of the Myc-to-p27Kip1 pathway. Myc appears to induce Cks1 expression largely through effects on its transcription, but the induction of Cks1 mRNA by Myc is E2f1 and FoxM1 independent and is indirect, suggesting that Myc regulates the expression of some other transcription factor that in turn controls Cks1 transcription. Ultimately, defining the mediators of the Myc-to-Cks1 pathway is important, as our findings establish a crucial role for Cks1 in tumorigenesis, suggesting this pathway could be targeted in cancer prevention and/or therapeutics.
In the response to Myc, Cks1 and perhaps Skp2 are the limiting components of the SCFSkp2 complex that are required to degrade p27Kip1, as Myc does not significantly affect the expression of Cullin1, Skp1 or Rbx1, at least in Eμ-Myc transgenic B cells. Cks1's critical role is underscored by the absolute requirement for Cks1 in mediating downregulation of p27Kip1 protein in Myc-expressing B cells, a function which is also shared by E2f1 in Myc-driven lymphomagenesis (Baudino et al, 2003). Since E2f1 and Cks1 are now shown to play independent and critical roles in the pathway(s) by which Myc downregulates p27Kip1, one might predict that they are critical for proper stoichiometry of the SCFSkp2 holoenzyme complex. Along these lines, it is intriguing that the retinoblastoma protein (Rb) interacts with the N-terminus of Skp2 and interferes with Skp2-p27Kip1 interactions, and with p27Kip1 ubiquitylation (Ji et al, 2004). In the context of E2f1 loss, where one should see reductions in active cyclin E/Cdk2 complexes (DeGregori et al, 1995), one might then expect Rb's functions to be augmented and to selectively inactivate Skp2 functions. Thus, Myc's ability to induce both Cks1 and E2f1 would ensure that proper levels of Cks1 and Skp2 are available to target p27Kip1 to the SCFSkp2 holoenzyme for degradation, and it will be important to test this model. Alternatively, it is possible that Myc somehow activates signaling pathways that lead to selective phosphorylation/dephosphorylation(s) of p27Kip1 that promote its recognition by the SCFSkp2 complex or by the KPC complex (Kamura et al, 2004).
The effect of Cks1 loss on Myc's proliferative, but not apoptotic, response indicates that Cks1 contributes to Myc-induced pathways that regulate the rates of cell division. In vivo, this response increases the size of the target population of immature B cells that are most susceptible to transformation, and this is rate-limiting for tumor development. Indeed, there were essentially no effects of Cks1 loss on Myc's apoptotic programs in B cells, as observed in Cks1-deficient animals (Spruck et al, 2001). Therefore, the effects of Cks1 loss upon lymphomagenesis appear restricted to their effects on the proliferative rates of Myc-expressing B cells.
A striking finding revealed by the analyses of lymphomas arising in both Eμ-Myc transgenic mice and in human Burkitt lymphoma was that alterations in Cks1, Skp2 and p27Kip1 expression were evident in virtually every tumor, a frequency much higher than that observed for alterations of Arf or p53 (∼55% of tumors in Eμ-Myc transgenic mice; Eischen et al, 1999). These findings underscore the importance of this pathway in tumor development and may indicate an important role in maintaining the tumorigenic state. Further, since typical reductions in p27Kip1 expression do not occur in lymphomas lacking Cks1, and since Eμ-Myc;Cks1−/− lymphomas are much less invasive (Figure 8), these findings underscore the potential of targeting the Myc-to-Cks1 pathway in cancer therapeutics.
