

Abstract
Vibrio cholerae RTX is a large multifunctional bacterial toxin that causes actin crosslinking. Due to its size, it was predicted to undergo proteolytic cleavage during translocation into host cells to deliver activity domains to the cytosol. In this study, we identified a domain within the RTX toxin that is conserved in large clostridial glucosylating toxins TcdB, TcdA, TcnA, and TcsL; putative toxins from V. vulnificus, Yersinia sp., Photorhabdus sp., and Xenorhabdus sp.; and a filamentous/hemagglutinin-like protein FhaL from Bordetella sp. In vivo transfection studies and in vitro characterization of purified recombinant protein revealed that this domain from the V. cholerae RTX toxin is an autoprocessing cysteine protease whose activity is stimulated by the intracellular environment. A cysteine point mutation within the RTX holotoxin attenuated actin crosslinking activity suggesting that processing of the toxin is an important step in toxin translocation. Overall, we have uncovered a new mechanism by which large bacterial toxins and proteins deliver catalytic activities to the eukaryotic cell cytosol by autoprocessing after translocation.
Keywords: Clostridium difficile Toxin B, cysteine protease, GTP, HCV NS2, V. cholerae RTX toxin
Introduction
Bacterial toxins utilize different mechanisms to facilitate entry of their catalytic domains into host cells. Type III and Type IV secretion systems accomplish cytosolic delivery of effectors through direct injection from the bacterium to the eukaryotic cytosol (Henderson et al, 2004). Other bipartite toxins utilize a secreted B-subunit that binds to a eukaryotic membrane receptor and delivers a bound catalytic A-subunit to the cytosol through a pore or channel (Falnes and Sandvig, 2000). Some larger bacterial toxins such as Bordetella pertussis adenylate cyclase (AC) utilize a poorly characterized self-translocation mechanism. AC self-inserts into the plasma membrane of host cells and then translocates the catalytic AC domain into the cell where it is held below the membrane to catalyze the transformation of ATP to cAMP (Rogel and Hanski, 1992). Similarly, the Clostridium difficile TcdB gains entry into cells through receptor-mediated endocytosis followed by self-translocation across the endosomal membrane. However, TcdB then undergoes proteolytic processing through an unidentified mechanism releasing the N-terminal catalytic domain to the cytosol (Pfeifer et al, 2003).
The 4545 amino acids Vibrio cholerae RTX toxin is a multifunctional toxin that was initially characterized for its ability to round epithelial cells (Lin et al, 1999). Further investigation revealed that the toxin causes depolymerization of the actin cytoskeleton through covalent actin crosslinking (Fullner and Mekalanos, 2000). A 48 kDa domain located at residues 1963–2419 was shown to be sufficient to crosslink actin in vitro (Cordero et al, 2006). In vivo experiments demonstrated that this actin crosslinking domain (ACD) must access the cytosol to crosslink actin (Cordero et al, 2006; Sheahan et al, 2004). In addition, a V. cholerae strain secreting RTX with an in-frame deletion removing the ACD still had the ability to round epithelial cells revealing that this toxin has multiple cytopathic activities (Sheahan et al, 2004). Thus, the RTX toxin is predicted to carry multiple activity domains, suggesting that the toxin undergoes processing after translocation to release individual domains into the cytosol. In particular, the ACD could be released to access actin at cellular locations other than directly below the membrane.
In this study, we identify a domain within RTX that is a cysteine protease responsible for autoproteolysis of the toxin. Activation of the protease requires binding of GTP indicating that processing would occur after translocation to the cytosol. Indeed, a conservative point mutation of the catalytic cysteine residue within the RTX holotoxin attenuates its actin crosslinking activity demonstrating that autoprocessing is important for cytopathic effects of this toxin. This domain is found in other known and putative large bacterial toxins suggesting that autoproteolysis by cysteine proteases is a conserved process for release and delivery of catalytic regions from large bacterial proteins.
Results
Identification of a conserved domain in large secreted bacterial proteins
Previously it was reported that amino acids 3376–3625 of V. cholerae RTX is 40% similar to a region of unknown function of C. difficile TcdA (Fullner and Mekalanos, 2000). Additional amino-acid sequence database searches revealed a total of 19 distinct copies of this domain (Figure 1A). Fifteen proteins, including nine Vibrio-type RTX toxins, four clostridial glucosyltransferase toxins, and two putative toxins from Yersinia sp., carry one copy of the domain while the filamentous/hemagglutinin-like protein FhaL from Bordetella sp. has four copies appearing in tandem in the C-terminus. Alignment of five diverse sequences by CLUSTALW revealed 13 highly conserved residues (Figure 1B). A CLUSTALW alignment of all 19 sequences showed that (according to consensus sequence in Figure 1B) Q49, K66, GHG122-4, GC175-6, and G228 were conserved in all copies of the domain. Other highly conserved residues AA59-60, L63, S71, and V215 contained only conservative substitutions in some copies (Supplementary Figure 1). Thus, this domain originally identified by alignment of the V. cholerae RTX toxin and TcdA appears to be a conserved domain of unknown function found in other large bacterial proteins that are predicted to be secreted and function either as toxins or adhesins.
