

Abstract
A guanine (G638) within the substrate loop of the VS ribozyme plays a critical role in the cleavage reaction. Replacement by any other nucleotide results in severe impairment of cleavage, yet folding of the substrate is not perturbed, and the variant substrates bind the ribozyme with similar affinity, acting as competitive inhibitors. Functional group substitution shows that the imino proton on the N1 is critical, suggesting a possible role in general acid–base catalysis, and this in accord with the pH dependence of the reaction rate for the natural and modified substrates. We propose a chemical mechanism for the ribozyme that involves general acid–base catalysis by the combination of the nucleobases of guanine 638 and adenine 756. This is closely similar to the probable mechanism of the hairpin ribozyme, and the active site arrangements for the two ribozymes appear topologically equivalent. This has probably arisen by convergent evolution.
Keywords: general acid–base catalysis, guanine, ribozyme mechanism, RNA catalysis
Introduction
The nucleolytic ribozymes bring about the site-specific cleavage or ligation of the backbone of RNA, with an acceleration of around a million-fold or greater. The chemical origins of catalysis are not fully understood, but significant progress has been made recently. The cleavage reaction requires the in-line attack of a 2′-O on the adjacent 3′-P with concomitant rupture of the bond to the 5′-O, in an SN2 reaction that passes through a trigonal bipyramidal phosphorane transition state. In principle, this could be catalysed by a number of processes, including substrate orientation and stabilization of the transition state. In addition, general acid–base catalysis could be important, increasing the strength of the attacking nucleophile by deprotonation and stabilization of the departing oxyanion by protonation. However, compared with proteins, the catalytic resources of RNA are rather limited, being restricted to the nucleobases and water coordinated to metal ions.
There is good evidence for the direct participation of nucleobases in a number of nucleolytic ribozymes. Crystal structures of the hairpin ribozyme (Rupert and Ferré-D'Amaré, 2001; Rupert et al, 2002) reveal the presence of guanine (G8) and adenine (A38) bases juxtaposed with the 2′-O and 5′-O, respectively, of the scissile phosphate, making a number of hydrogen bonds that should stabilize the transition state. They are well placed to act in general acid–base catalysis, consistent with the pH dependence of the reaction (Bevilacqua, 2003), and its variation with functional group modifications (Pinard et al, 2001; Kuzmin et al, 2004, 2005) and replacement by imidazole (Wilson et al, 2006). The positions of the bases are consistent with a role for guanine as the general base and adenine as the general acid in the cleavage reaction. Recent crystallographic data on the extended form of the hammerhead ribozyme suggest a similar role for a guanine base (G12) (Martick and Scott, 2006), also supported by chemical data (Han and Burke, 2005). General acid–base catalysis also seems to be very important in the hepatitis delta virus ribozyme, but the participants are different. A cytosine base (C75) is juxtaposed with the scissile phosphate (Ferré-d'Amaré et al, 1998; Ke et al, 2004); substitution of this base leads to a marked loss of activity that can be partially restored by exogenous imidazole (Perrotta et al, 1999). In this ribozyme, the second participant appears to be hydrated metal ion (Nakano et al, 2000). Experiments in which the leaving group has been labilized by phosphorothiolate substitution show that the cytosine nucleobase is the general acid in the catalysis (Das and Piccirilli, 2005).
Unlike the other nucleolytic ribozymes, there is no crystal structure for the VS ribozyme. Nevertheless, it has been possible to generate a model of the structure based on the known secondary structure (Beattie et al, 1995) (Figure 1), together with biophysical analysis of the component three-way helical junctions (Lafontaine et al, 2001a, 2002a). The structure is based upon the coaxial stacking of helices IV, III and VI, from which helices V and II extend laterally. This structure is in good agreement with recent studies by small-angle X-ray scattering in solution (unpublished data). The substrate stem-loop is connected through the end of helix II in the cis form of the ribozyme and makes a loop–loop interaction with helix V (Rastogi et al, 1996). These constrain both ends, and suggest that it is located in the cleft formed between helices II and VI (Lafontaine et al, 2002a). The internal loop containing A730 within helix VI has attracted particular interest as a candidate component of the active site (Lafontaine et al, 2001b; Sood and Collins, 2002). Moreover, one nucleotide in particular within the A730 loop appears to be critical for catalytic activity; any substitution of A756 leads to a loss of activity of three orders of magnitude (Lafontaine et al, 2001b; Sood and Collins, 2002), and functional group changes indicate that the Watson–Crick edge of the adenine nucleobase is especially important (Lafontaine et al, 2002b). Nucleotide analog interference mapping experiments showed that A756 is the single most sensitive nucleotide to substitution in the ribozyme (Jones and Strobel, 2003). The A730 loop may be readily juxtaposed with the substrate loop containing the scissile phosphate in the structural model of the ribozyme (Lafontaine et al, 2002a), and physical proximity was demonstrated by the observation of a UV-induced crosslink between a thiouridine placed adjacent to the scissile phosphate and A756 (Hiley et al, 2002). There is evidence consistent with a role for A756 in general acid–base chemistry. Substitution of the nucleobase of A756 with variants of altered pKA resulted in changes of ligation rate with pH, indicative of proton transfer in the transition state (Jones and Strobel, 2003), and substitution by a novel imidazole nucleoside led to a significant level of cleavage and ligation activity (Zhao et al, 2005).
Figure 1.
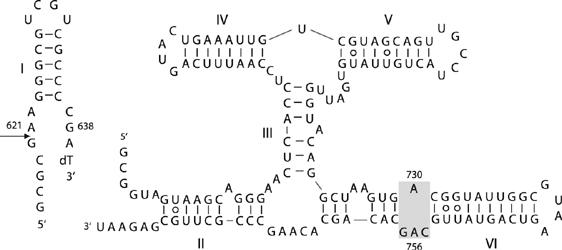
The VS ribozyme. The sequence of the ribozyme in its trans-acting form, comprising the substrate stem-loop (helix I) and the ribozyme (helices II–VI). The site of cleavage is arrowed. The A730 loop within helix VI (shaded) is the putative active site of the ribozyme.
The possibility that A756 could be acting as a general acid or base raises the question of what might be acting as a partner in the transfer of protons in the transition state. It is unlikely that a hydrated metal ion plays this role, analogous to that in the HDV ribozyme, since cleavage proceeds at a significant rate in the absence of divalent metal ions (Murray et al, 1998), unlike the HDV ribozyme. We therefore considered the possibility that a second nucleobase might participate in the catalytic chemistry, analogous to the situation in the hairpin ribozyme. We can exclude most of the ribozyme, as substitutions can be tolerated so long as secondary structure is preserved, together with the lengths of helices III and V (Lafontaine et al, 2001a, 2002a). Moreover, physical access seems improbable for much of the ribozyme in terms of present models of the structure. Strongly disabling substitutions have been identified in the three-way junctions, but these correlate well with resulting changes to the folding of the ribozyme. Even the base pairs flanking the A730 loop can be reversed without great loss of activity. Substitutions of G757 lead to significant loss of cleavage and ligation activity, but remain considerably more active than A756 variants, suggesting that G757 does not play a critical role in the catalytic chemistry. This leaves one region for consideration, the internal loop of the substrate stem-loop. We have therefore examined the effect of sequence substitutions within this region. In this study, we have identified a guanine base that is critical for catalysis and is a strong candidate for the second nucleobase acting in general acid–base catalysis.
Results
Identification of an important nucleobase in the substrate stem-loop
We have explored the importance of a number of nucleotides in the substrate stem-loop of the VS ribozyme, in the search for a catalytic nucleobase. The upper stem loop Ib is either regular duplex or involved in a loop–loop interaction with the loop of helix V in the ribozyme. We have therefore focused on the internal loop, which is believed to interact with the 730 loop of the ribozyme (Lafontaine et al, 2001b; Hiley et al, 2002). The cleavage activity under standard conditions (50 mM Tris (pH 8.0), 10 mM MgCl2, 25 mM KCl and 2 mM spermidine at 37°C) of sequence variants was examined using the trans form of the ribozyme under single-turnover conditions. (Table I). An A639G substitution had only a very small effect on cleavage rate. A621 is adjacent to the scissile phosphate, and substitution by guanine led to a 40-fold reduction in cleavage rate under standard conditions. A622U substitution led to a cleavage rate of 3.3 × 10−4 min−1. Although this is a significant loss of activity, it is likely to result from perturbation to the structure of the loop, as a uridine at position 622 could basepair with G638 in the five-nucleotide form of the loop (Andersen and Collins, 2000), extending helix Ib. Moreover, it would be difficult for a nucleobase at position 622 to interact with the scissile phosphate on the same strand.
