

Abstract
Sulfatase modifying factor 1 (SUMF1) is the gene mutated in multiple sulfatase deficiency (MSD) that encodes the formylglycine-generating enzyme, an essential activator of all the sulfatases. SUMF1 is a glycosylated enzyme that is resident in the endoplasmic reticulum (ER), although it is also secreted. Here, we demonstrate that upon secretion, SUMF1 can be taken up from the medium by several cell lines. Furthermore, the in vivo engineering of mice liver to produce SUMF1 shows its secretion into the blood serum and its uptake into different tissues. Additionally, we show that non-glycosylated forms of SUMF1 can still be secreted, while only the glycosylated SUMF1 enters cells, via a receptor-mediated mechanism. Surprisingly, following its uptake, SUMF1 shuttles from the plasma membrane to the ER, a route that has to date only been well characterized for some of the toxins. Remarkably, once taken up and relocalized into the ER, SUMF1 is still active, enhancing the sulfatase activities in both cultured cells and mice tissues.
Keywords: endoplasmic reticulum, protein secretion and uptake, sulfatases, SUMF1, trafficking
Introduction
The sulfatases are a family of enzymes that catalyze the hydrolysis of sulfate esters after they have been post-translationally activated (Diez-Roux and Ballabio, 2005). A consensus sequence in their catalytic domain contains a cysteine that is modified into formylglycine (FGly) within the endoplasmic reticulum (ER). The FGly is essential for sulfatase activity, as in its hydrate form, FGly is able to accept the sulfate group from the substrate and subsequently remove it from the enzyme (Schmidt et al, 1995; Boltes et al, 2001). We and others have identified the gene, sulfatase modifying factor 1 (SUMF1), that encodes the FGly-generating enzyme (Cosma et al, 2003; Dierks et al, 2003). In patients affected by multiple sulfatase deficiency (MSD), all of their sulfatase activities are reduced because SUMF1 is hampered in its function, and mutations in SUMF1 have been found in all MSD patients analyzed to date (Cosma et al, 2004). The crystal structure of SUMF1 has been solved and SUMF1 has been recognized as having an oxygenase function (Dierks et al, 2005). Overexpression of SUMF1 with sulfatases in cultured cells via transfection or viral delivery results in a strong enhancement of the sulfatase activities (Cosma et al, 2003; Fraldi et al, 2007). SUMF1 has been conserved through evolution and has retained a high homology with the bacterial SUMF1 proteins (Sardiello et al, 2005). Sequence comparisons led to the discovery of a paralogue of SUMF1 in the vertebrate genome, known as SUMF2 (Cosma et al, 2003; Dierks et al, 2003). The primary sequences of the SUMF1 and SUMF2 proteins are highly similar. SUMF2 colocalizes with SUMF1 within the ER and inhibits the enhancing effect of SUMF1 on the sulfatases (Zito et al, 2005).
SUMF1 is an ER-resident protein. It is glycosylated and its intracellular form contains high mannose-type oligosaccharides. SUMF1 is also secreted and the secreted form contains a family of complex-type oligosaccharides (Preusser-Kunze et al, 2005). Many secreted glycoproteins are cleared from the plasma through the mannose receptor (MR) (Lee et al, 2002). The MR is an endocytic receptor for glycans that is ubiquitously expressed. It contains carbohydrate recognition domains that bind complex-type oligosaccharides such as terminal fucose and acetylglucosamine residues (Taylor and Drickamer, 1993). Sulfatases are also secreted into the plasma and taken up via the mannose-6-phosphate receptor (MPR) (Ni et al, 2006). The MPR has two forms in the cell, MPR 46 and MPR 300, both of which can transport hydrolases from the trans-Golgi network to endosomes and/or lysosomes (Ni et al, 2006).
We now show that endogenous SUMF1 is also secreted. Furthermore, we demonstrate that both overexpressed and endogenous SUMF1 can be taken up from the medium by different cells and from the plasma in tissues of mice engineered to produce SUMF1 in the liver. The uptake of SUMF1 is principally mediated by the MR, and to some extent by the MPR. The secretion and activity of SUMF1 are independent of its glycosylation; in contrast, its uptake in cells is impaired when the protein lacks sugars. Finally, we show here that once SUMF1 has been taken up, it relocalizes to the ER in both cultured cells and mice tissues, where it is enzymatically active, as it can enhance the sulfatase activities.
Results
SUMF1 is secreted and taken up into the ER
SUMF1 is localized in the ER, where it activates all newly synthesized sulfatases during or shortly after cotranslational import (Cosma et al, 2003; Dierks et al, 2003). We have produced a stable HeLa cell clone that expresses SUMF1-3xFlag that we have named the HL3xFS1 clone. Three specific bands of about 42, 39 and 33 kDa that correspond to three forms of the SUMF1 protein were detected in cellular extracts from the HL3xFS1 clone using an anti-Flag antibody (Figure 1A). These different bands of SUMF1 correspond to its differently glycosylated and/or proteolytically processed forms, as has been previously demonstrated (Preusser-Kunze et al, 2005; Zito et al, 2005), and as shown by mass spectrometry analysis of protein extracts of the HL3xFS1 clone (Supplementary Figure 1). SUMF1 was predicted to be secreted through large-scale, bioinformatics, high-throughput screening (Clark et al, 2003). Recently, it was demonstrated that SUMF1 is secreted when it is overexpressed in HT1080 cells (Preusser-Kunze et al, 2005). As expected, Western blotting of conditioned medium collected from the HL3xFS1 clone showed secretion of SUMF1-3xFlag into the medium (Supplementary Figure 2A). In addition, by decorating the filter with antibodies against several different intracellular and secreted proteins, we have demonstrated that SUMF1-3xFlag is actively secreted into the medium, and have excluded that cell breakage was causing a leakage of SUMF1 into the medium. The overexpression of SUMF1-3xFlag also does not result in the nonspecific secretion of other endogenous proteins (Supplementary Figure 2A).
Figure 1.

Secretion and uptake of SUMF1. (A) SUMF1 expression in the HL3xFS1 clone. Three bands of about 42, 39 and 33 kDa were detected by Western blotting with an anti-Flag antibody. (B) SUMF1 is secreted from the HL3xFS1 clone and taken up into Cos7 and HeLa cells. Lanes 1–4: cellular extracts from recipient Cos7 and HeLa cells incubated with conditioned medium (HL3xFS1M) collected from the HL3xFS1 clone or with control HeLa cell medium, as analyzed by Western blotting. Lanes 5 and 6: 5% of SUMF1-Flag-conditioned and control medium used to culture the recipient cells. Lane 7: 10% of the total SUMF1-Flag-conditioned medium. Western blotting was carried out with anti-Flag and anti--tubulin antibodies. (C) SUMF1 is secreted from the HL3xFS1 clone and taken up in wild-type MEFs and Sumf1−/− MEFs. Lanes 1–4: cellular extracts from recipient cells cultured in SUMF1-Flag-conditioned and control medium were immunoblotted with anti-Flag and anti--tubulin antibodies. (D) Endogenous SUMF1 is secreted from HepG2 cells and taken up by MSD fibroblasts. HepG2 cells were cultured for 24, 36 and 48 h. Protein extracts and concentrated media were analyzed by Western blotting with an anti-human-SUMF1 antibody. MSD1 (p.A149-A173del+p.S359X) and MSD2 (p.M1R+rfs) fibroblasts were cultured for 24 h in concentrated HepG2 medium collected from two T75 flasks of confluent HepG2 cells. The protein extracts were analyzed using anti-SUMF1 and anti--tubulin antibodies.