Figure 1.

Alignment of putative cysteine protease domains. (A) Phylogenetic tree of 19 putative CPDs and (B) CLUSTALW alignment of five diverse sequences. †Symbol in phylogenetic tree indicates diverse sequences selected for alignment in (B). In (B), asterisks in consensus sequence represent conserved residues in 3/5 sequences and capital letters represent 100% identity. Outlined dipeptide sequence indicates cleavage sites determined experimentally. CPDs were identified within nine Vibrio-type RTX toxins from V. cholerae (VcRtx); V. vulnificus (VvRtx), V. splendidus (VsRtx), Xenorhabdus nematophila (XnRtx), X. bovienii (XbRtx), and Photorhabdus luminescens (Plu1344, Plu1341, Plu3217, and Plu3324); four clostridial toxins, specifically C. difficile toxin A (TcdA), toxin B (TcdB), C. sordellii cytotoxin L (TcsL), and C. noveyi alpha toxin (TcnA); two putative Yersinia toxins Y. pseudotuberculosis YPTB3219 (YpRtx) and Y. mollaretti Mfp2 (YmMfp2); and four domains arranged in tandem in B. pertussis putative adhesin FhaL (FhaL1-4).
Transient expression of amino acids 3376–3637 in COS-7 cells elicits cytotoxicity
To investigate the function of this domain, the region similar to TcdA corresponding to amino acids 3376–3637 of V. cholerae RTX was amplified by PCR and cloned into the eukaryotic expression vector pEGFP-N3 to create plasmid pCPDc. After transient transfection of this plasmid into COS-7 cells, cells expressing the EGFP fusion protein were identified by fluorescence microscopy and we observed that all transfected cells were rounded. The observed cell rounding was quite distinct from the cell rounding previously observed after transient expression of the ACD (Figure 2B). While cells expressing ACD were spherical and refractory to light (Sheahan et al, 2004), most cells expressing this region of RTX appeared apoptotic exhibiting condensed nuclei revealed by staining with Hoechst 33342. This phenotype was specific for cells expressing the EGFP fusion protein and was not observed in cells transiently expressing EGFP alone (Figure 2A). A Western blot of cell lysates with anti-GFP antibody revealed that the fusion protein ran on SDS–PAGE with an apparent molecular weight (mw) of 48 kDa, approximately 9 kDa smaller than the predicted mw 56.5 kDa (Figure 2E). These results initially suggested that this region of the RTX toxin carries cytotoxic activity and may undergo intracellular processing.
Figure 2.
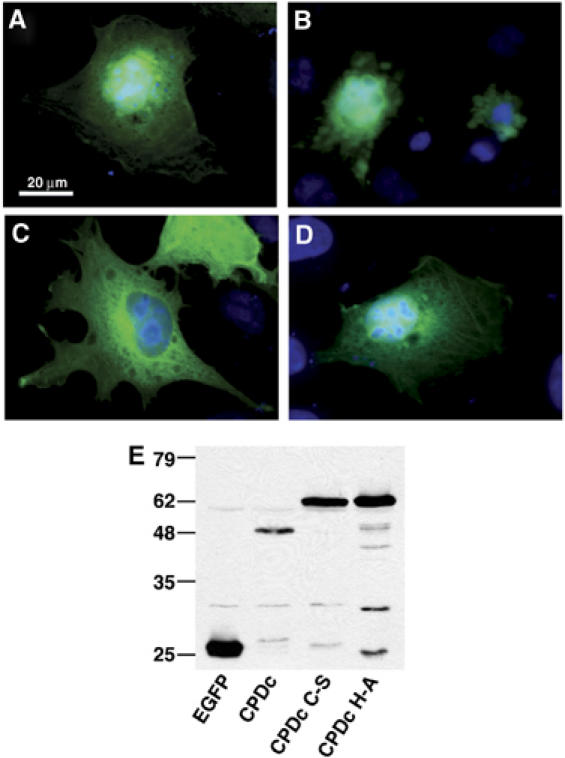
Expression of RTX amino acids 3376–3637 is cytotoxic to cells and the protein is processed intracellularly. COS-7 cells transiently expressing EGFP (A), CPDc-EGFP (B), CPDc C-S-EGFP (C), and CPDc H-A-EGFP (D) were observed by fluorescence microscopy. Representative images are displayed as an overlay of the fluorescence micrograph obtained at 550–575 nm to detect GFP and 440–470 nm to detect Hoechst staining. (E) At 24 h after transfection, cells were resuspended in SDS buffer, boiled, and subjected to SDS–PAGE and Western blotting with an anti-GFP antibody.