Table 1.
VS ribozyme cleavage rates measured under standard conditions for different substrates
| Substrate | Rate (min−1) | Error (min−1) | Relative decrease |
|---|---|---|---|
| Natural | 0.72 | 0.15 | 1 |
| A621G | 0.018 | 0.002 | 40 |
| A622U | 3.3 × 10−4 | 7 × 10−5 | 2200 |
| A639G | 0.42 | 0.06 | 1.7 |
| G638A | 9.9 × 10−5 | 9 × 10−6 | 7300 |
| G638C | 9 × 10−5 | 1 × 10−5 | 8400 |
| G638U | 6 × 10−5 | 1 × 10−5 | 12 000 |
| G638 purine | 9.0 × 10−5 | 9 × 10−6 | 8000 |
| G638 diaminopurine | 4.6 × 10−4 | 2 × 10−5 | 1600 |
| G6382 aminopurine | 8.5 × 10−5 | 7 × 10−6 | 8500 |
| G638 inosine | 0.026 | 0.004 | 27 |
| G638A, C755Aa | 8 × 10−5 | 1 × 10−5 | 9100 |
| G638A, C755Ga | 7 × 10−5 | 2 × 10−5 | 9800 |
| G638A, C755Ua | 9 × 10−5 | 3 × 10−5 | 8200 |
| The rate of cleavage of the G638A substrate was measured using variant ribozyme in which C755 was replaced by adenine, guanine or uracil. | |||
The largest effects on the rate of cleavage resulted from changes at position 638. Replacement of this guanine by any other nucleotide led to markedly impaired cleavage, with very little cleavage detectable after an hour (Figure 2). Incubation of the G638A substrate over much longer time courses revealed that the rate of cleavage is 9.9 × 10−5 min−1, reduced 104-fold compared with the natural sequence. The magnitude of this effect is even greater than that arising from an A756G substitution (Lafontaine et al, 2001b), and suggests an important role of G638 in the function of the ribozyme.
Figure 2.
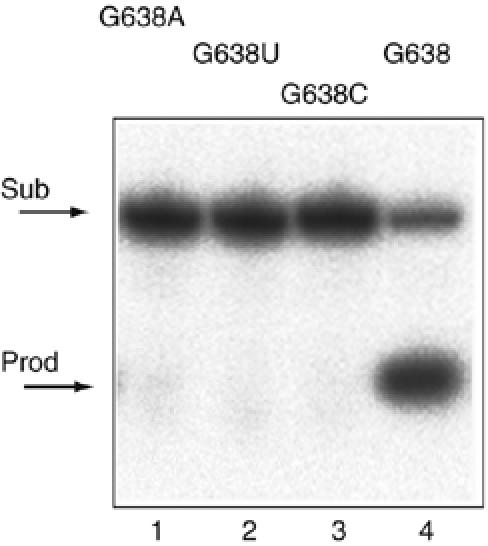
Sequence substitutions at position 638 in the substrate strongly impair cleavage by the VS ribozyme. Products of VS ribozyme cleavage in trans on the substrate with the natural G638, and substitution by A, U or C. Cleavage reactions were performed on radioactively 5′-32P-labelled substrates under single-turnover conditions at 37°C in standard VS buffer, that is, 50 mM Tris (pH 8.0), 10 mM MgCl2, 25 mM KCl, 2 mM spermidine. The natural substrate was reacted for 5 min and the three variants for 60 min. Substrate and product were separated by electrophoresis in 20% polyacrylamide gels under denaturing conditions and visualized by phosphorimaging. Tracks: 1, G638A; 2, G638U; 3, G638C and 4, G638 natural sequence substrate.
The secondary structure of the G638A substrate is not significantly altered
It was important to examine the trivial possibility that the impairment of VS ribozyme cleavage of the G638A substrate results from altered folding. We compared the secondary structure of the natural and G638A substrates using the method of in-line probing (Soukup and Breaker, 1999). In this approach, radioactively 5′-32P-labelled RNA is subjected to a prolonged incubation in the presence of buffer and Mg2+ ions. Where the backbone is relatively flexible, the RNA can locally sample a conformation in which the 2′-OH may carry out an in-line nucleophilic attack on the 3′-phosphate, but this is hindered in more rigid parts of the molecule, including duplex regions. We performed the analysis under conditions similar to those used for most of the ribozyme cleavage experiments (Figure 3). The helical regions of both species were clearly protected from cleavage, whereas the terminal loop and the formally single-stranded regions at the lower end of the helix were subject to significant cleavage. Evidently the general secondary structure of the substrate is unaltered by the G638A substitution. Cleavage immediately adjacent to position 638 is enhanced in the G638A substrate relative to the natural sequence. This is a very local effect, and increased sensitivity at this position is also observed in hydroxide cleavage of the same RNA under fully denaturing conditions. But the in-line probing indicates that no gross conformational change results from the substitution.
Figure 3.
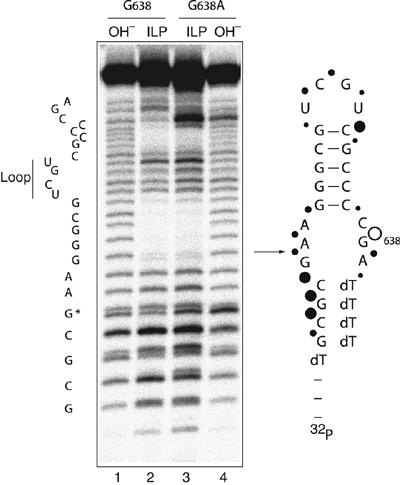
Comparison of the conformations of the natural and G638A substrates by in-line probing. Versions of natural sequence and G638A substrates with 5′ and 3′ terminal extensions of deoxyribonucleotides were synthesized to improve the electrophoretic resolution of the RNA sections. Radioactively 5′-32P-labelled substrates were incubated in standard VS buffer at 25°C for 40 h. Cleavages were analysed by electrophoresis in a 20% polyacrylamide gel under denaturing conditions. Tracks 1 and 4, base cleavage of natural and G638A substrates, respectively and tracks 2 and 3, in-line probing analysis of natural and G638A substrates, respectively. The scheme shows the sequence of the natural substrate, with the arrow indicating the position of ribozyme cleavage. The positions of sensitivity to in-line probing are indicated by filled circles, the size of which reflects the extent of cleavage. The open circle shows the position of the phosphodiester linkage that is more sensitive in the G638A substrate; note that this position also exhibits enhanced base cleavage. The shorter fragments migrate as doublets, due to resolution of the cyclic 2′3′-phosphates and their products of hydrolysis during the long incubation.
A G638A substrate exhibits unperturbed binding to the VS ribozyme
Another possible explanation for the low rate of cleavage of a G638A substrate by the VS ribozyme is impaired binding. If the G638A substrate is able to bind to the ribozyme in a manner that is similar to that of the normal substrate, it is likely to act as a competitive inhibitor, described by the scheme shown in Figure 4A. We therefore carried out cleavage reactions under standard conditions, in the presence of different concentrations of G638A substrate. The progress curves (Figure 4B) show a marked reduction in cleavage rate with increased concentration of G638A substrate. The observed rates, kobs, have been fitted to the equation for competitive inhibition that is, 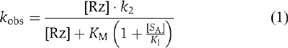
Figure 4.