The sulfatases are secreted proteins that can be taken up into all cells of the body via the ubiquitously expressed MPR. Starting from this observation, we asked whether SUMF1 itself can also be taken up into cells from the medium. We have in the laboratory, mouse embryonic fibroblasts (MEFs) from a Sumf1−/− mouse model that we recently generated (Settembre et al, 2007). Thus, Cos7 and HeLa cells, and MEFs from wild-type and Sumf1−/− mice were incubated in conditioned medium collected from the confluent HL3xFS1 clone after 12 h of culture. The protein extracts from the recipient cells were analyzed by Western blotting with an anti-Flag antibody. SUMF1 was seen to be taken up in all three cell types (Figure 1B and C, lanes 1–4). Here, the controls included conditioned and non-conditioned medium (Figure 1B, lanes 5 and 6) and a sample of the conditioned medium that was not applied to recipient cells (Figure 1B, lane 7). Protein loading controls were also performed using an anti--tubulin antibody (Figure 1B and C). To confirm these results, we performed mass spectrometry analysis of SUMF1-Flag following its uptake into recipient HeLa cells incubated in conditioned medium collected from the HL3xFS1 clone. Protein extracts of the recipient cells were immunoprecipitated with an anti-Flag antibody and MALDIMS and LC-MS/MS analysis of the excised, deglycosylated and trypsin-digested bands were carried out. We clearly detected SUMF1 in bands 1 and 2 and did not detect it in band 3 (Supplementary Figure 1).
These data clearly show that the overexpressed SUMF1 that is secreted into the medium by the HL3xFS1 clone is taken up into all the cell lines analyzed. By Western blotting, we detected primarily the uptake of the 42-kDa SUMF1 form, although under some conditions (e.g. when more protein is accumulated in the conditioned medium), the 39-kDa SUMF1 form can also be taken up, as shown in the mass spectrometry analysis (Supplementary Figure 1).
We then investigated whether secretion and uptake of SUMF1 occurs only when SUMF1 is overexpressed, or whether the endogenous protein can also follow the same route. For this, we used HepG2 cells, which are a human hepatoma cell line that has been described as being highly secretory (Knowles et al, 1980). HepG2 cells were cultured for 8 h and the conditioned medium was analyzed. A specific, albeit faint, band was detected by Western blotting using an anti-human-SUMF1 polyclonal antibody (Supplementary Figure 2B). Thus, we performed time courses by collecting the conditioned medium after 24, 36 and 48 h of HepG2 cultivation. A positive signal for SUMF1 was detected in the cellular pellet and in the concentrated media from these HepG2 cells using an anti-human-SUMF1 polyclonal antibody (Figure 1D, top panels). These data demonstrate that endogenous SUMF1 is secreted from the HepG2 cells.
Furthermore, for the uptake of this endogenous SUMF1, confluent recipient MSD fibroblasts were incubated in the concentrated conditioned medium (collected after 8 h of culture of 10 million confluent HepG2 cells). Of note, these two specifically chosen MSD fibroblast cell lines harbor mutations that cause a frameshift or the early truncation of SUMF1, and thus they did not have detectable levels of the endogenous protein. The endogenous SUMF1 of the HepG2 cells was indeed taken up by these MSD cells, as shown by Western blotting (Figure 1D, lower panels). A cellular extract of wild-type fibroblasts was also analyzed to indicate the control levels of SUMF1 in these cells (Figure 1D, lower panels, WT). The total proteins were normalized using an anti--tubulin antibody (Figure 1D, lower panels). Thus, the endogenous SUMF1 in HepG2 cells is secreted into the medium and can be taken up by human fibroblast cell lines.
We next analyzed the subcellular localization of SUMF1 after its uptake. Sumf1−/− MEFs, Cos7 cells and MSD fibroblasts were incubated with SUMF1-Flag-conditioned medium. Indirect immunofluorescence using an anti-Flag antibody demonstrated that SUMF1-Flag localized in the perinuclear region of Sumf1−/− MEFs and Cos7 cells (Figure 2A and Supplementary Figure 3), with an ER localization confirmed in Sumf1−/− MEFs, Cos7 cells and MSD cells by its colocalization with the ER marker ERAB using anti-Flag, anti-SUMF1 and anti-ERAB antibodies (Figure 2B; Supplementary Figure 3, and data not shown). In addition, SUMF1 colocalized partly with the endosome markers LAMP1 and LAMP2, indicating that this uptake could be mediated via the endosomal compartment (Figure 2B, and data not shown). The localization of SUMF1 after its uptake resembles the localization of the endogenous SUMF1 protein in wild-type MEFs that colocalizes with the ERAB marker (Figure 2C). Vice versa, a SUMF1-specific signal was not seen in Sumf1−/− MEFs (Figure 2D). These results are striking since they show for the first time that apart from some of the toxins (Sandvig and van Deurs, 2000), other proteins appear to be taken up by cells and localized to the ER.
Figure 2.

Subcellular localization of SUMF1 taken up into Sumf1−/− MEFs cultured in the conditioned medium for 12 h. (A) SUMF1-Flag is taken up and localizes to the perinuclear region of Sumf1−/− MEFs. Internalized SUMF1-Flag is revealed using an anti-Flag antibody. DAPI is shown as a nuclear marker. (B) Subcellular localization of SUMF1 following its uptake into Sumf1−/− MEFs. SUMF1-Flag uptake in Sumf1−/− MEFs was revealed using an anti-SUMF1 antibody. SUMF1-Flag colocalizes with ERAB (an ER marker) and LAMP1 (an endosome marker), as seen by confocal microscopy. (C, D) Wild-type and Sumf1−/− MEFs were decorated with anti-SUMF1 and anti-ERAB antibodies. Scale bar, 5 μm.