Cys3568 is essential for processing indicating the domain is an autoproteolytic cysteine protease
The CLUSTALW alignment showed that Cys3568 is conserved among all 19 copies of this domain of unknown function (Figure 1B) and analysis of the entire RTX sequence revealed that there are only two Cys residues present in all 4545 amino acids of the toxin. Therefore, we hypothesized that Cys3568 was important for cytotoxicity. To test this hypothesis, the Cys3568 codon in pCPDc was altered to encode a Ser residue. When the resulting plasmid pCPDc C-S was transfected into COS-7 cells, cell rounding and nuclear condensation were not observed (Figure 2C). Western blotting with anti-GFP antibody revealed that the EGFP fusion protein expressed by the pCPDc C-S plasmid ran at the predicted mw of 56.5 kDa showing that C3568 is essential for protein processing (Figure 2E).
A longer gene fusion that extended the expressed portion of RTX by 275 amino acids on the N-terminus also yielded an EGFP fusion protein that resolved at the same apparent mw as the protein expressed from pCPDc and processing was dependent upon Cys3568 (Figure 3B). Finally, expression of the plasmid pCPDn with an EGFP fusion to the N-terminus was cleaved between EGFP and this domain produced a fragment of 32 kDa corresponding to the predicted mw of EGFP with only a small portion of this domain attached (Figure 3B). Based on the observations from these three constructs, we concluded that amino acids 3376–3637 of RTX represents a cysteine protease domain (CPD) that catalyzes autoprocessing of the toxin to the N-terminal side of the CPD and the observed cytotoxic activity is an artifact of protease overexpression. A deletion analysis of the N-terminus of the CPD predicted that cleavage occurs within residues 3411–3441 of RTX (data not shown). Cotransfection studies showed that active CPD cleaved approximately 6% of CPD C-S, demonstrating that while a trans-acting processing event can occur, CPD predominantly undergoes a cis-acting proteolytic event (Supplementary Figure 2).
Figure 3.
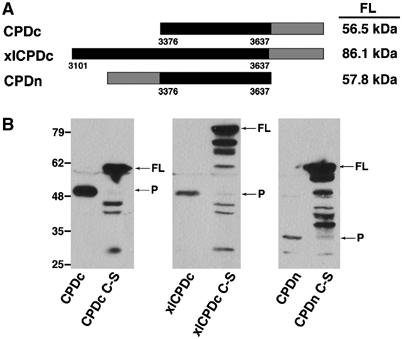
Transiently expressed CPD in cells is autoprocessed within the N-terminus. (A) Schematic representation of fusion proteins with the predicted mw of each full-length (FL) fusion protein indicated at right. Numbers along the bottom correspond to the amino-acid sequence of translated products according to the RTX toxin annotation of Lin et al (1999) (GenBank accession no. gi ∣4455065). (B) Expression of the CPD and CPD C-S fusion proteins was detected in transiently transfected COS-7 cells by Western blotting for GFP. Arrows mark full-length (FL) and the processed (P) forms of CPD.
His3519 is critical for CPD activity
The catalytic dyad of cysteine proteases generally requires a His residue (Rawlings and Barrett, 1994). His3519 was identified as a putative critical residue since it is conserved among all 19 proteins with this domain (Figure 1B). To determine if this residue is essential for proteolysis, pCPDc was mutagenized to convert the codon for His3519 to Ala. Transient expression of this plasmid in COS-7 cells also did not cause cytotoxicity (Figure 2D) and Western blotting revealed that the fusion protein was expressed at its predicted mw (Figure 2E). Some, but not all, cysteine proteases require a Glu to complete a catalytic triad. Glu3543 is conserved in 10 of the 19 copies of CPD; however, mutation of Glu3543 to an Ala on the pCPDc plasmid did not disrupt the autoprocessing activity of CPD (Supplementary Figures 1 and 4). These data confirm that both Cys3568 and His3519 are critical for CPD autoprocessing.
Recombinant CPD (rCPD) requires a heat-stable host factor for autoprocessing
In order to further characterize the CPD protease activity in vitro, recombinant CPD (rCPD) was purified for biochemical analysis. Both the CPD and CPD C-S were cloned into the His-tag vector pMCSG7 (Stols et al, 2002) to encode amino acids 3378–3637 of RTX with 6 × His tag fusions on both the N- and C-termini (Figure 4A). After expression in Escherichia coli, rCPD and rCPD C-S were purified by Ni-affinity chromatography from the soluble fraction. The proteins eluted from the column at the full length and resolved on SDS–PAGE at the predicted mw of 34 kDa demonstrating that rCPD was not cleaved in E. coli or during purification. This observation initially suggested that a eukaryotic host factor is necessary to stimulate the autoproteolytic activity of CPD.