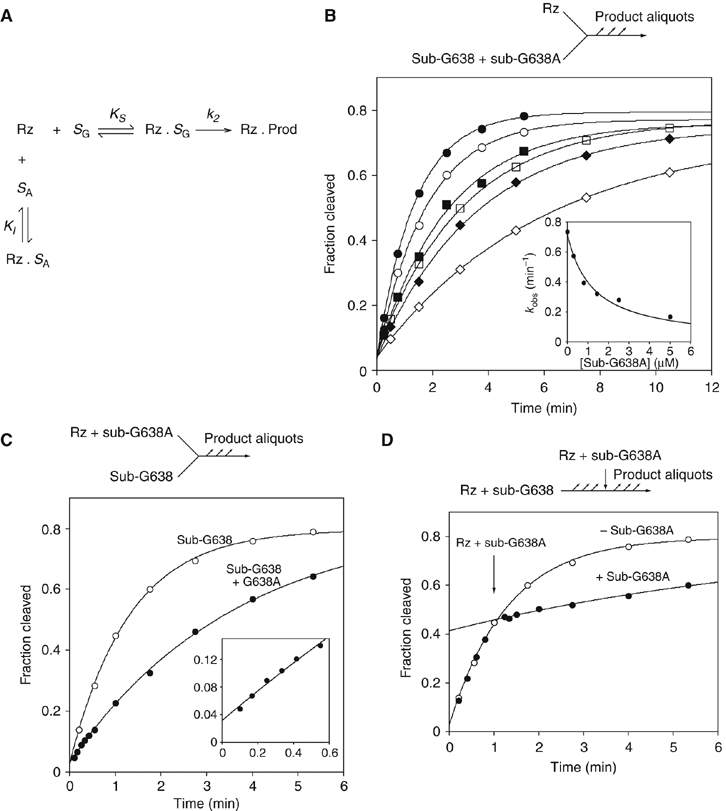
Affinity and rate of substrate binding in trans to the VS ribozyme. (A) Reaction scheme for the ribozyme cleavage in the presence of the variant substrate. The natural sequence substrate (SG) binds to the ribozyme (Rz) with an affinity KS to form a non-covalent complex that undergoes the cleavage reaction at rate k2. The G638A substrate binds to the ribozyme with an affinity KI. The variant substrate undergoes a negligible amount of cleavage during the incubation, and therefore simply acts as an inhibitor of the reaction. (B) Cleavage of a natural sequence substrate by VS ribozyme was performed in trans under standard single-turnover conditions in the presence of different concentrations of G638A substrate. Progress curves are shown for reactions carried out in the presence of the following concentrations of G638A substrate: 0 (filled circles), 0.3 (open circles), 0.8 (filled squares), 1.4 (open squares), 2.5 (filled diamonds) and 5 μM (open diamonds). In the inset, the observed rate constants (kobs) are plotted as a function of G638A substrate concentration and fitted to equation 1, appropriate for competitive inhibition by the variant substrate. (C) Substrate cleavage by VS ribozyme that had been preincubated with G638A variant substrate. Ribozyme (1 μM, with or without 2.8 μM G638A substrate) and substrate were separately incubated followed by mixing together at 0 time. The progress of both reactions is shown: no G638A substrate (open circles); plus G638A substrate (filled circles) and fitted to single exponentials, yielding rates of 0.77±0.03 and 0.28±0.01 min−1. The data for the first 0.6 min are shown expanded in the inset. Note that no lag phase is discernible. (D) A VS cleavage reaction interrupted by addition of G638A substrate. A cleavage reaction was initiated by addition of 10 μl of 1 μM ribozyme to 10 μl of natural sequence substrate, followed by addition of 10 μl of 30 μM G638A substrate, 1 μM ribozyme after 60 s. Progress curves are plotted for the interrupted reaction (filled circles) and one reaction allowed to continue normally (open circles; these data are taken from panel C), and fitted to single exponentials. There is no discernible intermediate phase, and the curves intersect at 64.8 s.
where [Rz] is the ribozyme concentration, [SA] is the concentration of the G638A substrate (treated as an inhibitor in this analysis), k2 is the rate of conversion of substrate to product complex, KM is the Michaelis constant for the substrate and KI is the dissociation constant for the G638A substrate. Values of KM=0.54 μM and KMapp=1 μM have been measured by multiple- (Tzokov et al, 2002) and single-turnover (Lafontaine et al, 2001b) experiments, respectively, and for the present analysis, we have assumed that KM=1 μM. The data are well fitted by this equation, consistent with the G638A substrate acting as a competitive inhibitor of the cleavage of the natural substrate, and yielding a value of KI=0.6±0.1 μM. Thus, the binding affinities of the natural and G638A substrates are closely similar.
To investigate the rates of substrate binding, we compared the rate of cleavage of the natural substrate by 1 μM VS ribozyme under standard conditions and by 1 μM VS ribozyme that had been preincubated with 2.8 μM G638A substrate (Figure 4C). The progress curves show the expected reduction in cleavage rate in the presence of the G638A substrate, while the absence of a lag in the early part of the time course indicates that dissociation of the G638A substrate is either very fast or very slow. To distinguish between these possibilities, the progress of the cleavage reaction of the natural substrate was interrupted by the addition of an excess of G638A substrate after 60 s (Figure 4D). The adjustment of cleavage rate occurs within 5 s, showing that the dissociation of the natural substrate and the association of the G638A substrate are rapid. These experiments demonstrate that the rate-limiting step for cleavage must be subsequent to binding.
Potential basepairing by G638
A further possibility is that G638 is required to basepair with a cytosine to allow the substrate and A730 loops to interact productively, analogous perhaps to the basepairing between G+1 and C25 in the hairpin ribozyme (Rupert and Ferré-D'Amaré, 2001). Were this to be the case, it would be necessary to identify the participating cytosine. We have previously shown that the base pairs flanking the A730 loop can be reversed with relatively low penalties to cleavage rate (Lafontaine et al, 2001b), so these are unlikely to be involved. This leaves C755 within the A730 loop as the only candidate nucleotide. Cleavage is only weakly affected by any change other than C755G (Lafontaine et al, 2001b), but a C755A variant is significantly impaired in ligation (McLeod and Lilley, 2004). We examined the rates of cleavage of the G638A substrate by VS ribozymes with C755 replaced by either A, G or U. In all cases, extremely slow rates of cleavage were obtained (Table I), with no evidence of restoration of activity when an A–U basepair might replace one between G638 and C755.
Importance of functional groups at position 638
The data presented above demonstrate that G638 plays a direct role in the central conversion of substrate to product. We therefore explored the importance of different functional groups at position 638. It has been shown that the 2′-OH is not important in the cleavage reaction (Tzokov et al, 2002), and we therefore focused on the nucleobase (Figure 5). Cleavage rates are collected in Table I. Removal of the exocyclic carbonyl and amine groups (purine) resulted in a similarly low rate of cleavage as G638A, as did removal of the C6 carbonyl group alone (2-aminopurine). Replacement of the C6 carbonyl group by an amine (2,6-diaminopurine) also led to very impaired cleavage. By contrast, removal of the C2 amine with retention of the carbonyl (hypoxanthine, that is, an inosine nucleoside) resulted in a significant rate of cleavage, 27-fold slower than the natural sequence. The results suggest that the main requirement for the nucleobase at position 638 for cleavage under standard conditions is the C6 carbonyl, or the imino proton on N1, or both.
Figure 5.
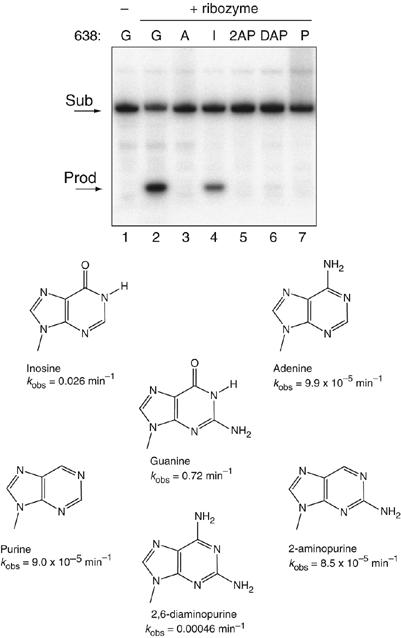
Effect of functional group changes at position 638 on the rate of VS ribozyme cleavage. Variant substrates were synthesized with alternative nucleotide bases or analogs at position 638 as shown and radioactively 5′-32P labelled. These were subjected to cleavage in trans by VS ribozyme under single-turnover conditions for 30 min at 37°C in standard buffer. Substrate and product were separated by electrophoresis in 20% polyacrylamide gels under denaturing conditions, and visualized by phosphorimaging. Tracks 1, natural substrate before cleavage; tracks 2–8, incubation with VS ribozyme; track 2, natural substrate; track 3, substitution by adenine; track 4, substitution by inosine; track 5, substitution by 2-aminopurine; track 6, substitution by 2,6-diaminopurine and track 7, substitution by purine.