Materials and methods
Interbreeding of mice and tumor surveillance
Cks1-null mice (mixed background) (Spruck et al, 2001) were bred with Eμ-Myc transgenic mice (C57BL/6) (Adams et al, 1985). F1 offspring were then bred to obtain Cks1+/+, Cks1+/− and Cks1−/−-Eμ-Myc transgenic littermates. E2f1-null mice (C57BL/6) were bred with Eμ-Myc transgenics (C57BL/6). F1 offspring were bred to obtain E2f1+/+, E2f1+/− and E2f1−/− Eμ-Myc transgenic littermates. Animals were observed for signs of morbidity and tumor development. Tumors were harvested after killing mice, snap-frozen in liquid nitrogen and processed for analysis of DNA, RNA and protein.
Burkitt lymphoma samples
With Institutional Review Board approval and following informed consent, tumors from 17 Burkitt lymphoma patients were banked. RNA and protein were extracted from these tumors. As a control, pooled peripheral blood mononuclear cells from a healthy donor were enriched using CD19-MicroBeads (Miltenyi Biotech) and RNA and protein were prepared.
Cell culture
Primary bone marrow-derived pre-B cells were cultured as described (Eischen et al, 1999). MEFs from E13.5–E14.5 embryos, or primary B cells, were cultured and infected with retroviruses as previously described (Eischen et al, 1999; Nilsson et al, 2004). To evaluate the consequences of Myc activation, cells were treated with 2 μM 4-HT±1μg/ml Chx (Sigma Chemicals) and harvested for protein and RNA preparation.
FACS analysis and magnetic-activated cell sorting (MACS) of B cells
Rates of proliferation of B220+sIgM+ and B220+sIgM− cells were determined using a Flow kit as described (Baudino et al, 2003). The remainder of the bone marrow and spleen cells were incubated with B220 MicroBeads and enriched by magnetic cell sorting for B cells (Miltenyi Biotech) and used for immunoblot or real-time PCR analysis.
Methods for RNA, protein and histological analyses can be found in the Supplementary data online.
Supplementary Material
Supplemental Material
Acknowledgments
We thank Sara Norton, Lottie Peppers and Elsie White for expert technical assistance, the Animal Resource Center, the Hartwell Center and the FACS Core of St Jude Children's Research Hospital (SJCRH), Mihaela Onciu and John T Sandlund (SJCRH) for providing Burkitt lymphoma samples, Charles Sherr and Martine Roussel (SJRCH) for providing p19Arf antibody, Kristian Helin (Biotech Research & Innovation Centre, Copenhagen, Denmark) for providing ER-E2f1 and the laboratory of Robert Costa (University of Illinois-Chicago) for providing FoxM1−/− MEFs. This work was supported by NIH grant CA76379 (JLC), Cancer Center Core Grant CA21765 and by the American Lebanese Syrian Associated Charities (ALSAC) of SJCRH. UK was funded by the Deutsche Forschungsgemeinschaft (grant KE222/5-1) and JBO was supported by NRSA grant F32 CA099478.
References
- Adams JM, Harris AW, Pinkert CA, Corcoran LM, Alexander WS, Cory S, Palmiter RD, Brinster RL (1985) The c-myc oncogene driven by immunoglobulin enhancers induces lymphoid malignancy in transgenic mice. Nature 318: 533–538 [DOI] [PubMed] [Google Scholar]
- Alt JR, Greiner TC, Cleveland JL, Eischen CM (2003) Mdm2 haplo-insufficiency profoundly inhibits Myc-induced lymphomagenesis. EMBO J 22: 1442–1450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askew DS, Ashmun RA, Simmons BC, Cleveland JL (1991) Constitutive c-myc expression in an IL-3-dependent myeloid cell line suppresses cell cycle arrest and accelerates apoptosis. Oncogene 6: 1915–1922 [PubMed] [Google Scholar]