Figure 4.
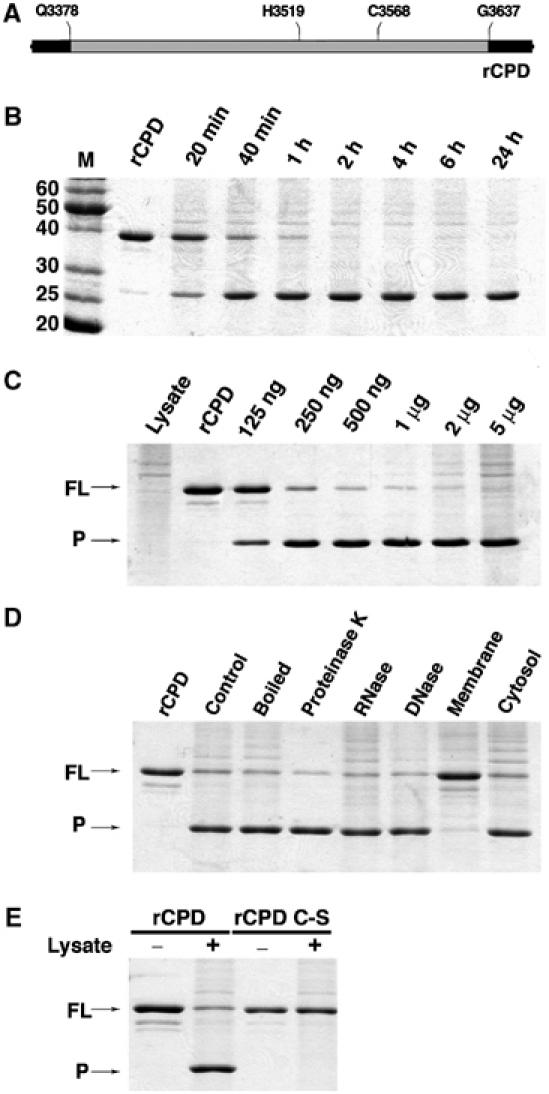
rCPD undergoes in vitro processing after addition of host cell lysate. (A) Schematic representation of rCPD (gray shading) with N- and C-terminal 6 × His tag fusions (black shading). (B–E) In vitro processing reactions were performed with 2 μg rCPD incubated in the presence of lysate and samples were separated by SDS–PAGE and stained with Coomassie R250. (B) rCPD was incubated with 5 μg of total cell protein and the reaction was stopped at the indicated time points. (C) Overnight incubation of rCPD with increasing amounts of lysate as indicated. (D) rCPD was incubated with equivalent volumes of lysate, boiled lysate, or lysate pretreated for 15 min with 200 μg/ml of Proteinase K, DNase, or RNase. rCPD was also incubated with membrane and cytosolic fractions (5 μg of total cell protein) obtained from subcellular fractionation of the lysate. (E) rCPD and rCPD C-S were incubated for 2 h in the absence (lanes 1 and 3) or presence of 5 μg of total cell protein (lanes 2 and 4). Arrows mark full-length (FL) and the processed (P) forms of rCPD.
The CPD cleavage activity was indeed stimulated by the addition of a nuclear-free cell lysate confirming that a host factor is necessary. As depicted in Figure 4B, the addition of 5 μg of total cell protein to 2 μg of rCPD stimulated processing as rapidly as 20 min with essentially all of the protein cleaved within 2 h. Further analysis of the reaction revealed that as little as 125 ng of total cell protein was sufficient to stimulate the cleavage of 2 μg of rCPD after overnight incubation (Figure 4C). Increasing the ratio of cell lysate to rCPD was able to drive the cleavage of rCPD to completion demonstrating that the lysate contained a limiting component of the reaction. Cell lysate that was boiled for 5 min or incubated with Proteinase K, DNase or RNase prior to addition to rCPD also stimulated the processing activity (Figure 4D). Subcellular fractionation of the lysate revealed that the host cell factor stimulating cleavage of rCPD resides in the cytosolic fraction (Figure 4D). Thus, the autoprocessing activity of RTX requires a heat-stable cytosolic host cell factor that is not a protein, DNA, or RNA. The addition of cell lysate to the rCPD C-S protein did not produce the processed protein confirming that the cysteine is necessary for activity and processing did not depend on a protease in the cell lysate (Figure 4E).