SUMF1 is enzymatically active after its uptake
To determine if SUMF1 is enzymatically active after its uptake, the activities of different sulfatases were tested in recipient cells. First, we transfected Cos7 cells with IDS (iduronate sulfatase), SGSH (sulfamidase) and ARSA (arylsulfatase A) cDNAs. These cells were then incubated with SUMF1-conditioned medium or with a conditioned medium containing the fully inactive SUMF1C336R mutant (Cosma et al, 2004; Dierks et al, 2005). These conditioned media were recovered from HeLa cells that had been transfected with either wild-type SUMF1 cDNA or the SUMF1C336R mutant cDNA. The sulfatase activities were measured as previously described (Cosma et al, 2004), using specific substrates (Figure 3A). Enhancement of sulfatase activity was detected after the uptake of SUMF1 and not after the uptake of the SUMF1C336R mutant, with respect to the basal levels of the transfected sulfatases. Since IDS, SGSH and other sulfatases are also secreted and taken up, we wanted to be certain that these enhanced sulfatase activities measured in the Cos7 cells were actually due to uptake of SUMF1 and not to the uptake of endogenous sulfatases secreted into the conditioned medium by the HeLa cells. We thus used a lentiviral vector expressing SUMF1-3xFlag to transduce two different human MPSII and MPSIIIA fibroblast lines. MPSII and MPSIIIA are two mucopolysaccharidoses in which the IDS and SGSH enzymes, respectively, are inactive (Neufeld and Muenzer, 2001). The transduced fibroblasts were used as the SUMF1-producing cell lines so as to obtain medium containing active SUMF1 and inactive IDS or SGSH. MSD-recipient cells were incubated with these conditioned media. IDS activity was measured in the cell line cultured in the SUMF1-conditioned medium produced from the MPSII (II-1S1 and II-2S1)-transduced cells, whereas SGSH activity was measured in the MSD cell lines incubated in the SUMF1-conditioned medium produced from the MPSIIIA-transduced cells (IIIA-1S1 and IIIA-2S1). A partial rescue of both enzymatic activities was detected (Figure 3B). The IDS and SGSH activities measured in the MSD cell lines after uptake were higher with respect to the basal levels for both of these enzymes. SUMF1 expression in MPSII and MPSIIIA fibroblasts and its uptake into the MSD cell lines was also investigated by Western blotting (Figure 3B). In addition, to be further sure that the effects seen were due to SUMF1 uptake, we investigated the rescue of a sulfatase activity that is not secreted, ARSC (Ballabio and Shapiro, 1995). We thus incubated the MSD cell lines with SUMF1-conditioned medium from the HL3xFS1 clone. We measured a high level of ARSC activity after uptake in the MSD cell line. The levels detected were also much higher than the endogenous enzymatic ARSC activity of the MSD fibroblasts incubated with control HeLa cell-conditioned medium (Figure 3B). Finally, we performed uptake experiments using MEFs from Sumf1−/− mice as recipient cells. These cells have the main advantage of not having any residual sulfatase activity (Settembre et al, 2007). SUMF1 was produced from the MPSII- and MPSIIIA-producing cells and from the HL3xFS1 clone. A partial rescue of the IDS, SGSH and ARSC activities in the extracts from the recipient cells was seen (Figure 3C). The activities of IDS and SGSH were higher with respect to the basal levels in the Sumf1−/− MEFs; however, they were lower with respect to the levels measured in the wild-type MEFs. Of note, after the uptake of SUMF1, the ARSC activity was higher with respect to ARSC levels measured in both the Sumf1−/− and wild-type MEFs (Figure 3C). Likewise, although we clearly detected a rescue of the activity of different sulfatases in the MSD fibroblasts (Figure 3B), the measured enzymatic levels in the complemented MSD cell lines were lower with respect to the mean activities in wild-type fibroblasts (IDS was about 20% with respect to wild type, SGSH about 27% and ARSC about 80%; data not shown). Thus, to better detect the enhancing activity of internalized SUMF1, we decided to overexpress one sulfatase, IDS, in recipient MSD and Sumf1−/− cell lines using a lentiviral vector. The IDS-transduced MSD cells were incubated with SUMF1-conditioned media from each of the two different MPSII-producing cell lines: the IDS activities were increased by 30 and 76% in the recipient MSD cells after SUMF1 uptake (Figure 3D, II-2S1 and II-1S1, respectively). Similarly, the IDS-transduced Sumf1−/− MEFs were cultured with SUMF1-conditioned medium produced from Ids−/− MEFs transduced with a lentivirus expressing SUMF1. After SUMF1 uptake, the IDS activity was increased by 90% in the recipient Sumf1−/− cells (Figure 3D, Ids−/−S1). Altogether, these data demonstrate that SUMF1 is active after its uptake, as it can still enhance sulfatase activities.
Figure 3.
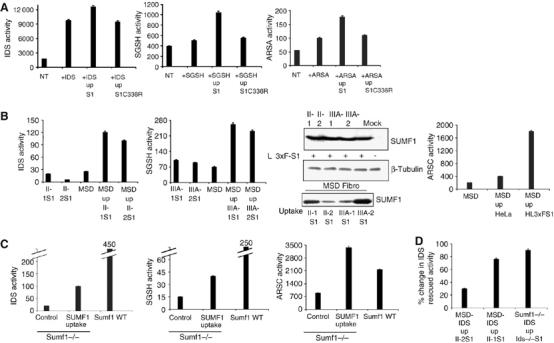
SUMF1 is enzymatically active following its uptake. (A) After its uptake, SUMF1 enhances the activity of transfected sulfatases. Cos7 cells were transfected with IDS, SGSH and ARSA cDNAs, and after 24 h, were incubated with medium containing SUMF1 or the inactive SUMF1C336R mutant. Sulfatase activities were measured in cellular extracts and compared with the activities in the extracts prepared from non-transfected cells (NT) and from sulfatase-transfected cells without SUMF1 uptake. (B) SUMF1 is taken up and can partially rescue sulfatase activities in MSD fibroblasts. Two different MPSII (II-1, II-2) and MPSIIIA (IIIA-1, IIIA-2) cell lines were transduced with a lentiviral vector carrying SUMF1-3xFlag cDNA to generate the conditioned-medium-producing II-1S1, II-2S1, IIIA-1S1 and IIIA-2S1 cells. The IDS and SGSH activities were analyzed in cellular pellets of producing cells and of MSD (p.S155P+S155P)-recipient fibroblasts. For ARSC activity, the producer cell line was the HL3xFS1 clone, and ARSC activity was evaluated in recipient cells and in control cells (MSD incubated in HeLa medium). Expression of SUMF1-Flag (producing cells, upper panels) and its uptake (recipient cells, lower panel) was also evaluated by Western blotting using an anti-Flag antibody. Western blotting with an anti--tubulin antibody provided the loading controls. (C) Uptake of SUMF1 in Sumf1−/− MEFs cultured in media collected from II-2S1 (for IDS activity), from IIIA-2S1 (for SGSH activity) and from HL3xFS1 (for ARSC activity) cells. The IDS, SGSH and ARSC activities in Sumf1−/− and wild-type MEFs are shown. (D) Uptake of SUMF1 in MSD fibroblasts and Sumf1−/− MEFs overexpressing IDS. The MSD+IDS-recipient cells were cultured in SUMF1-conditioned medium collected from II-2S1 and II-1S1-producing cells. The Sumf1−/−+IDS-recipient cells were cultured in SUMF1-conditioned medium collected from Ids−/− MEFs transduced with a lentivirus overexpressing SUMF1 cDNA. The IDS activities are expressed as percentages of the increased activities of the recipient cells cultured in the SUMF1-conditioned medium versus the activity measured in the recipient cells cultured in non-conditioned medium.