- Bartek J, Lukas J (2001) p27 destruction: Cks1 pulls the trigger. Nat Cell Biol 3: E95–E98 [DOI] [PubMed] [Google Scholar]
- Bashir T, Dorrello NV, Amador V, Guardavaccaro D, Pagano M (2004) Control of the SCF(Skp2–Cks1) ubiquitin ligase by the APC/C(Cdh1) ubiquitin ligase. Nature 428: 190–193 [DOI] [PubMed] [Google Scholar]
- Baudino TA, McKay C, Pendeville-Samain H, Nilsson JA, Maclean KH, White EL, Davis AC, Ihle JN, Cleveland JL (2002) c-Myc is essential for vasculogenesis and angiogenesis during development and tumor progression. Genes Dev 16: 2530–2543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudino TA, Maclean KH, Brennan J, Parganas E, Yang C, Aslanian A, Lees JA, Sherr CJ, Roussel MF, Cleveland JL (2003) Myc-mediated proliferation and lymphomagenesis, but not apoptosis, are compromised by E2f1 loss. Mol Cell 11: 905–914 [DOI] [PubMed] [Google Scholar]
- Bello-Fernandez C, Packham G, Cleveland JL (1993) The ornithine decarboxylase gene is a transcriptional target of c-Myc. Proc Natl Acad Sci USA 90: 7804–7808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black EP, Hallstrom T, Dressman HK, West M, Nevins JR (2005) Distinctions in the specificity of E2F function revealed by gene expression signatures. Proc Natl Acad Sci USA 102: 15948–15953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouchard C, Thieke K, Maier A, Saffrich R, Hanley-Hyde J, Ansorge W, Reed S, Sicinski P, Bartek J, Eilers M (1999) Direct induction of cyclin D2 by Myc contributes to cell cycle progression and sequestration of p27. EMBO J 18: 5321–5333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boxer LM, Dang CV (2001) Translocations involving c-myc and c-myc function. Oncogene 20: 5595–5610 [DOI] [PubMed] [Google Scholar]
- Carrano AC, Eytan E, Hershko A, Pagano M (1999) SKP2 is required for ubiquitin-mediated degradation of the CDK inhibitor p27. Nat Cell Biol 1: 193–199 [DOI] [PubMed] [Google Scholar]
- DeGregori J, Kowalik T, Nevins JR (1995) Cellular targets for activation by the E2F1 transcription factor include DNA synthesis- and G1/S-regulatory genes. Mol Cell Biol 15: 4215–4224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egle A, Harris AW, Bouillet P, Cory S (2004) Bim is a suppressor of Myc-induced mouse B cell leukemia. Proc Natl Acad Sci USA 101: 6164–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Weber JD, Roussel MF, Sherr CJ, Cleveland JL (1999) Disruption of the ARF-Mdm2-p53 tumor suppressor pathway in Myc-induced lymphomagenesis. Genes Dev 13: 2658–2669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eischen CM, Woo D, Roussel MF, Cleveland JL (2001) Apoptosis triggered by Myc-induced suppression of Bcl-X(L) or Bcl-2 is bypassed during lymphomagenesis. Mol Cell Biol 21: 5063–5070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evan GI, Vousden KH (2001) Proliferation, cell cycle and apoptosis in cancer. Nature 411: 342–348 [DOI] [PubMed] [Google Scholar]
- Fero ML, Randel E, Gurley KE, Roberts JM, Kemp CJ (1998) The murine gene p27Kip1 is haplo-insufficient for tumour suppression. Nature 396: 177–180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fero ML, Rivkin M, Tasch M, Porter P, Carow CE, Firpo E, Polyak K, Tsai LH, Broudy V, Perlmutter RM, Kaushansky K, Roberts JM (1996) A syndrome of multiorgan hyperplasia with features of gigantism, tumorigenesis, and female sterility in p27(Kip1)-deficient mice. Cell 85: 733–744 [DOI] [PubMed] [Google Scholar]