The pH dependence of cleavage rates
Drawing analogies with other nucleolytic ribozymes, G638 may participate in general acid–base catalysis of the transesterification reactions, probably in concert with A756 in the light of previous data. If so, the ribozyme is required to be in the correct state of protonation to be active, and this could be reflected by the pH dependence of the observed rate of cleavage.
We were concerned that a dependence of reaction rate upon divalent ion concentration might distort the measured pH dependence, and therefore made an initial study of the dependence of cleavage upon Mg2+ concentration. We found that the rate of cleavage was near-maximal in the presence of 200 mM Mg2+ at both pH 5.5 and 8 (data not shown). We therefore performed all our studies of pH dependence in the presence of in 200 mM MgCl2, 25 mM KCl and 50 mM buffer at the required pH; under these conditions, the natural sequence substrate was cleaved at a rate of ∼5 min−1 at pH 8.
We have found it helpful to simulate the pH dependence expected for the participation of nucleobases of different pKA to aid the interpretation of experimental data. Figure 6A shows the protonated and deprotonated fractions of an acid (fraction fA) of pKA 5.5 and base (fB) of pKA 9.0, which might correspond to an adenine base (with its pKA raised by the electronegative environment) and guanine base respectively. The product fA·fB is then the fraction of ribozyme in the correct form to perform catalysis. The observed rate of cleavage should follow the pH dependence determined by fA·fB. The shaded regions correspond to the extreme pH values inaccessible to experimental study. The rate of reaction rises at lower pH, reaching a maximum around neutrality due to deprotonation of the acid. At increased pH, the rate declines again, due to almost complete deprotonation of the base. This may or may not be within the accessible region; in the hairpin ribozyme, this is barely observable (Kuzmin et al, 2004; Wilson et al, 2006). The shape of the fA·fB curve is unaltered if the acid and base pKA values are exchanged (Bevilacqua, 2003).
Figure 6.
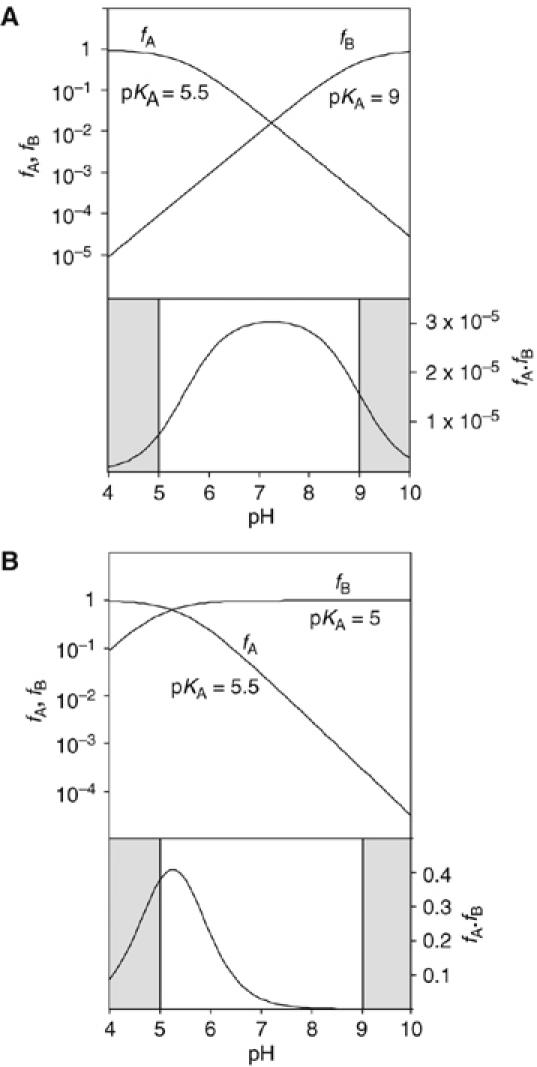
Calculated pH dependence of the cleavage reaction of the VS ribozyme as a function of base pKA, assuming general acid–base catalysis. The fractions of protonated acid fA, unprotonated base fB and their product fA·fB have been calculated and plotted as a function of pH, following the approach of Bevilacqua (2003). The shaded sections are the regions of pH not accessible to experimental study. Reaction rate should be proportional to the fraction of ribozyme in the appropriate state of protonation, that is, fA·fB. Note that in these graphs, fA and fB are plotted on a log10 scale (left), whereas fA·fB is plotted on a linear scale (right). (A) Plot for pKA values of 5.5 and 9 for the acid and base, respectively. This might correspond to the natural ribozyme, assuming that the acid is an adenine with an elevated pKA and the base is a guanine with a slightly reduced pKA. The predicted reaction rate profile is a broad bell shape, with a maximum close to neutrality. (B) Plot for pKA values of 5.5 and 5 for the acid and base, respectively. This situation could emerge if the guanine were replaced by a nucleobase of significantly lower pKA (e.g. adenine or DAP). The reaction rate is predicted to exhibit a marked increase at low pH, with a maximum that is just detectable for these values.
The pH dependence of the cleavage rate of the natural substrate (i.e. with guanine at position 638) is shown in Figure 7A. An increase of rate with pH is clearly observable up to pH 7, and declines with further increase in pH. The data have been fitted to a double-ionization model (Bevilacqua, 2003), giving apparent pKA values of 5.2±0.1 and 8.4±0.1. While other explanations are possible (see Discussion), this pH profile is consistent with the hypothesis that A756 and G638 participate in acid–base catalysis. The lower pKA is significantly higher than free adenosine mononucleotide (3.7; Dawson et al, 1986), but corresponds well to values measured previously by NMR for adenine bases in catalytic RNA molecules (Legault and Pardi, 1997; Ravindranathan et al, 2000). A value of 5.6 was found for the trans ligation reaction of the VS ribozyme under standard buffer conditions (McLeod and Lilley, 2004). That study, and an earlier one of the cleavage reaction (Guo and Collins, 1995), did not show a significant decrease in rate at high pH, consistent with an upper pKA lying outside the observable range; the pKA of free GMP is 9.4 (Dawson et al, 1986). The lower apparent pKA values observed under present conditions could be a consequence of the 20-fold higher concentration of Mg2+ ions used in this study.
Figure 7.