Autoprocessing of CPD is inhibited by N-ethylmaleimide but not other protease inhibitors
To further define the mechanism of the CPD-mediated cleavage, rCPD was pre-incubated with various protease inhibitors for 30 min, followed by the addition of cell lysate and continued incubation overnight. Only N-ethylmaleimide (NEM) completely inhibited in vitro cleavage of rCPD (Figure 5). This inhibitor irreversibly alkylates the thiol group of cysteine and is a potent inhibitor of all cysteine protease families (Anderson and Vasini, 1970). The small molecules E64 and calpeptin, inhibitors of the CA clan of cysteine proteases (Hanada et al, 1978; Sasaki et al, 1990), did not inhibit cleavage of rCPD consistent with the His-Cys arrangement of the catalytic dyad of rCPD (Figure 1B) rather than the Cys-His arrangement found in clan CA cysteine proteases. Phenylmethylsulphonylfluoride (PMSF), which is predominantly a serine protease inhibitor but has also been shown to inhibit some cysteine proteases (Whitaker and Perez-Villase nor, 1968), partially inhibited cleavage and this inhibition was not due to the addition of isopropanol used to solubilize PMSF (data not shown). Other protease inhibitors including aspartate protease inhibitor pepstatin and serine protease inhibitor leupeptin did not inhibit cleavage of rCPD. Altogether, these inhibitor studies confirm that rCPD is an autoprocessing cysteine protease unrelated to the Clan CA proteases.
Figure 5.
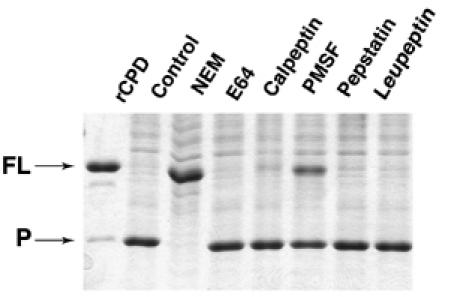
Autoprocessing of rCPD is inhibited by NEM. rCPD (2 μg) was preincubated for 30 min with 1 mM NEM, E-64, Calpeptin, or PMSF, 100 μM Pepstatin, or 200 μM Leupeptin. Then, 5 μg of total cell protein was added to stimulate processing followed by overnight incubation. Proteins were separated by SDS–PAGE and stained with Coomassie R250. Arrows mark full-length (FL) and the processed (P) forms of rCPD.
GTP binding activates rCPD
To pursue the chemical nature of the cytosolic stimulatory factor, common small molecule enzymatic co-factors present in the cytosol were added to rCPD and cleavage was assessed at 2 h (Figure 6A). Addition of 5 mM guanine nucleotide molecules GTP, GDP, or the non-hydrolyzable GTP analogs guanosine 5′-[β,γ-imido]triphosphate (GMP-PNP) and guanosine 5′-[γ-thiol]triphosphate (GTPγS) initiated processing of rCPD (Figure 6A and B). No other nucleotides stimulated autoprocessing of rCPD at 2 h (Figure 6A), although ATP partially stimulated cleavage after 20 hr incubation (data not shown). rCPD processing initiated by GTPγS was more efficient than processing initiated by GTP possibly due to the rapid hydrolysis of GTP in solution (Supplementary Figure 3). Indeed, autoprocessing of rCPD could be initiated by addition of only 0.2 mM GTPγS, did not require addition of MgCl2 to the reaction, and reached completion by 2 h similar to initiation of processing with a cytosolic protein preparation (Figure 6C). These results demonstrated that autoprocessing is stimulated by binding of physiologically relevant concentrations of GTP, but hydrolysis of GTP is not required for enzyme activity.
Figure 6.
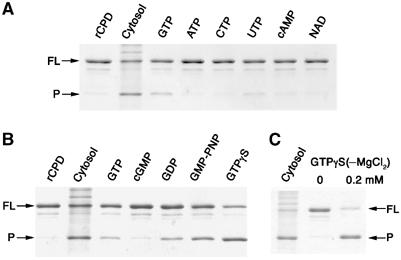
rCPD processing is stimulated by guanine nucleotides. (A, B) rCPD (2 μg) was incubated alone (rCPD), with 5 μg of cytosolic protein preparation (Cytosol), or with 5 mM MgCl2 plus 5 mM of the indicated compound. (C) Incubation of 2 μg rCPD with the indicated concentration of GTPγS in the absence of MgCl2. All reactions were terminated after 2 h at 37°C. Proteins were separated by SDS–PAGE and stained with Coomassie R250. Arrows mark full-length (FL) and the processed (P) forms of rCPD.