- Ganoth D, Bornstein G, Ko TK, Larsen B, Tyers M, Pagano M, Hershko A (2001) The cell-cycle regulatory protein Cks1 is required for SCF(Skp2)-mediated ubiquitinylation of p27. Nat Cell Biol 3: 321–324 [DOI] [PubMed] [Google Scholar]
- Hao B, Zheng N, Schulman BA, Wu G, Miller JJ, Pagano M, Pavletich NP (2005) Structural basis of the Cks1-dependent recognition of p27(Kip1) by the SCF(Skp2) ubiquitin ligase. Mol Cell 20: 9–19 [DOI] [PubMed] [Google Scholar]
- Hemann MT, Zilfou JT, Zhao Z, Burgess DJ, Hannon GJ, Lowe SW (2004) Suppression of tumorigenesis by the p53 target PUMA. Proc Natl Acad Sci USA 101: 9333–9338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iritani BM, Eisenman RN (1999) c-Myc enhances protein synthesis and cell size during B lymphocyte development. Proc Natl Acad Sci USA 96: 13180–13185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishida N, Kitagawa M, Hatakeyama S, Nakayama K (2000) Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem 275: 25146–25154 [DOI] [PubMed] [Google Scholar]
- Jeffers JR, Parganas E, Lee Y, Yang C, Wang J, Brennan J, MacLean KH, Han J, Chittenden T, Ihle JN, McKinnon PJ, Cleveland JL, Zambetti GP (2003) Puma is an essential mediator of p53-dependent and -independent apoptotic pathways. Cancer Cell 4: 321–328 [DOI] [PubMed] [Google Scholar]
- Ji P, Jiang H, Rekhtman K, Bloom J, Ichetovkin M, Pagano M, Zhu L (2004) An Rb–Skp2–p27 pathway mediates acute cell cycle inhibition by Rb and is retained in a partial-penetrance Rb mutant. Mol Cell 16: 47–58 [DOI] [PubMed] [Google Scholar]
- Kamura T, Hara T, Matsumoto M, Ishida N, Okumura F, Hatakeyama S, Yoshida M, Nakayama K, Nakayama KI (2004) Cytoplasmic ubiquitin ligase KPC regulates proteolysis of p27(Kip1) at G1 phase. Nat Cell Biol 6: 1229–1235 [DOI] [PubMed] [Google Scholar]
- Kim SY, Herbst A, Twokowski KA, Salghetti SE, Tansey WP (2003) Skp2 regulates Myc protein stability and activity. Mol Cell 11: 1177–1188 [DOI] [PubMed] [Google Scholar]
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, Khanam D, Hayday AC, Frohman LA, Koff A (1996) Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 85: 721–732 [DOI] [PubMed] [Google Scholar]
- Kotake Y, Nakayama K, Ishida N, Nakayama KI (2005) Role of Serine-10 phosphorylation in p27 stabilization revealed by analysis of p27 knock-in mice harboring a Serine-10 mutation. J Biol Chem 280: 1095–1102 [DOI] [PubMed] [Google Scholar]
- Lewis BC, Shim H, Li Q, Wu CS, Lee LA, Maity A, Dang CV (1997) Identification of putative c-Myc-responsive genes: characterization of rcl, a novel growth-related gene. Mol Cell Biol 17: 4967–4978 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Levens D (2006) Making myc. Curr Top Microbiol Immunol 302: 1–32 [DOI] [PubMed] [Google Scholar]
- Maclean KH, Keller UB, Rodriguez-Galindo C, Nilsson JA, Cleveland JL (2003) c-Myc augments gamma irradiation-induced apoptosis by suppressing Bcl-XL. Mol Cell Biol 23: 7256–7270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek NP, Sundberg H, McGrew S, Nakayama K, Kyriakides TR, Roberts JM (2001) A mouse knock-in model exposes sequential proteolytic pathways that regulate p27Kip1 in G1 and S phase. Nature 413: 323–327 [DOI] [PubMed] [Google Scholar]