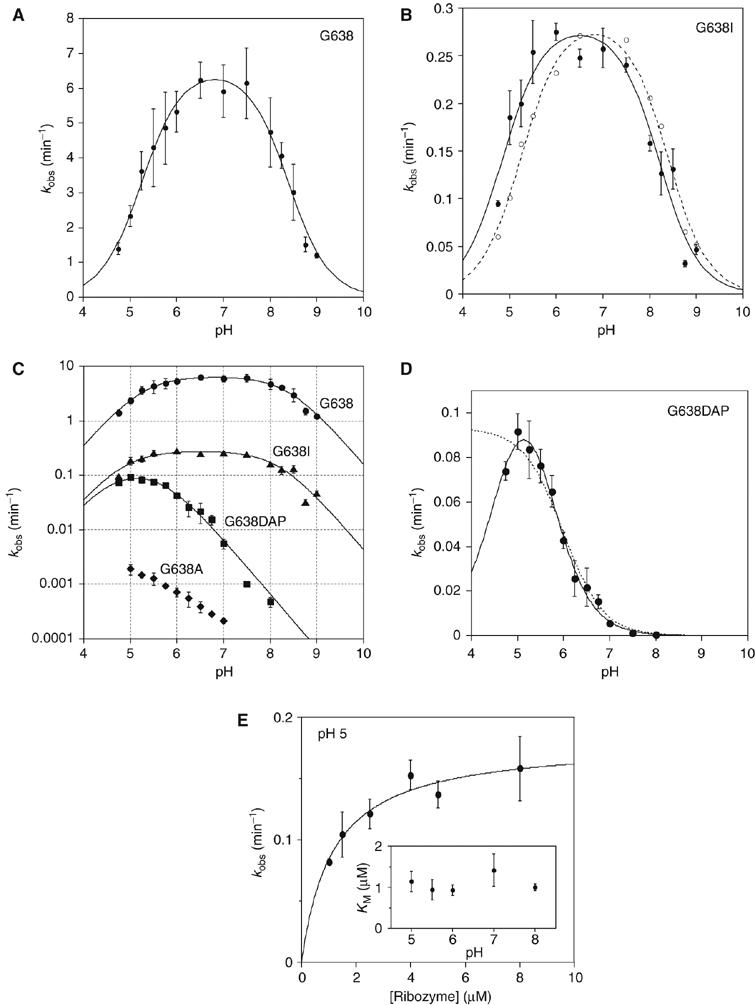
pH dependence of observed rates for VS ribozyme cleavage reactions in trans with the natural and modified substrates. Cleavage reaction rates were measured under single-turnover conditions in 50 mM MES, HEPES or TAPS of required pH containing 200 mM MgCl2 and 25 mM KCl. All rates are averages of ⩾3 measurements, and the error bars indicate one standard deviation. All data have been fitted to a double-ionization model in which there is a requirement for one protonated and one deprotonated form (equation 2), from which apparent pKA values have been determined. (A) The pH dependence of the rate of cleavage of the natural sequence substrate. The data are well fitted by the double-ionization model model, giving apparent pKA values of 5.2±0.1 and 8.4±0.1, and an intrinsic rate of 6.6 min−1. (B) The pH dependence of the rate of cleavage of the G638I substrate. The data (filled circles) are fitted (continuous line) to the double-ionization model model, giving pKA values of 4.8±0.1 and 8.2±0.1, and an intrinsic rate of 0.29 min−1. The data (open circles) and fit (broken line) for the natural sequence substrate (scaled to the intrinsic rate of the G638I reaction) reveal significant shifts of reaction pKA values for the modified substrate. (C) The data for four substrates plotted as the log10 of the observed rate as a function of pH. The grid facilitates estimation of the gradients. Natural sequence substrate, circles; G638I substrate, triangles; G638DAP, squares and G638A, diamonds. (D) The pH dependence of the rate of cleavage of the G638DAP substrate. The reaction is accelerated at lower pH values, and a clear maximum is observed at pH 5. The data have been fitted to single- (broken line; equation 3) and double (unbroken line)-ionization models. The fit to the double-ionization model gives pKA values of 4.6±0.2 and 5.6±0.1 and an intrinsic rate of 0.15 min−1. (E) The KM for the G638DAP substrate. Rates of cleavage by the trans-acting VS ribozyme were measured as a function of ribozyme concentration over a range of buffer pH. Each rate was plotted and fitted to equation 4, from which values of KM and k2 were calculated. The data for pH 5 are shown. The variation of KM over the range of pH between 5 and 8 is plotted (inset).
Altered pKA values for cleavage of a G638 inosine substrate
As noted above, substitution of G638 by inosine (G638I) leads to a cleavage rate that is only reduced by a factor of 27 at pH 8. The expected pKA of inosine is ∼0.5 pH units lower than that of guanine, and we therefore analysed the pH dependence of the cleavage rate of the G638I substrate (Figure 7B). The shape of the profile is quite similar to that of the natural sequence substrate, but the maximum of the bell is shifted to lower pH. Fitting the experimental data to the double-ionization model gave apparent pKA values of 4.8±0.1 and 8.2±0.1. The upper value is lower than that of IMP by ∼0.7 pH units (Dawson et al, 1986), and represents a shift of 0.2 pH units compared with the natural guanine containing sequence. The lower pKA is also reduced with respect to that for the natural sequence substrate (0.4 pH units), suggesting that the substitution of guanine by inosine has altered the pKA of the putative adenine in the ribozyme. This is consistent with close proximity of these two nucleobases in the active site.
The rates of cleavage of the G638A and G638DAP substrates are elevated at low pH
We have simulated the expected effect of replacing guanine by a nucleobase of low pKA, shown in Figure 6B. This leads to a predicted bell-shaped pH dependence centered near pH 5; however the low-pH wing of the bell may occur at a pH that is too low to be observable. While the rate of cleavage of the G638A substrate is very low at pH 8, a significant increase in rate was observed at low pH (Figure 7C). Since there is no evidence of an approach to a maximum rate, it is only possible to conclude that this pH dependence is consistent with one or more groups with a pKA less than 5. This implies that substitution of guanine by adenosine has also reduced the pKA of the putative adenine in the ribozyme.
In contrast to adenine, the normal pKA of 2,6-diaminopurine (DAP) of 5.1 (Dawson et al, 1986) lies within the experimentally accessible range of pH and the cleavage rate of G638DAP substrate is maximal at pH ∼5 (Figure 7D). Over the same pH range, the KM of the G638DAP substrate is constant (Figure 7E), and competition experiments similar to those in Figure 4 demonstrate that substrate binding is rapid (data not shown). Thus, the observed rate is a measure of the central conversion of substrate to product by the ribozyme. The data can be fitted by a single-ionization model (equation 3), yielding an apparent pKA of 6.0±0.1. This could plausibly be the pKA of DAP, or even that of an adenine in the ribozyme. While this model cannot be excluded, a four-fold improvement in χ2 was obtained when the double-ionization model was used to fit the data, yielding apparent pKA values of 4.6±0.2 and 5.6±0.1. These values are too close to be assigned with confidence, but we assume that the higher value corresponds to DAP, which is the weaker acid in the free state. The lower value is similar to that of the proposed adenine in the cleavage of the G638I substrate, providing further evidence that the nucleotide at position 638 influences the pKA of the second nucleobase. The maximum rate determined for the G638DAP substrate is only two-fold lower than that for the G638I substrate, indicating that the carbonyl group at position 6 is not critical to the catalytic mechanism, leaving the protonation of N1 as the main requirement for activity.
Discussion
In this study, we have identified the nucleotide G638 as being critical to the function of the VS ribozyme. Replacement by any other natural nucleotide leads to almost 10 000-fold slower rates of substrate cleavage under standard conditions, and a number of functional group changes were similarly disabling. The only other nucleotide in the ribozyme of comparable importance is A756, for which there is good evidence indicative of a role in catalysis (Lafontaine et al, 2001b, 2002b; Hiley et al, 2002; Sood and Collins, 2002; Jones and Strobel, 2003; Zhao et al, 2005).
The large impairment of cleavage activity arising from sequence substitution at position 638 is unlikely to result from incorrect folding of the substrate stem-loop. In-line probing indicates that the secondary structure is unaltered by a G638A substitution. Moreover, the G638A substrate acts as a competitive inhibitor of cleavage of the regular substrate, with a KI that is closely similar to the KM of the reaction. Competition experiments have also established that binding and dissociation of both the natural and variant substrates is rapid, establishing an equilibrium within seconds (Figure 4). Thus, under standard conditions, the rate-limiting step lies in the central conversion of substrate to product. Other competition experiments and the single-exponential dependence of reaction progress show that this remains true under all experimental conditions utilized in this study (data not shown). Since the G638A substrate binds to the ribozyme in an apparently normal manner, but is catalytically impaired, it suggests that nucleotide G638 plays a direct role in the function of the ribozyme, and since the 2′-hydroxyl group can be substituted by a proton at this position (Tzokov et al, 2002), it is probable that the nucleobase performs this function.