Glycosylation of SUMF1 is not essential for its secretion and function
SUMF1 is N-glycosylated on its asparagine 141 (N141; Preusser-Kunze et al, 2005). To analyze the role of the glycosyl residues in the secretion and activity of SUMF1, we mutated N141 to alanine in the SUMF1-3xFlag cDNA. The SUMF1N141A mutant protein was detected in cellular extracts of HeLa cells transfected with the mutated cDNA and showed a profile of migrating bands slower with respect to wild-type SUMF1 (Figure 4A, lanes 1 and 3). SUMF1N141A is not sensitive to EndoH digestion (Figure 4A, lane 4) and the Western blotting showed a pattern of bands equivalent to those of SUMF1 digested with EndoH (Figure 4A, compare lanes 1 and 4), further demonstrating that SUMF1N141A is not glycosylated. In addition, the protein correctly localized in the ER when the mutant cDNA was transfected in HeLa cells (Figure 4B). Surprisingly, SUMF1N141A was still secreted into the medium, and furthermore, the secreted mutant was insensitive to the digestion with PNGase F, as expected (Figure 4C, upper panel). This enzyme cleaves between the innermost GlcNAC and asparagine residues of the glycosylated content and has previously been demonstrated to digest the secreted SUMF1 forms (Preusser-Kunze et al, 2005). Finally, we did not detect uptake of SUMF1N141A in HeLa cells cultured in medium containing the mutant SUMF1 form (Figure 4C, lower panel).
Figure 4.
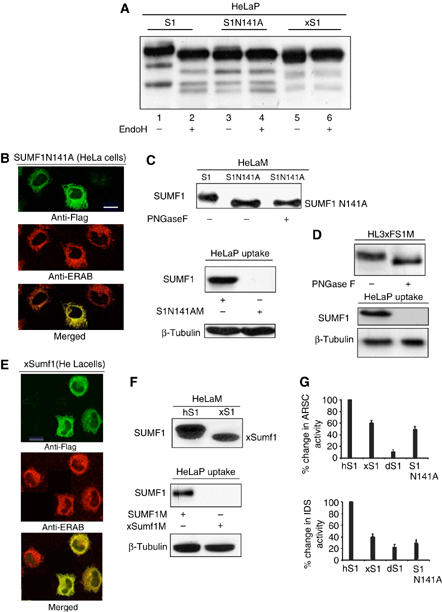
SUMF1 secretion and activity is independent of its glycosylation. (A) Glycosylation analysis. HeLa cells were transfected with SUMF1-3xFlag, SUMF1421G>A,422C>A-3xFlag (transduced to SUMF1N141A-3xFlag) and xSumf1-3xFlag. After 48 h, the cells were harvested and the cellular extracts were treated with EndoH, as indicated. The samples were analyzed by Western blotting and the filters were decorated with an anti-Flag antibody. The pattern of bands of SUMF1 in the second lane shows EndoH-sensitive glycosylation. (B) SUMF1N141A localizes to the ER. HeLa cells transfected with SUMF1421G>A,422C>A-3xFlag were processed for indirect immunofluorescence using anti-Flag and anti-ERAB antibodies. SUMF1N141A colocalizes with ERAB (an ER marker), as seen by confocal microscopy. (C) SUMF1N141A is secreted from transfected HeLa cells. Media were collected and concentrated. The conditioned medium containing SUMF1N141A was digested with PNGase F. The samples were analyzed by Western blotting using an anti-Flag antibody (upper panels). For uptake, HeLa cells were cultured for 12 h in concentrated medium collected from one T75 flask of confluent cells transfected with SUMF1-3xFlag or SUMF1421G>A,422C>A-3xFlag. The protein extracts were analyzed using anti-Flag and anti--tubulin antibodies (lower panels). (D) SUMF1 secreted from HL3xFS1 contains PNGase F-sensitive glycosylation. Media collected from the HL3xFS1 clone were treated with PNGase F, as indicated (upper panel). HeLa cells were cultured in media treated and not treated with PNGase F (lower panel). The samples were analyzed by Western blotting. (E) xSumf1 localizes to the ER. HeLa cells transfected with xSumf1-3xFlag were analyzed by indirect immunofluorescence using anti-Flag and anti-ERAB antibodies. xSumf1 colocalized with the ERAB marker, as seen by confocal microscopy. (F) xSumf1 is secreted from transfected HeLa cells. Media were collected, concentrated and analyzed by Western blotting using an anti-Flag antibody (upper panels). For uptake, HeLa cells were cultured for 12 h in concentrated conditioned medium collected from one T75 flask of confluent cells transfected with SUMF1-3xFlag or xSumf1-3xFlag. The protein extracts were analyzed using an anti-Flag antibody (lower panels). (G) SUMF1N141A and xSumf1 retain the enhancing activity on IDS and ARSC. ARSC and IDS were transfected alone or in combination with SUMF1-3xFlag, xSumf1-3xFlag, dSumf1-3xFlag or SUMF1421G>A,422C>A-3xFlag in HeLa cells. The IDS and ARSC activities are expressed as percentages of the increased activities in cells expressing only the sulfatase versus the activities measured in cells expressing the sulfatase plus SUMF1, xSumf1, dSumf1 or SUMF1N141A.
To investigate whether the uptake of SUMF1 is dependent on its glycosylation, we digested the conditioned medium recovered from the HL3xFS1 clone with the PNGase F enzyme. Western blotting of the treated and non-treated media was performed with an anti-Flag antibody and a fast migrating band corresponding to the digested SUMF1 form was detected (Figure 4D, upper panel). Thus, we cultured HeLa-recipient cells in the HL3xFS1-conditioned medium that had been digested and not been digested with PNGase F. Western blotting of the protein extracts demonstrated the uptake of the non-digested SUMF1 exclusively (Figure 4D, lower panel). The mass spectrometry analysis is also of note, where we saw that in the HeLa-recipient cells, the glycosylated SUMF1 forms corresponding to bands 1 and 2 were present, whereas band 3, corresponding to non-glycosylated SUMF1, was not seen (Supplementary Figure 1). Thus, SUMF1 can be taken up into the cells only if its glycosylation is preserved. These results demonstrate that non-glycosylated SUMF1 can be secreted into the medium, but it cannot be taken up, at least under these experimental conditions. Finally, to further confirm these observations, we expressed Xenopus Sumf1 (xSumf1) cDNA in HeLa cells. xSumf1 does not contain N-glycosylation since N141 is not conserved (Dierks et al, 2005). The xSumf1 protein was well expressed in the HeLa cells, as shown by Western blotting (Figure 4A, lane 5) and it is not sensitive to EndoH digestion, further demonstrating that it is not glycosylated (Figure 4A, lane 6). Furthermore, the xSumf1 protein localized to the ER, as shown by the merged signal with ERAB (Figure 4E). Thus, we checked its secretion and uptake by Western blotting. We found efficient secretion from HeLa cells (Figure 4F, upper blot), but no uptake into HeLa-recipient cells (Figure 4F, lower blot).
Finally, since we discovered that both SUMF1N141A and xSumf1 localize to the ER, we wondered if they were still active and thus, if they retained their enhancing activities on the sulfatases. Transient transfections were carried out in HeLa cells of SUMF1N141A and xSumf1, or as controls, (Drosophila) dSumf1 and (human) hSUMF1, with either ARSC or IDS cDNAs. SUMF1N141A and xSumf1 retained 50 and 62% of enhancing activity on ARSC, and 35 and 40% for IDS, respectively, with respect to wild-type hSUMF1 (Figure 4G). As a control, dSumf1 had 17% activity on ARSC and 25% on IDS, with respect to wild-type hSUMF1, similar to our previous findings (Cosma et al, 2003). These results demonstrate that not only does non-glycosylated SUMF1 localize to the ER, but it also still retains its enhancing activity on the sulfatases.