- Martins CP, Berns A (2002) Loss of p27(Kip1) but not p21(Cip1) decreases survival and synergizes with MYC in murine lymphomagenesis. EMBO J 21: 3739–3748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masuda TA, Inoue H, Nishida K, Sonoda H, Yoshikawa Y, Kakeji Y, Utsunomiya T, Mori M (2003) Cyclin-dependent kinase 1 gene expression is associated with poor prognosis in gastric carcinoma. Clin Cancer Res 9: 5693–5698 [PubMed] [Google Scholar]
- Montagnoli A, Fiore F, Eytan E, Carrano AC, Draetta GF, Hershko A, Pagano M (1999) Ubiquitination of p27 is regulated by Cdk-dependent phosphorylation and trimeric complex formation. Genes Dev 13: 1181–1189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller D, Bouchard C, Rudolph B, Steiner P, Stuckmann I, Saffrich R, Ansorge W, Huttner W, Eilers M (1997) Cdk2-dependent phosphorylation of p27 facilitates its Myc-induced release from cyclin E/cdk2 complexes. Oncogene 15: 2561–2576 [DOI] [PubMed] [Google Scholar]
- Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, Horii I, Loh DY, Nakayama K (1996) Mice lacking p27(Kip1) display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell 85: 707–720 [DOI] [PubMed] [Google Scholar]
- Nakayama K, Nagahama H, Minamishima YA, Matsumoto M, Nakamichi I, Kitagawa K, Shirane M, Tsunematsu R, Tsukiyama T, Ishida N, Kitagawa M, Nakayama K, Hatakeyama S (2000) Targeted disruption of Skp2 results in accumulation of cyclin E and p27(Kip1), polyploidy and centrosome overduplication. EMBO J 19: 2069–2081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsson JA, Cleveland JL (2003) Myc pathways provoking cell suicide and cancer. Oncogene 22: 9007–9021 [DOI] [PubMed] [Google Scholar]
- Nilsson JA, Maclean KH, Keller UB, Pendeville H, Baudino TA, Cleveland JL (2004) Mnt loss triggers Myc transcription targets, proliferation, apoptosis, and transformation. Mol Cell Biol 24: 1560–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Hagan RC, Ohh M, David G, de Alboran IM, Alt FW, Kaelin WG Jr, DePinho RA (2000) Myc-enhanced expression of Cul1 promotes ubiquitin-dependent proteolysis and cell cycle progression. Genes Dev 14: 2185–2191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oster SK, Ho CS, Soucie EL, Penn LZ (2002) The myc oncogene: marvelously complex. Adv Cancer Res 84: 81–154 [DOI] [PubMed] [Google Scholar]
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, Yew PR, Draetta GF, Rolfe M (1995) Role of the ubiquitin–proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 269: 682–685 [DOI] [PubMed] [Google Scholar]
- Pelengaris S, Littlewood T, Khan M, Elia G, Evan G (1999) Reversible activation of c-Myc in skin: induction of a complex neoplastic phenotype by a single oncogenic lesion. Mol Cell 3: 565–577 [DOI] [PubMed] [Google Scholar]
- Perez-Roger I, Kim SH, Griffiths B, Sewing A, Land H (1999) Cyclins D1 and D2 mediate myc-induced proliferation via sequestration of p27(Kip1) and p21(Cip1). EMBO J 18: 5310–5320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel MF, Cleveland JL, Shurtleff SA, Sherr CJ (1991) Myc rescue of a mutant CSF-1 receptor impaired in mitogenic signalling. Nature 353: 361–363 [DOI] [PubMed] [Google Scholar]
- Schmitt CA, McCurrach ME, de Stanchina E, Wallace-Brodeur RR, Lowe SW (1999) INK4a/ARF mutations accelerate lymphomagenesis and promote chemoresistance by disabling p53. Genes Dev 13: 2670–2677 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sgambato A, Cittadini A, Faraglia B, Weinstein IB (2000) Multiple functions of p27(Kip1) and its alterations in tumor cells: a review. J Cell Physiol 183: 18–27 [DOI] [PubMed] [Google Scholar]