In both the hairpin and hammerhead ribozymes, guanine nucleobases (G+1 and G8, respectively) play key roles by basepairing with remote cytosine bases. A similar role for G638 is unlikely in the VS ribozyme, however. In the hairpin ribozyme, G+1 is immediately adjacent to the scissile phosphate and basepairing with C25 rotates the nucleotide to generate a near in-line geometry for nucleophilic attack (Rupert and Ferré-D'Amaré, 2001). By contrast, in the VS ribozyme G638 is located on the opposing strand to the scissile phosphate, so cannot play a similar role. G8 of the hammerhead ribozyme is present at the point of strand exchange of the three-way helical junction, and pairing with C3 can be regarded as a one-step branch migration contributing to the remodelling and concomitant activation of the ribozyme (Martick and Scott, 2006). The position of G638 in the VS ribozyme precludes such a role. If G638 were to pair with a remote cytosine base, the only probable candidate seems to be C755. However, we find that cleavage activity of the G638A substrate is not significantly increased by the C755U substitution that could restore basepairing between these positions.
This leaves two further potential roles for G638. First, it could directly participate in catalysis. This might occur through stabilization of the transition state, general acid–base catalysis, or perhaps both. In the absence of crystallographic data, it is difficult to evaluate transition state stabilization. However, our data are fully consistent with the hypothesis that G638 participates in general acid–base catalysis. Significant rates of cleavage require only a purine nucleus and an imino proton at N1. Neither the amine at C2 nor the carbonyl at C6 are essential for significant levels of activity under all conditions. Assuming that the apparent pKA values reflect the titration of functional groups contributing to catalysis, all our data on the variation of cleavage rates with pH are consistent with protonation–deprotonation events occurring at the nucleobase at position 638 (guanine, adenine, 2,6-diaminopurine or inosine) in concert with another nucleobase of relatively low pKA, that is, the putative A756. Furthermore, a plot of log kobs as a function of pH (Figure 7D) shows that the rate of cleavage of the G638DAP substrate is linear with unit gradient between pH 6–8. This indicates that general acid–base catalysis contributes more than two orders of magnitude to the catalytic power of the ribozyme.
A chemical mechanism consistent with the proposed roles of G638 and A756 is shown in Figure 8A. The present data do not provide any basis for deciding which nucleobase plays the role of base and which is the acid; either would be consistent with the pH dependence of cleavage rates. Clearly these mechanisms require close juxtaposition of the scissile phosphate and the nucleobases of G638 and A756. The pH dependence of the cleavage rate of the natural sequence substrate indicates that the pKA value of the putative adenine nucleobase of the ribozyme is shifted toward neutrality in the environment of the active site. Each chemical substitution made at position 638 reduced this effect, consistent with their close proximity. Although there are no atomic resolution data on the structure of the ribozyme, the isolated substrate stem-loop has been the subject of a number of studies by NMR in various forms (Michiels et al, 2000; Flinders and Dieckmann, 2001; Hoffmann et al, 2003). In the putative active form with a five-nucleotide loop (Hoffmann et al, 2003), the Watson–Crick edge of G638 is not involved in hydrogen bonding, and the N1 proton is free of any interactions and 6.5 Å from the scissile phosphate. There appears to be little hindrance to moving the nucleobase upon complex formation with the A730 loop in order to juxtapose G638 and the scissile phosphate. NMR chemical shift mapping has identified magnesium ion binding in the substrate loop; this might contribute to perturbation of pKA values.
Figure 8.
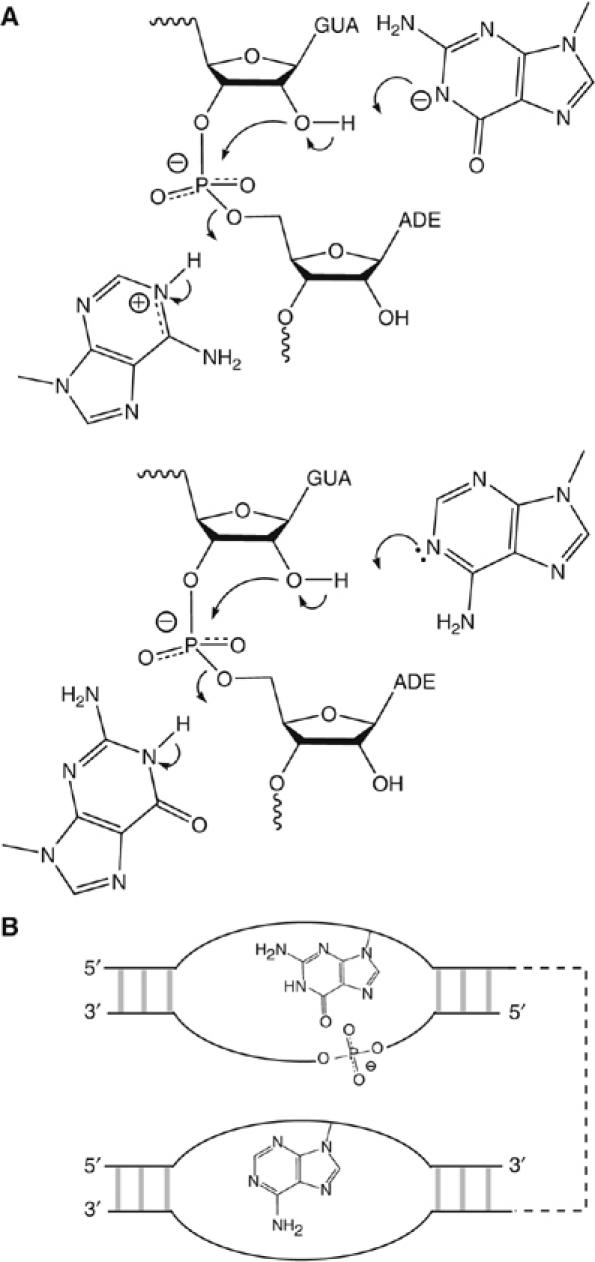
Probable catalytic mechanisms, and a strong similarity between the VS and hairpin ribozymes. (A) Two catalytic mechanisms based on the participation of G638 and A756 in general acid–base catalysis. In one possibility (upper), the guanine acts as the general base deprotonating the 2′-OH, whereas the adenine protonates the 5′-oxyanion leaving group. In the other (lower), the roles of the two nucleobases are reversed. At the present time we have no way to distinguish these possibilities; the pH dependence predicted on the basis of the two models would be identical. (B) The VS and hairpin ribozymes may be closely similar in catalytic mechanism. The active geometry of both is generated by loop–loop interaction, and the topological organization of scissile phosphate and the putative catalytic nucleobases is identical in both ribozymes. The hairpin ribozyme utilizes the upper catalytic mechanism shown in (A).
The second possible role for G638 consistent with our data is that the imino proton at N1 is required for a conformational change essential to cleavage. In this case, titration of the imino proton would yield rate versus pH curves indistinguishable from those resulting from the participation of G638 in general acid–base catalysis. Is there evidence for such a conformational change? The substrate undergoes a conformational change on binding to the ribozyme (Andersen and Collins, 2000); however this has been shown to require the loop–loop interaction with helix V that is critical to binding (Andersen and Collins, 2001). Perturbation of this conformational change would be expected to result in an increased KM (Zamel and Collins, 2002), which is not observed when G638 is substituted with adenine or DAP. Up to 100-fold faster rates of cleavage have been reported for modified cis VS ribozymes (Zamel et al, 2004), which could suggest that chemistry is not rate-limiting for our trans VS experiments. But such high rates have only been reported for substrates with additional nucleotides inserted into the substrate loop, which may alter the energetics of the reaction. cis VS ribozymes without insertions have two to three-fold faster reported cleavage rates, which may be due to differences in sequence or the extent of saturation of the ribozyme.
While there is no strong evidence allowing us to distinguish between roles for G638 in either conformational change or catalysis, if it is assumed that G638 participates only in a conformational change then the question remains as to what contributes to the considerable catalytic power of the VS ribozyme. All the available structural data point to an interaction between the 730 loop of the ribozyme and the internal loop of the substrate creating the active site for catalysis (see Introduction). Every nucleotide within these two loops has been subjected to mutagenesis, and the nucleobases of G638 and A756 identified as being particularly critical. Excluding functional groups on the substrate strand containing the scissile phosphate on structural grounds, the only other modifications that have a significant effect are the nucleobases of A730 and G757. Changes to the former must perturb the secondary structure of the 730 loop, whereas changes to the latter reduce k2 by only 10-fold (Lafontaine et al, 2001b), and the pH dependence of a G757A ribozyme is bell-shaped with apparent pKA values close to those of the natural ribozyme (unpublished data). Therefore, we favor the hypothesis that G638 and A756 directly contribute to catalysis by the VS ribozyme through general acid–base catalysis.