SUMF1 is secreted and taken up in vivo
Having shown the secretion and uptake of SUMF1 in cultured cells, we next tested whether this effect applies also in vivo. Thus, we produced an adeno-associated viral vector, AAV2/8, expressing SUMF1-3xFlag from the thyroxine-binding globulin (TBG) promoter. The AAV2/8 serotype transduces hepatocytes with high efficiency and the TBG promoter is liver specific; therefore, SUMF1 can only be expressed by the liver (Cardone et al, 2006). We transduced 2-month-old wild-type mice (n=3) by tail-vein injection with a total of 2.8 × 1011 viral particles. Ten days after transduction, the mice were bled and killed. Western blotting was performed using an anti-Flag antibody on the liver homogenates and on the immunoprecipitates from the serum. A SUMF1 signal was clearly detected in the liver and serum of the transduced animals (Figure 5A, left-hand panels). To determine if SUMF1 was taken up in the non-transduced organs of these mice, we analyzed the presence of the protein in bone marrow cells after immunoprecipitation with an anti-Flag antibody. A clear signal was detected in all of the transduced animals (Figure 5A, right-hand panel). Furthermore, we analyzed the uptake of SUMF1 by immunofluorescence in the non-transduced tissues, including spleen, lung and kidney, using an anti-Flag monoclonal antibody. We detected positive signals in the liver, the producer organ, and also in the spleen, kidney and lung, indicating that SUMF1 was taken up into these tissues from the serum. Again, SUMF1 localized to the ER, as seen by the merge with the signals that were detectable with the ER marker ERAB, by both indirect light and confocal microscopy techniques (Figure 5B). To determine if SUMF1 was also active after its uptake, we temporal-vein injected one wild-type and two Sumf1−/− mice littermates at 2 days of age. Ten days after the injections, the transduced and control littermates (non-transduced mice) were killed and the activity of ARSC, the non-secreted sulfatase, was measured. The use of ARSC here was specifically chosen, again since it is not secreted, and this allows the conclusion that the rescue is due to the uptake of SUMF1 and not to the uptake of the secreted sulfatases produced in the liver. The transduction efficiencies were different in the two injected Sumf1−/− mice, as shown by the activities measured in the livers (Figure 5C). Surprisingly, we detected a significant rescue of ARSC activity in the kidneys of the two Sumf1−/− knock-out mice and an increased activity in the kidney of the treated wild-type mouse (Figure 5C). These data demonstrate that once secreted from a producing organ into the serum, SUMF1 is not only taken up by other distant tissues, but it also relocalizes to the ER, while enhancing the activity of the sulfatases.
Figure 5.
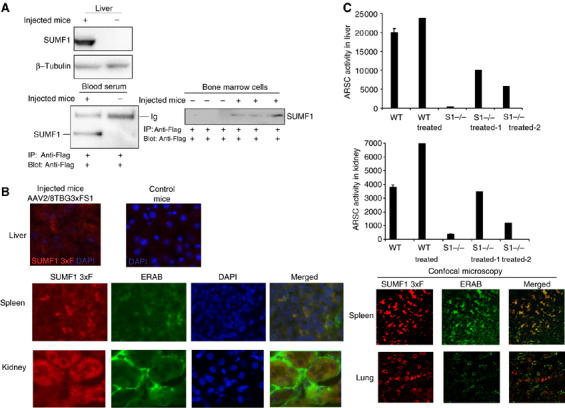
SUMF1 is taken up into mice tissues. (A) Uptake and secretion of SUMF1 in tissues of wild-type mice. AAV2/8TBG-3xFS1 particles (2.8 × 1011 total) were tail-vein injected into three C56BL/6 mice. Ten days later, control (non-injected) and injected mice were killed and their tissues were analyzed. Expression of SUMF1 was detected in the transduced livers by Western blotting and secretion of SUMF1-Flag was detected in the plasma by immunoprecipitation (one transduced mouse shown). The uptake of SUMF1-Flag in bone marrow cells was evaluated by immunoprecipitating cell extracts, and is shown for all of the injected mice. Anti-Flag and anti--tubulin antibodies were used. (B) Immunofluorescence of sections from injected mice. The expression of SUMF1-Flag was analyzed in the transduced liver. Uptake and subcellular localization of SUMF1-Flag were analyzed in the spleen, kidney and lung by fluorescence and confocal microscopy techniques. Anti-Flag polyclonal and anti-ERAB monoclonal antibodies were used. DAPI is shown as a nuclear marker. (C) ARSC activity following uptake of SUMF1 in Sumf1−/− mice. AAV2/8TBG-3xFS1 particles (5.6 × 1011 total) were temporal-vein injected into two Sumf1−/− mice and one wild-type mouse at p2. Ten days later, transduction and ARSC activities were assayed in their liver and kidney homogenates.
The uptake of SUMF1 is receptor mediated
We next investigated how SUMF1 is taken up into cells. The sulfatases are internalized from the plasma into cells through the MPR, and by a low-affinity process through the MR (Lee et al, 2002; Ni et al, 2006). Therefore, we wanted to see if SUMF1 follows the same path, by using receptor-competition assays in the uptake experiments. Human wild-type fibroblasts were cultured in SUMF1-conditioned medium containing two concentrations of yeast mannan (2 and 3 mg/ml), a specific competitor for binding to the MR (Taylor and Drickamer, 1993). Alternatively, these cells were cultured in medium containing SUMF1 plus anti-MR-antibody (1 mg/ml). The uptake of SUMF1 was drastically reduced under both of these conditions (Figure 6A, compare lane 1 with lanes 2–4). In contrast, when the cells were cultured in SUMF1-conditioned medium plus mannose 6-phosphate (10 mM), which competes for the MPR (Hiesberger et al, 1998), almost no reduction in uptake was detected (Figure 6A, compare lanes 5 and 8). Finally, when the cells where cultured in SUMF1-conditioned medium plus both mannan (3 mg/ml) and mannose 6-phosphate (10 mM), the uptake of SUMF1 was fully abolished (Figure 6A, compare lanes 5 and 7). The loading control used here was hybridization with an anti--tubulin antibody. These data demonstrate that in human fibroblasts, SUMF1 is taken up via the MR, although a part is also taken up via the MPR, albeit with low affinity.
Figure 6.
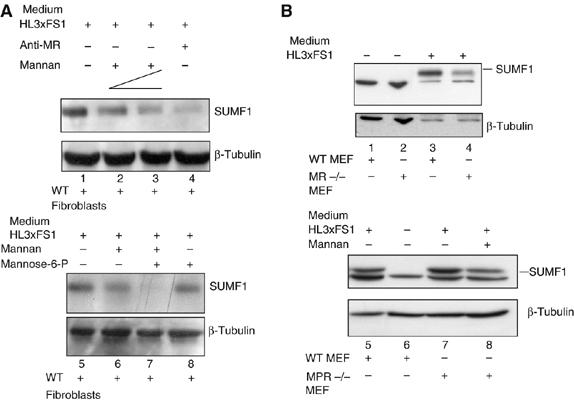
Uptake of SUMF1 is receptor mediated. (A) MR and MPR competition assays in human fibroblasts. Wild-type fibroblasts were cultured in SUMF1-Flag-conditioned medium collected from the HL3xFS1 clone, in the absence and presence of mannan (2 and 3 mg/ml), mannose 6-phospate (10 mM) and an anti-MR antibody (1 mg/ml). The uptake of SUMF1-Flag was determined by Western blotting using anti-Flag and anti--tubulin antibodies. (B) Uptake of SUMF1 in wild-type, MR−/− and MPR300−/−MPR46−/− MEFs. Recipient cells were cultured in SUMF1-Flag-conditioned medium from the HL3xFS1 clone in the absence and presence of mannan (2 mg/ml).