- Shapira M, Ben-Izhak O, Bishara B, Futerman B, Minkov I, Krausz MM, Pagano M, Hershko DD (2004) Alterations in the expression of the cell cycle regulatory protein cyclin kinase subunit 1 in colorectal carcinoma. Cancer 100: 1615–1621 [DOI] [PubMed] [Google Scholar]
- Sherr CJ (2004) Principles of tumor suppression. Cell 116: 235–246 [DOI] [PubMed] [Google Scholar]
- Sherr CJ, Roberts JM (1999) CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 13: 1501–1512 [DOI] [PubMed] [Google Scholar]
- Slingerland J, Pagano M (2000) Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 183: 10–17 [DOI] [PubMed] [Google Scholar]
- Slotky M, Shapira M, Ben-Izhak O, Linn S, Futerman B, Tsalic M, Hershko DD (2005) The expression of the ubiquitin ligase subunit Cks1 in human breast cancer. Breast Cancer Res 7: R737–R744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spruck C, Strohmaier H, Watson M, Smith AP, Ryan A, Krek TW, Reed SI (2001) A CDK-independent function of mammalian Cks1: targeting of SCF(Skp2) to the CDK inhibitor p27Kip1. Mol Cell 7: 639–650 [DOI] [PubMed] [Google Scholar]
- Strasser A, Harris AW, Bath ML, Cory S (1990) Novel primitive lymphoid tumours induced in transgenic mice by cooperation between myc and bcl-2. Nature 348: 331–333 [DOI] [PubMed] [Google Scholar]
- Tedesco D, Lukas J, Reed SI (2002) The pRb-related protein p130 is regulated by phosphorylation-dependent proteolysis via the protein-ubiquitin ligase SCF(Skp2). Genes Dev 16: 2946–2957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trumpp A, Refaeli Y, Oskarsson T, Gasser S, Murphy M, Martin GR, Bishop JM (2001) c-Myc regulates mammalian body size by controlling cell number but not cell size. Nature 414: 768–773 [DOI] [PubMed] [Google Scholar]
- Vigo E, Muller H, Prosperini E, Hateboer G, Cartwright P, Moroni MC, Helin K (1999) CDC25A phosphatase is a target of E2F and is required for efficient E2F-induced S phase. Mol Cell Biol 19: 6379–6395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang IC, Chen YJ, Hughes D, Petrovic V, Major ML, Park HJ, Tan Y, Ackerson T, Costa RH (2005) Forkhead box M1 regulates the transcriptional network of genes essential for mitotic progression and genes encoding the SCF (Skp2–Cks1) ubiquitin ligase. Mol Cell Biol 25: 10875–10894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welcker M, Orian A, Jin J, Grim JA, Harper JW, Eisenman RN, Clurman BE (2004) The Fbw7 tumor suppressor regulates glycogen synthase kinase-3 phosphorylation-dependent c-Myc protein degradation. Proc Natl Acad Sci USA 101: 9085–9090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlach J, Hennecke S, Alevizopoulos K, Conti D, Amati B (1996) Growth arrest by the cyclin-dependent kinase inhibitor p27Kip1 is abrogated by c-Myc. EMBO J 15: 6595–6604 [PMC free article] [PubMed] [Google Scholar]
- von der Lehr N, Johansson S, Wu S, Bahram F, Castell A, Cetinkaya C, Hydbring P, Weidung I, Nakayama K, Nakayama KI, Soderberg O, Kerppola TK, Larsson LG (2003) The F-box protein Skp2 participates in c-Myc proteosomal degradation and acts as a cofactor for c-Myc-regulated transcription. Mol Cell 11: 189–2000 [DOI] [PubMed] [Google Scholar]
- Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, Kim DW, Hofmann CS, Pianetti S, Romieu-Mourez R, Freedman LP, Sonenshein GE (2001) Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 20: 1688–1702 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material