The proposed mechanism for the VS ribozyme brings out a striking similarity with the hairpin ribozyme. Figure 8B shows a schematic representation to illustrate the common organization of both species. Each generates the active geometry by the intimate association between two internal loops. In both cases, the scissile phosphate lies on the opposing strand within the same loop as the active guanine (the substrate loop of the VS and the A loop of the hairpin ribozymes), while the adenine is provided by the other loop (the A730 and B loops of the VS and hairpin ribozymes respectively). In fact, the topology of the two ribozymes is identical when the polarity of the strands is included. Moreover, the relative positioning of the scissile phosphate and guanine is quite similar in both ribozymes. The probable key role of G638 in the VS ribozyme could be regarded as a form of substrate assistance, although it may be misleading to continue to refer to the ‘substrate' stem-loop, as we must now consider it to be an integral part of the ribozyme.
The likely topological identity of the VS and hairpin ribozymes is probably the result of convergent evolution, since the two ribozymes share little structural similarity otherwise. Rather, it probably arises because there are relatively few ways to build an active site for nucleolysis in RNA. And while these ribozymes seem to have adopted very similar solutions to the problem, the HDV ribozyme uses alternative functionalities to carry out otherwise similar general acid–base catalysis. The glmS ribozyme appears to have adopted a yet different solution, using a glucosamine-6-phosphate effector in its catalytic chemistry (McCarthy et al, 2005; Klein and Ferré-d'Amaré, 2006). Yet the probable mechanistic similarities between the different ribozymes are more important than the differences, suggesting rather common mechanisms to achieve their function.
Materials and methods
Chemical synthesis of oligoribonucleotides
Oligonucleotides were synthesized using t-BDMS phosphoramidite chemistry (Beaucage and Caruthers, 1981), deprotected in 25% ethanol/ammonia solution at 55°C and evaporated to dryness. Oligonucleotides were redissolved in 300 μl 1 M TBAF (Aldrich) in THF to remove t-BDMS protecting groups and agitated at 20°C for 16 h before desalting by G25 Sephadex (Pharmacia). All oligoribonucleotides were purified by gel electrophoresis in polyacrylamide and recovered from gel fragments by electroelution. The sequence of the substrate was (all sequences written 5′–3′): GCGCGAAGGGCGUCGUCGCCCCGAT, including deoxyribothymine at the 3′-terminus (underlined) added for synthetic convenience, which does not affect the rate of cleavage. Versions of the substrate with alternative nucleotides or non-natural nucleotides substituted at specific positions were synthesized as indicated in the text.
Preparation of VS ribozyme by transcription
Templates for transcription of ribozymes were prepared by recursive PCR from synthetic DNA oligonucleotides. RNA was synthesized using T7 RNA polymerase (Milligan et al, 1987) and purified by electrophoresis in 5% polyacrylamide gels containing 7 M urea. RNA was recovered by electroelution into ammonium acetate. The sequence of the trans-acting ribozyme was: GCGGUA GUAAGCAGGGAACUCACCUCCAAUUUCAGUACUGA AAUUGUCGU AGCAGUUGACUACUGUUAUGUGAUUGGUAGAGGCU AAGUGACGG UAUUGGCGUAAGUCAGUAUUGCAGCACAGCACAAG CCCGCUUGC GAGAAU
Analysis of ribozyme kinetics
Cleavage kinetics were studied under single-turnover conditions. Equal volumes containing ribozyme and 5′-32P-labelled substrate were incubated at 37°C for 20 min in reaction buffer and the reaction initiated by mixing the two. The final reaction contained 1 μM ribozyme and 10 nM substrate. Standard reaction conditions were 50 mM Tris (pH 8), 10 mM MgCl2, 25 mM KCl and 2 mM spermidine. For the study of the pH dependence of rates, the cleavage reactions were performed in 200 mM MgCl2, 25 mM KCl and 50 mM buffer at the required pH, that is, MES, pH 4.75–6.75, HEPES, pH 6.75–8.0 and TAPS, pH 8.0–9.0. Slow reactions requiring long incubations (up to 48 h) were carried out under mineral oil to prevent evaporation. Aliquots (2 μl) were removed at intervals and the reaction terminated by addition to 8 μl of a mixture containing 95% (v/v) formamide, 50 mM EDTA and electrophoresis dyes. Substrate and product were separated by electrophoresis in 20% polyacrylamide gels containing 7 M urea and quantified by phosphorimaging. Progress curves were fitted by non-linear regression analysis to single- or double-exponential functions using Kalaidagraph (Abelbeck Software).
Analysis of pH dependence of reaction rate
The pH dependence of observed cleavage rates, kobs, was fitted to a double-ionization model assuming a requirement for one protonated and one deprotonated form, that is, 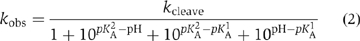
where kcleave is the intrinsic rate of the cleavage reaction and pKA1 and pKA2 are the acid dissociation constants of two titrating functional groups. This model is appropriate for analysis of general acid–base catalysis. The pH dependence of cleavage rate of the G638DAP substrate was also fitted to a single-ionization model in which the protonated form is assumed to be active, that is, 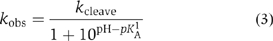
where the parameters are as defined above.
Determination of KM
The pH dependence of KM for the G638DAP substrate was determined under single-turnover conditions by measuring the rate of the cleavage reaction as a function of ribozyme concentration and fitting to the equation 
where [Rz] is the ribozyme concentration, k2 is the rate of conversion of substrate to product complex and KM is the Michaelis constant for the substrate.
In-line probing of RNA structure
Variant substrates were synthesized with DNA extensions at the 5′ and 3′ ends, so that the entire RNA section would be resolved by gel electrophoresis. The sequences were:
natural GTCTCAATCTGCGCGAAGGGCGUCGUCGCCCCGATTTT
G638A GTCTCAATCTGCGCGAAGGGCGUCGUCGCCCCAATTTT
where deoxyribose nucleotides are underlined. A modified version of the probing method of Breaker (Soukup and Breaker, 1999) was used. A 1 pmol solution of 5′-32P-labelled substrate RNA was incubated in 20 μl standard VS buffer for 40 h at 25°C. Aliquots were diluted with two volumes of formamide, heated to 90°C and placed on ice. Uniformly cleaved RNA was generated by heating 10 pmol 5′-32P-labelled substrate RNA in 40 mM NaOH and 40% formamide at 90°C for 2 min before addition of an equal volume of 500 mM Tris–HCl (pH 7.5) and placing on ice.
Acknowledgments
We thank Cancer Research UK and the BBSRC for financial support.