We obtained similar results in MEFs. Wild-type and MR−/− MEFs were incubated with SUMF1-conditioned medium. We detected a reduction in the uptake of SUMF1 into the MR−/− MEFs, with respect to the wild-type cells (Figure 6B, lanes 1–4). In contrast, there was no reduction in SUMF1 uptake detected in the double knock-out MPR46−/−MPR300−/− MEFs, with respect to wild-type cells (Figure 6B, lanes 5–7). However, a reduction in SUMF1 uptake was detected when the MPR46−/−MPR300−/− MEFs were cultured in SUMF1-conditioned medium plus mannan (2 mg/ml; Figure 6B, lane 8). In conclusion, in MEFs and human fibroblasts, uptake of SUMF1 is mediated principally by the MR, and to some extent by the MPR.
Discussion
Here, we have demonstrated that SUMF1 is secreted into and taken up from the medium in different types of cultured cells, and from the plasma in tissues of AAV-transduced mice. Furthermore, by using the SUMF1N141A mutant and the Xenopus Sumf1 cDNAs, both coding for the proteins lacking glycosylation, we have demonstrated that SUMF1 secretion is independent of its sugar content. We also show that the uptake, which is impaired by the lack of glycosylation, is mainly mediated via the MR, and, to some extent, by the MPR. Strikingly, once taken up, SUMF1 travels through the cell until it reaches the ER. Finally, we demonstrate that in Cos7 cells, MSD fibroblasts, Sumf1−/− MEFs and in vivo in Sumf1−/− mice, SUMF1 can enhance at least four different sulfatase activities after being taken up.
To date, the only proteins, which have been extensively characterized as being able to move from the external environment into cells via the plasma membrane to reach the ER are some of the toxins (Sandvig and van Deurs, 2000). In addition, autocrine motility factor has also been seen to be endocytosed to the smooth ER (Lagana et al, 2005), and ER-mediated phagocytosis of red blood cells has been described as a source of iron from the internalized hemoglobin (Desjardins, 2003). In the case of toxin endocytosis, they are obviously pirating of a physiological route in cells. Here, however, we are facing a mammalian protein that is apparently using its own route to reach the ER compartment. Two principal questions thus arise: (i) how does SUMF1 get secreted from the ER of cells and reach the ER of other cells? (ii) Does SUMF1 behave as a paracrine agent, or in other words, is the transit of SUMF1 through the cells a physiological condition?
For the former, we have only just started to determine how SUMF1 leaves the ER to reach the extracellular environment, and then how it gets back into the ER. SUMF1 does not have an ER-retention signal, although it resides in the lumen of the ER. Given the extraction conditions that are needed to recover SUMF1 from cells, we would predict that SUMF1 is associated with membranes. Therefore, our hypothesis would be that since SUMF1 does not have an ER-retention signal, it must be associated to ER membranes via an unknown interactor that acts as a receptor to retain it in the ER. It has been suggested that transport of glycoproteins out of the ER is regulated by lectins, which specifically bind to glycoproteins and function as receptors. The role of these receptor-like lectins is to retain glycoproteins rather than transport them actively (Helenius, 1994; Nyfeler et al, 2006). Thus, many proteins would not move out of the ER using a bulk-flow mechanism. In contrast, their trafficking might be regulated by receptors, which harmonize their retention and escape. We have demonstrated that non-glycosylated Xenopus Sumf1 and an N-glycosylation mutant of human SUMF1 can still be secreted. Thus, our prediction is that glycosylation is important for SUMF1 to be retained in the ER by an as yet unknown receptor-like interactor, although a fraction of the SUMF1 can be secreted. It will be interesting to demonstrate if non-glycosylated SUMF1 escapes the ER more efficiently with respect to its glycosylated form. Interestingly, non-glycosylated SUMF1 is still able to activate the sulfatases, as shown for the SUMF1N141A mutant and for the natural xSumf1. Thus, we would predict that non-glycosylated SUMF1 folds correctly and glycosylation has a role in the retention of SUMF1 in the ER exclusively, rather than for its activity. It has been postulated that although most glycoproteins need their N-linked oligosaccharides during folding, the degree of dependence is variable. Some proteins display total or only partial misfolding in the absence of sugars; however, there are numerous proteins, which fold correctly without their N-linked sugars (Helenius, 1994).
Once SUMF1 is secreted, it can be taken up by other cells, including immortalized cell lines, MEFs and human fibroblasts, as we have also confirmed by mass spectrometry analysis. Our receptor-competition and uptake data in the MR−/− knock-out MEFs and in the MPR46−/−MPR300−/− double knock-out MEFs show that SUMF1 is internalized via the MR, and to some extent, via the MPR. Under our cellular conditions, the lack of sugars, that is, of the mannose content, is crucial to abrogate the uptake. However, we cannot exclude that in different cell lines, alternative compensatory mechanisms can allow partial uptake of SUMF1. The MR mediates endocytosis of glycoproteins with terminal mannose, fucose, N-acetylglucosamine and/or glucose residues (Stahl, 1990). Furthermore, the MR mediates the clearance of glycoproteins with high mannose oligosaccharides, such as lysosomal enzymes (Stahl, 1990). A deficiency in the MR in mice affects the uptake of multiple glycoproteins from plasma into tissues (Lee et al, 2002). In addition, the MR mediates uptake of the toxin ricin (Simmons et al, 1986). Here, we have seen that SUMF1 is also taken up via the MR, which appears to be the route that is used by ricin, which enters the cells via the MR, travels across the Golgi complex and reaches the ER. In contrast, MPR 300 mediates the uptake of lysosomal enzymes, including the sulfatases, which bear the mannose-6-phosphate marker (Ni et al, 2006). From the crystal structure and mass spectrometry, it appears that SUMF1 does not contain mannose 6-phosphate (Dierks et al, 2005); furthermore, SUMF1 was not included in a recent study in which all of the lysosomal and non-lysosomal proteins with mannose-6-phosphate residues were fished out by affinity chromatography (Sleat et al, 2006). Our data clearly demonstrate a very low affinity of SUMF1 for the MPR, suggesting that the MPR might only have a compensatory role. There is also the possibility that SUMF1 is internalized via a piggyback mechanism, along with the sulfatases. We have recently demonstrated that a stable complex can be formed between SUMF1, SUMF2 and the sulfatases; however, we have never been able to detect a stable complex between SUMF1 and the sulfatases (Zito et al, 2005). To exclude the possibility of such a piggyback mechanism, we also examined the uptake of SUMF1 in MEFs that we established from an Sumf2−/− mouse (Annunziata et al, unpublished data). We chose the Sumf2−/− MEFs since the triple complex (SUMF1/SUMF2/sulfatase) is abrogated here, while it would be impossible to have a cell line without all of the sulfatases. In these Sumf2−/− MEFs, the uptake of SUMF1 is completely normal, as SUMF1 is taken up as in wild-type cells, thus excluding a piggyback mechanism of uptake (Supplementary Figure 4). However, the question as to how SUMF1 travels from the plasma membrane to the ER remains completely open. The toxins were the first molecules that were demonstrated to enter the cell via the endosomes and to get transported ‘back' to the Golgi complex, and thence to the ER (Sandvig and van Deurs, 2000). This retrograde transport from the Golgi complex to the ER could be either COP-I dependent or independent. The COP-I-independent pathway is regulated by the GTPase Rab6, and it is used by Golgi glycosylation enzymes, Shiga toxin (ST) and ricin (Girod et al, 1999). In contrast, the lectin ERGIC-53, which is a cargo-receptor for glycoproteins, and cholera toxin use a COP-I-dependent pathway to reach the ER (Girod et al, 1999; Sandvig and van Deurs, 2000). It will be fascinating to identify which pathway SUMF1 uses to travel across the ER and the Golgi complex, and thus to dissect out its anterograde and retrograde transport.