To further investigate GTP activation of CPD, binding of GTP to the protein was monitored using a fluorescence-based assay. For these assays, the fluorophore-labeled nucleotide is quenched by interaction between guanosine and the fluorophore; however, when the nucleotide binds to a protein, there is a detectable increase in fluorescence due to the stabilization of the fluorophore (Kimple et al, 2001). Preliminary experiments with mant-GTP demonstrated binding of 0.5 μM mant-GTP to 50 μM rCPD (I=33 972±1078; FIC=0.31±0.04) and rCPD C-S (I=32 345±470; FIC=0.25±0.02), suggesting a slight decrease in the binding affinity of rCPD C-S compared to rCPD. However, we were unable to establish a binding constant for rCPD with mant-GTP due to the high background fluorescence of rCPD at 440 nm, especially at high protein concentrations, and the potential for GTP hydrolysis and protein processing during the binding reaction. Therefore, kinetic binding experiments were performed with BODIPY FL-GTPγS since rCPD does not fluoresce at the BODIPY FL emission maximum of 512 nm and hydrolysis of the nucleotide is prevented. In addition, rCPD C-S was used to avoid protein cleavage during the binding reaction. As depicted in Figure 7A, upon excitation at 488 nm the emission spectra revealed that the addition of 199 μM rCPD C-S to 0.25 μM BODIPY FL-GTPγS resulted in an increase in the fluorescence intensity (I) with maximum emission at 512 nm. At 50 μM rCPD C-S, the measured FIC (Iobs–Ifree)/Ifree was similar to that measured for binding of mant-GTP (Figure 7B and C). The interaction of varied concentrations of rCPD with 0.25 μM BODIPY FL-GTPγS showed standard binding kinetics with a Kd of 130 μM (Figure 7B). Competitive inhibition studies demonstrated a decrease in FIC with increasing concentrations of unlabeled GTPγS with a Ki of 0.3 μM demonstrating that the interaction of GTPγS with the protein is specific and not due to a nonspecific interaction of the protein with fluorophore (Figure 7C). These studies show that GTP and GTPγS bind to rCPD and rCPD C-S and indicate binding and autoprocessing readily occur under cellular concentrations of GTP, which are 10-fold above the binding constant.
Figure 7.
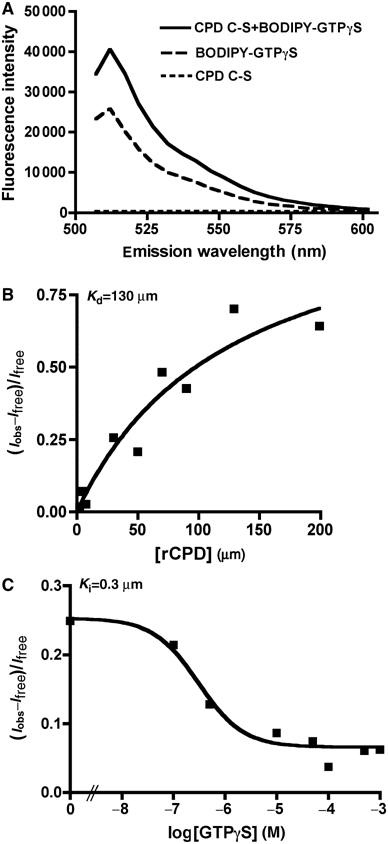
rCPD C-S binds GTPγS. (A) Representative emission scan after excitation at 488 nm of 200 μM rCPD C-S alone, 0.25 μM BODIPY FL- GTPγS alone, or rCPD mixed with BODIPY FL- GTPγS showing maximum fluorescence intensity at 512 nm and the absence of background fluorescence from the protein. (B) Binding curve of varying concentration of rCPD incubated with 0.25 μM BODIPY FL- GTPγS. (C) Competitive inhibition curve of 50 μM CPD incubated with 0.25 μM BODIPY FL- GTPγS with varying concentrations of unlabeled GTPγS. Kd and Ki were determined as described in Materials and methods.
CPD cleaves between L3428 and A3429 of the RTX toxin
Fourier transform mass spectrometry (FT-MS) is an analytical method that can determine the mw of proteins under 30 000 Da to within 0.1 mass units. Since the mw of rCPD is known to be 34157.2 Da, the exact cleavage site can be deduced from the FT-MS determined mw of the peptides obtained after stimulation of the protease. After overnight incubation, an rCPD cleavage reaction was directly injected onto a C-8 reverse phase column and peptides eluted in two peaks at 10.56 and 10.88 min. The samples were then analyzed by LTQ-FT and the mass of the proteins in the two peaks was measured as 25 854.08 Da and 8320.86 Da (Figure 8). Calculation of the predicted masses using PROTPARAM for all potential N-terminal and C-terminal rCPD cleavage products indicated that the observed mw would correspond to a cleavage between Leu3428 and Ala3429 resulting in predicted fragments of 25854.4 Da and 8320.8 Da (Figure 8). This cleavage site falls within the region between residues 3411–3441 identified by deletion analysis using the transfection assay.