References
- Andersen AA, Collins RA (2000) Rearrangement of a stable RNA secondary structure during VS ribozyme catalysis. Mol Cell 5: 469–478 [DOI] [PubMed] [Google Scholar]
- Andersen AA, Collins RA (2001) Intramolecular secondary structure rearrangement by the kissing interaction of the Neurospora VS ribozyme. Proc Natl Acad Sci USA 98: 7730–7735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beattie TL, Olive JE, Collins RA (1995) A secondary-structure model for the self-cleaving region of Neurospora VS RNA. Proc Natl Acad Sci USA 92: 4686–4690 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beaucage SL, Caruthers MH (1981) Deoxynucleoside phosphoramidites—a new class of key intermediates for deoxypolynucleotide synthesis. Tetrahedron Lett 22: 1859–1862 [Google Scholar]
- Bevilacqua PC (2003) Mechanistic considerations for general acid–base catalysis by RNA: revisiting the mechanism of the hairpin ribozyme. Biochemistry 42: 2259–2265 [DOI] [PubMed] [Google Scholar]
- Das SR, Piccirilli JA (2005) General acid catalysis by the hepatitis delta virus ribozyme. Nat Chem Biol 1: 45–52 [DOI] [PubMed] [Google Scholar]
- Dawson RMC, Elliott DC, Elliott WH, Jones KM (1986) Data for Biochemical Research. Oxford: OUP [Google Scholar]
- Ferré-d'Amaré AR, Zhou K, Doudna JA (1998) Crystal structure of a hepatitis delta virus ribozyme. Nature 395: 567–574 [DOI] [PubMed] [Google Scholar]
- Flinders J, Dieckmann T (2001) A pH controlled conformational switch in the cleavage site of the VS ribozyme substrate RNA. J Molec Biol 308: 665–679 [DOI] [PubMed] [Google Scholar]
- Guo HCT, Collins RA (1995) Efficient trans-cleavage of a stem-loop RNA substrate by a ribozyme derived from Neurospora VS RNA. EMBO J 14: 368–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J, Burke JM (2005) Model for general acid–base catalysis by the hammerhead ribozyme: pH–activity relationships of G8 and G12 variants at the putative active site. Biochemistry 44: 7864–7870 [DOI] [PubMed] [Google Scholar]
- Hiley SL, Sood VD, Fan J, Collins RA (2002) 4-thio-U crosslinking identifies the active site of the VS ribozyme. EMBO J 21: 4691–4698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann B, Mitchell GT, Gendron P, Major F, Andersen AA, Collins RA, Legault P (2003) NMR structure of the active conformation of the Varkud satellite ribozyme cleavage site. Proc Natl Acad Sci USA 100: 7003–7008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones FD, Strobel SA (2003) Ionization of a critical adenosine residue in the Neurospora Varkud Satellite ribozyme active site. Biochemistry 42: 4265–4276 [DOI] [PubMed] [Google Scholar]
- Ke A, Zhou K, Ding F, Cate JH, Doudna JA (2004) A conformational switch controls hepatitis delta virus ribozyme catalysis. Nature 429: 201–205 [DOI] [PubMed] [Google Scholar]
- Klein DJ, Ferré-d'Amaré AR (2006) Structural basis of glmS ribozyme activation by glucosamine-6-phosphate. Science 313: 1752–1756 [DOI] [PubMed] [Google Scholar]
- Kuzmin YI, Da Costa CP, Cottrell JW, Fedor MJ (2005) Role of an active site adenine in hairpin ribozyme catalysis. J Mol Biol 349: 989–1010 [DOI] [PubMed] [Google Scholar]
- Kuzmin YI, Da Costa CP, Fedor MJ (2004) Role of an active site guanine in hairpin ribozyme catalysis probed by exogenous nucleobase rescue. J Mol Biol 340: 233–251 [DOI] [PubMed] [Google Scholar]
- Lafontaine DA, Norman DG, Lilley DMJ (2001a) Structure, folding and activity of the VS ribozyme: Importance of the 2–3–6 helical junction. EMBO J 20: 1415–1424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DA, Norman DG, Lilley DMJ (2002a) The global structure of the VS ribozyme. EMBO J 21: 2461–2471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lafontaine DA, Wilson TJ, Norman DG, Lilley DMJ (2001b) The A730 loop is an important component of the active site of the VS ribozyme. J Mol Biol 312: 663–674 [DOI] [PubMed] [Google Scholar]
- Lafontaine DA, Wilson TJ, Zhao Z-Y, Lilley DMJ (2002b) Functional group requirements in the probable active site of the VS ribozyme. J Mol Biol 323: 23–34 [DOI] [PubMed] [Google Scholar]
- Legault P, Pardi A (1997) Unusual dynamics and pKa shift at the active site of a lead-dependent ribozyme. J Am Chem Soc 119: 6621–6628 [Google Scholar]
- Martick M, Scott WG (2006) Tertiary contacts distant from the active site prime a ribozyme for catalysis. Cell 126: 309–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy TJ, Plog MA, Floy SA, Jansen JA, Soukup JK, Soukup GA (2005) Ligand requirements for glmS ribozyme self-cleavage. Chem Biol 12: 1221–1226 [DOI] [PubMed] [Google Scholar]
- McLeod AC, Lilley DMJ (2004) Efficient, pH-dependent RNA ligation by the VS ribozyme in trans. Biochemistry 43: 1118–1125 [DOI] [PubMed] [Google Scholar]
- Michiels PJA, Schouten CHJ, Hilbers CW, Heus HA (2000) Structure of the ribozyme substrate hairpin of Neurospora VS RNA: a close look at the cleavage site. RNA 6: 1821–1832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milligan JF, Groebe DR, Witherall GW, Uhlenbeck OC (1987) Oligoribonucleotide synthesis using T7 RNA polymerase and synthetic DNA templates. Nucleic Acids Res 15: 8783–8798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murray JB, Seyhan AA, Walter NG, Burke JM, Scott WG (1998) The hammerhead, hairpin and VS ribozymes are catalytically proficient in monovalent cations alone. Chem Biol 5: 587–595 [DOI] [PubMed] [Google Scholar]
- Nakano S, Chadalavada DM, Bevilacqua PC (2000) General acid–base catalysis in the mechanism of a hepatitis delta virus ribozyme. Science 287: 1493–1497 [DOI] [PubMed] [Google Scholar]
- Perrotta AT, Shih I, Been MD (1999) Imidazole rescue of a cytosine mutation in a self-cleaving ribozyme. Science 286: 123–126 [DOI] [PubMed] [Google Scholar]
- Pinard R, Hampel KJ, Heckman JE, Lambert D, Chan PA, Major F, Burke JM (2001) Functional involvement of G8 in the hairpin ribozyme cleavage mechanism. EMBO J 20: 6434–6442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rastogi T, Beattie TL, Olive JE, Collins RA (1996) A long-range pseudoknot is required for activity of the Neurospora VS ribozyme. EMBO J 15: 2820–2825 [PMC free article] [PubMed] [Google Scholar]
- Ravindranathan S, Butcher SE, Feigon J (2000) Adenine protonation in domain B of the hairpin ribozyme. Biochemistry 39: 16026–16032 [DOI] [PubMed] [Google Scholar]
- Rupert PB, Ferré-D'Amaré AR (2001) Crystal structure of a hairpin ribozyme–inhibitor complex with implications for catalysis. Nature 410: 780–786 [DOI] [PubMed] [Google Scholar]
- Rupert PB, Massey AP, Sigurdsson ST, Ferré-D'Amaré AR (2002) Transition state stabilization by a catalytic RNA. Science 298: 1421–1424 [DOI] [PubMed] [Google Scholar]
- Sood VD, Collins RA (2002) Identification of the catalytic subdomain of the VS ribozyme and evidence for remarkable sequence tolerance in the active site loop. J Mol Biol 320: 443–454 [DOI] [PubMed] [Google Scholar]
- Soukup GA, Breaker RR (1999) Relationship between internucleotide linkage geometry and the stability of RNA. RNA 5: 1308–1325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzokov AB, Murray IA, Grasby JA (2002) The role of magnesium ions and 2′-hydroxyl groups in the VS ribozyme–substrate interaction. J Mol Biol 324: 215–226 [DOI] [PubMed] [Google Scholar]
- Wilson TJ, Ouellet J, Zhao ZY, Harusawa S, Araki L, Kurihara T, Lilley DM (2006) Nucleobase catalysis in the hairpin ribozyme. RNA 12: 980–987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamel R, Collins RA (2002) Rearrangement of substrate secondary structure facilitates binding to the Neurospora VS ribozyme. J Mol Biol 324: 903–915 [DOI] [PubMed] [Google Scholar]
- Zamel R, Poon A, Jaikaran D, Andersen A, Olive J, De Abreu D, Collins RA (2004) Exceptionally fast self-cleavage by a Neurospora Varkud satellite ribozyme. Proc Natl Acad Sci USA 101: 1467–1472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Z, McLeod A, Harusawa S, Araki L, Yamaguchi M, Kurihara T, Lilley DMJ (2005) Nucleobase participation in ribozyme catalysis. J Am Chem Soc 127: 5026–5027 [DOI] [PubMed] [Google Scholar]