A further important question is to understand why SUMF1 acts as a paracrine agent, and if the movement of SUMF1 from the ER of one cell to the ER of another cell is a physiological condition. We have seen that endogenous SUMF1 secretion occurs and the secreted SUMF1 can be taken up by MSD fibroblasts. At present, we cannot demonstrate that this also applies in vivo, since we would need to generate a tissue-specific Sumf1−/− mouse to follow the trafficking of SUMF1 from the wild-type to the knock-out tissues. However, what is clear is that SUMF1 is enzymatically active after its uptake, a situation that we have demonstrated in several cell lines and in Sumf1−/− mice. Thus, although many proteins that reach the ER get degraded by the proteasome, including the toxins, this is not the destiny of SUMF1: we can detect SUMF1 in the ER by Western blotting and indirect immunofluorescence, and most importantly, we have demonstrated that the internalized SUMF1 can enhance sulfatase activities within the recipient cell. A provocative hypothesis would be that SUMF1 works as a hormone-like factor in the regulation of sulfatase activities, while working as a paracrine agent. In this respect, it will be intriguing to determine if SUMF1 modulates the function of the sulfatases in different tissues and cells in a paracrine manner, or if it mediates signalling pathways.
Finally, our findings have potential application in the therapy of MSD. We have demonstrated that engineering the liver to produce SUMF1 in the Sumf1−/− knock-out mouse model allows secretion of the protein and cross-correction of sulfatase activities in non-transduced tissues. Thus, it will be possible in the near future to set up gene therapy and enzyme replacement therapy protocols to treat first the Sumf1−/− mouse model, and then, in the long term, patients suffering from this severe disease.
Materials and methods
Cell lines, cDNAs, AAV and lentivirus production, Western blotting and immunoprecipitation, mass spectrometry analysis and endoglycosidase treatment of cellular extracts, and media
For details, see Supplementary data.
Cells and tissues immunofluorescence, transfection and sulfatase enzymatic analysis
These experiments were carried out as previously described (Cosma et al, 2004; Zito et al, 2005; Cardone et al, 2006). All animal experiments were carried out in accordance with Italian law.
SUMF1 uptake assay
Recipient cells, MEFs, human fibroblasts, Cos7 and HeLa cells were grown to 90% confluency in DMEM (Invitrogen, NY, USA), 10% fetal calf serum (Euroclone, Pero, MI, Italy) at 37°C, 5% CO2 in six-well plates (Costar, Cambridge, MA, USA). The cells were incubated for 12 h in SUMF1-conditioned medium collected from different subconfluent producing cells (HL3xFS1, HepG2, II-1-S1, II-2-S1, IIIA-1-S1, IIIA-2-S1,) cultured in T75 flasks (Costar). The recipient cells were washed twice with PBS, trypsinized and lysed in RIPA buffer (150 mM NaCl, 50 mM Tris–HCl, pH 7.5, 500 μM EDTA, 0.1% SDS, 1% Triton-X100 and 1% sodium deoxycholate containing a proteinase inhibitor cocktail; Sigma, St Louis, MO, USA). Protein concentrations were measured in total homogenates by the Bradford assay (Bio-Rad, Hercules, CA). SUMF1 uptake was analyzed by Western blotting and by testing the sulfatase activities. Additional details are provided in the Supplementary data.
Receptor-competition assay
Recipient cells were cultured for 12 h in SUMF1-conditioned medium collected from the HL3xFS1 clone in the presence of limiting amounts (100 ng/ml, final concentration) of cycloheximide (Sigma), and in the absence or presence of the following receptor inhibitors: 2 or 3 mg/ml yeast mannan (Sigma) (competitive inhibitor of the MR), 10 mM mannose 6-phosphate (Sigma) (competitor for the MPR) or 1 mg/ml anti-MR IgG. After 12 h, the recipient cells were washed twice with PBS and trypsinized. The SUMF1 uptake in the cell extracts was analyzed by Western blotting.
Supplementary Material
Supplementary data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4
Acknowledgments
We are particularly grateful to Blanche Schwappach, Hans-Peter Hauri and Frederic Lluis Viñas for discussions on the results. We thank Marinella Pirozzi for help with the immunofluorescence microscopy, Michel C Nussenzweig for providing the MR−/− mice and Wellcome Trust Sanger Institute for clone TEgg007D12 (xSumf1), the TIGEM AAV Vector Core Facility, the Telethon cell-line bank, the EMBO YIP and Telethon Foundation for support. The authors declare that they have no competing financial interests.