Figure 8.
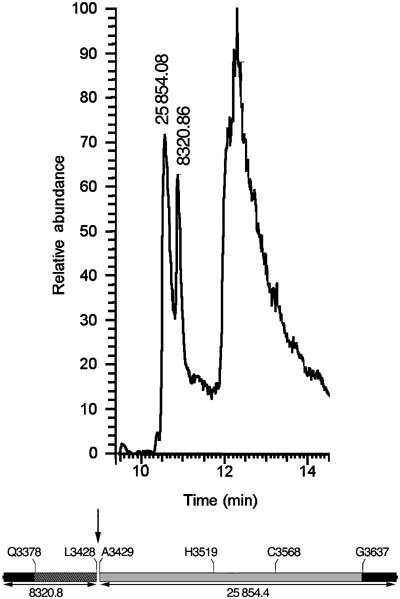
rCPD is cleaved between L3428 and A3429. Processed rCPD eluted from C8 reverse phase column in two peaks at 10.56 and 10.88 min. The third peak at 12.31 min consists of poorly resolved materials such a polymers, small molecules, and peptides. The mass of the proteins contained within the peptide fragment peaks was determined by LTQ-FT analysis and the results are shown above the peak. Schematic representation of rCPD shows the determined cleavage site as well as the computer-predicted mass of the two fragments.
A C3568S mutation in the RTX holotoxin is defective for actin crosslinking
In order to investigate whether autocleavage of the RTX toxin was important to overall function of the toxin, a point mutation of the catalytic Cys codon was introduced onto the bacterial chromosome by double homologous recombination using the sacB-lethality counterselection method (Metcalf et al, 1996). The resulting mutant strain, KSV10, carried a Ser residue in place of Cys3568 in the rtxA gene. To assess the effect of the C3568S point mutation on RTX toxin function, actin crosslinking activity was measured. As expected, the actin from cells exposed to the V. cholerae strain KFV119 expressing the wild-type RTX toxin appeared as a ladder of covalently crosslinked actin at 90 min with the actin progressing to only higher multimers at 180 min, while exposure to the rtxA null isogenic V. cholerae strain KFV92 did not generate crosslinked forms of actin (Figure 9). By contrast, cells incubated with KSV10 expressing the C3568S mutant RTX protein had a defect in actin crosslinking activity. As shown in Figure 9, crosslinked dimer was barely detectable after 90 min and the normal actin laddering pattern usually observed after exposure to the RTX toxin was not generated even after 180 min. It is unlikely that this defect was due to a disruption of the actin crosslinking activity of the toxin since the ACD is a distinct domain that can crosslink actin independent of the remainder of the toxin when expressed as a transgene (Sheahan et al, 2004). Thus, these results support a model wherein this defect in the actin crosslinking activity of the RTX toxin is due to the inability of the toxin to access substrate in the absence of autocleavage. Similarly, inactivation of Rho by the newly discovered Rho-inactivation domain (RID) of RTX was inhibited by mutation of Cys3568 indicating that this domain also must be released by autoprocessing to access its substrate (Sheahan and Satchell, 2007 and data not shown). Overall we conclude that autocleavage of the RTX toxin plays an important role in the delivery of activity domains as part of the mechanism of action of this toxin.
Figure 9.
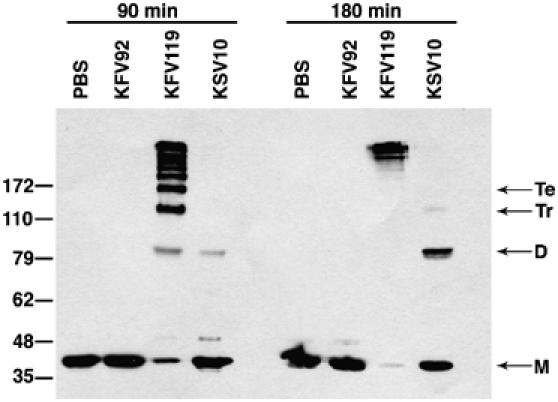
Cys3568 is important for RTX toxin actin crosslinking activity. COS-7 cells were incubated with PBS or V. cholerae strains with an intact rtxA gene (KFV119), a null mutation in rtxA (KFV92), or an rtxA gene with a C3568S point mutation (KSV10). Cells were harvested after 90 and 180 min of incubation, and actin crosslinking was measured by Western blotting with an anti-actin antibody. Lines at right mark monomer (M), dimer (D), trimer (Tr), and tetramer (Te) forms of actin.