References
- Ballabio A, Shapiro LJ (1995) STS deficiency and X-linked ichthyosis. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds.) The Metabolic and Molecular Bases of Inherited Disease. McGraw-Hill: New York Vol. II, chapter 96, pp. 2999–3022 [Google Scholar]
- Boltes I, Czapinska H, Kahnert A, von Bulow R, Dierks T, Schmidt B, von Figura K, Kertesz MA, Uson I (2001) 1.3 A structure of arylsulfatase from Pseudomonas aeruginosa establishes the catalytic mechanism of sulfate ester cleavage in the sulfatase family. Structure 9: 483–491 [DOI] [PubMed] [Google Scholar]
- Cardone M, Polito VA, Pepe S, Mann L, D'Azzo A, Auricchio A, Ballabio A, Cosma MP (2006) Correction of Hunter syndrome in the MPSII mouse model by AAV2/8-mediated gene delivery. Hum Mol Genet 15: 1225–1236 [DOI] [PubMed] [Google Scholar]
- Clark HF, Gurney AL, Abaya E, Baker K, Baldwin D, Brush J, Chen J, Chow B, Chui C, Crowley C, Currell B, Deuel B, Dowd P, Eaton D, Foster J, Grimaldi C, Gu Q, Hass PE, Heldens S, Huang A, Kim HS, Klimowski L, Jin Y, Johnson S, Lee J, Lewis L, Liao D, Mark M, Robbie E, Sanchez C, Schoenfeld J, Seshagiri S, Simmons L, Singh J, Smith V, Stinson J, Vagts A, Vandlen R, Watanabe C, Wieand D, Woods K, Xie MH, Yansura D, Yi S, Yu G, Yuan J, Zhang M, Zhang Z, Goddard A, Wood WI, Godowski P, Gray A (2003) The secreted protein discovery initiative (SPDI), a large-scale effort to identify novel human secreted and transmembrane proteins: a bioinformatics assessment. Genome Res 13: 2265–2270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Annunziata I, Newbold RF, Grompe M, Parenti G, Ballabio A (2003) The multiple sulfatase deficiency gene encodes an essential and limiting factor for the activity of sulfatases. Cell 113: 445–456 [DOI] [PubMed] [Google Scholar]
- Cosma MP, Pepe S, Parenti G, Settembre C, Annunziata I, Wade-Martins R, Di Domenico C, Di Natale P, Mankad A, Cox B, Uziel G, Mancini GM, Zammarchi E, Donati MA, Kleijer WJ, Filocamo M, Carrozzo R, Carella M, Ballabio A (2004) Molecular and functional analysis of SUMF1 mutations in multiple sulfatase deficiency. Hum Mutat 23: 576–581 [DOI] [PubMed] [Google Scholar]
- Desjardins M (2003) ER-mediated phagocytosis: a new membrane for new functions. Nat Rev Immunol 3: 280–291 [DOI] [PubMed] [Google Scholar]
- Dierks T, Dickmanns A, Preusser-Kunze A, Schmidt B, Mariappan M, von Figura K, Ficner R, Rudolph MG (2005) Molecular basis for multiple sulfatase deficiency and mechanism for formylglycine generation of the human formylglycine-generating enzyme. Cell 121: 541–552 [DOI] [PubMed] [Google Scholar]
- Dierks T, Schmidt B, Borissenko LV, Peng J, Preusser A, Mariappan M, von Figura K (2003) Multiple sulfatase deficiency is caused by mutations in the gene encoding the human C(alpha)-formylglycine generating enzyme. Cell 113: 435–444 [DOI] [PubMed] [Google Scholar]
- Diez-Roux G, Ballabio A (2005) Sulfatases and human disease. Annu Rev Genomics Hum Genet 6: 355–379 [DOI] [PubMed] [Google Scholar]
- Fraldi A, Biffi A, Lombardi A, Visigalli I, Pepe S, Settembre C, Nusco E, Auricchio A, Naldini L, Ballabio A, Cosma MP (2007) SUMF1 enhances sulfatase activities in vivo in five sulfatase deficiencies. Biochem J 403: 305–312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girod A, Storrie B, Simpson JC, Johannes L, Goud B, Roberts LM, Lord JM, Nilsson T, Pepperkok R (1999) Evidence for a COP-I-independent transport route from the Golgi complex to the endoplasmic reticulum. Nat Cell Biol 1: 423–430 [DOI] [PubMed] [Google Scholar]
- Helenius A (1994) How N-linked oligosaccharides affect glycoprotein folding in the endoplasmic reticulum. Mol Biol Cell 5: 253–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiesberger T, Huttler S, Rohlmann A, Schneider W, Sandhoff K, Herz J (1998) Cellular uptake of saposin (SAP) precursor and lysosomal delivery by the low density lipoprotein receptor-related protein (LRP). EMBO J 17: 4617–4625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knowles BB, Howe CC, Aden DP (1980) Human hepatocellular carcinoma cell lines secrete the major plasma proteins and hepatitis B surface antigen. Science 209: 497–499 [DOI] [PubMed] [Google Scholar]
- Lagana A, Goetz JG, Y N, Altschuler Y, Nabi IR (2005) pH-specific sequestration of phosphoglucose isomerase/autocrine motility factor by fibronectin and heparan sulphate. J Cell Sci 118: 4175–4185 [DOI] [PubMed] [Google Scholar]
- Lee SJ, Evers S, Roeder D, Parlow AF, Risteli J, Risteli L, Lee YC, Feizi T, Langen H, Nussenzweig MC (2002) Mannose receptor-mediated regulation of serum glycoprotein homeostasis. Science 295: 1898–1901 [DOI] [PubMed] [Google Scholar]
- Neufeld EF, Muenzer J (2001) The mucopolysaccharidoses. In: Scriver CR, Beaudet AL, Sly WS, Valle D (eds.) The Metabolic and Molecular Basis of Inherited Disease. Mc Graw-Hill: New York pp. 3421–3452 [Google Scholar]
- Ni X, Canuel M, Morales CR (2006) The sorting and trafficking of lysosomal proteins. Histol Histopathol 21: 899–913 [DOI] [PubMed] [Google Scholar]
- Nyfeler B, Zhang B, Ginsburg D, Kaufman RJ, Hauri HP (2006) Cargo selectivity of the ERGIC-53/MCFD2 transport receptor complex. Traffic 7: 1473–1481 [DOI] [PubMed] [Google Scholar]
- Preusser-Kunze A, Mariappan M, Schmidt B, Gande SL, Mutenda K, Wenzel D, von Figura K, Dierks T (2005) Molecular characterization of the human Calpha-formylglycine-generating enzyme. J Biol Chem 280: 14900–14910 [DOI] [PubMed] [Google Scholar]
- Sandvig K, van Deurs B (2000) Entry of ricin and Shiga toxin into cells: molecular mechanisms and medical perspectives. EMBO J 19: 5943–5950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sardiello M, Annunziata I, Roma G, Ballabio A (2005) Sulfatases and sulfatase modifying factors: an exclusive and promiscuous relationship. Hum Mol Genet 14: 3203–3217 [DOI] [PubMed] [Google Scholar]
- Schmidt B, Selmer T, Ingendoh A, von Figura K (1995) A novel amino acid modification in sulfatases that is defective in multiple sulfatase deficiency. Cell 82: 271–278 [DOI] [PubMed] [Google Scholar]
- Settembre C, Annunziata I, Spampanato C, Zarcone D, Cobellis G, Nusco E, Zito E, Tacchetti C, Cosma MP, Ballabio A (2007) Systemic inflammation and neurodegeneration in a mouse model of multiple sulfatase deficiency. Proc Natl Acad Sci USA 104: 4506–4511 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simmons BM, Stahl PD, Russell JH (1986) Mannose receptor-mediated uptake of ricin toxin and ricin A chain by macrophages. Multiple intracellular pathways for a chain translocation. J Biol Chem 261: 7912–7920 [PubMed] [Google Scholar]
- Sleat DE, Wang Y, Sohar I, Lackland H, Li Y, Li H, Zheng H, Lobel P (2006) Identification and validation of mannose 6-phosphate glycoproteins in human plasma reveals a wide range of lysosomal and non-lysosomal proteins. Mol Cell Proteomics 5: 1942–1956 [DOI] [PubMed] [Google Scholar]
- Stahl PD (1990) The macrophage mannose receptor: current status. Am J Respir Cell Mol Biol 2: 317–318 [DOI] [PubMed] [Google Scholar]
- Taylor ME, Drickamer K (1993) Structural requirements for high affinity binding of complex ligands by the macrophage mannose receptor. J Biol Chem 268: 399–404 [PubMed] [Google Scholar]
- Zito E, Fraldi A, Pepe S, Annunziata I, Kobinger G, Di Natale P, Ballabio A, Cosma MP (2005) Sulphatase activities are regulated by the interaction of sulphatase-modifying factor 1 with SUMF2. EMBO Rep 6: 655–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary data
Supplementary Figure 1
Supplementary Figure 2
Supplementary Figure 3
Supplementary Figure 4