Discussion
Previously, we proposed a model of V. cholerae RTX toxin entry wherein the toxin self-inserts into the host cell plasma membrane and translocates the ACD to the cytosol similar to translocation of Bordetella pertussis AC. Once inside the cell, the ACD would either remain anchored to the membrane or undergo proteolytic processing to release the ACD into the cytosol (Sheahan et al, 2004). In this study, we identified a domain within the RTX toxin that is conserved among many large secreted bacterial proteins. Characterization of this domain both in vivo and in vitro revealed that it is a cysteine protease responsible for autoprocessing of the V. cholerae RTX between L3428 and A3429. This processing event was essential to the function of this toxin as shown by a dramatic decrease in the efficiency of actin crosslinking when the catalytic cysteine was mutated. Therefore, we put forward a new model for V. cholerae toxin translocation wherein N- and C-terminal repeat regions function to translocate internal activity domains across the eukaryotic cell membrane (Figure 10B). The translocated regions would necessarily include the ACD, RID, and the CPD, which would then cleave the toxin between L3428 and A3429. Since even after processing, the ACD, RID, and other putative activity domains would be contained within a >350 kDa protein, it seems reasonable that the CPD cleaves the RTX toxin at multiple sites releasing activity domains to the cytosol.
Figure 10.
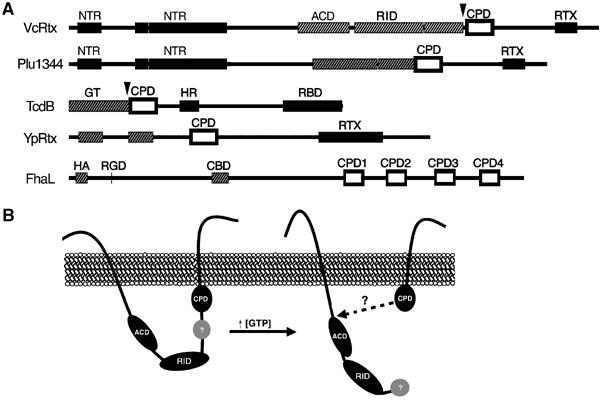
Autoprocessing by CPD is a common mechanism for processing large bacterial toxins. (A) Schematic representation of CPD location within large bacterial proteins. CPD is marked as an outlined open box. Black boxes represent regions predicted to be critical for toxin entry (NTR, N-terminal repeats; RTX, RTX repeats; HR, hydrophobic region; RBD, receptor binding domain). Grey hatched boxes represent known or predicted functional regions (ACD, actin crosslinking domain; GT, glucosyltransferase; HA, hemagglutinin; CBD, carbohydrate binding domain; RGD, integrin binding; RID, Rho-inactivation domain). Arrowheads mark cleavage sites determined experimentally. (B) Proposed model of RTX toxin processing.
Our studies indicate that cleavage of rCPD was stimulated by a cytosolic host cell factor. Additional experiments established that binding of GTP or the non-hydrolyzable GTP analog GTPγS was sufficient to stimulate autoprocessing of rCPD at physiological concentrations. The binding of GTPγS to rCPD C-S showed classic binding and competitive inhibition kinetics indicating a specific protein-ligand interaction. Therefore, processing of V. cholerae RTX toxin would be stimulated after transitioning to the high GTP intracellular environment of the eukaryotic cytosol (Figure 10B). Premature cleavage of the toxin in the bacterial cytoplasm would be prevented since the V. cholerae RTX toxin would remain unfolded until after secretion by the type I secretion system (Boardman and Satchell, 2004).
Unfortunately, further characterization of RTX holotoxin processing after translocation is hindered due to a lack of purified toxin for in vitro study. However, processing in vivo and in vitro has been demonstrated for a different bacterial toxin that we identified to carry a CPD. C. difficile TcdB is known to be proteolytically processed at a Leu-Gly pair to the N-terminal side of the TcdB CPD (Figure 1) after translocation across the endosomal membrane. Cleavage then releases the N-terminal glucosyltransferase domain to the cytosol where it can UDP-glucosylate the target Rho (Pfeifer et al, 2003; Rupnik et al, 2005). Preliminary data from our lab indicates this cleavage can be initiated by addition of GTPγS and inhibited by NEM suggesting autoprocessing of the toxin by the TcdB CPD (our unpublished results). Consistent with this model, mutation of the putative catalytic His and Cys residues within the TcdB CPD decreased cell rounding activity of TcdB by 90–99% (Barroso et al, 1994). Thus it seems reasonable to suggest that TcdB also undergoes GTP-stimulated autoprocessing after translocation and this processing is essential for function of the toxin. The CPDs in other clostridial toxins and putative Yersinia toxins could function in a similar manner. Indeed, in 16 identified large bacterial proteins, the CPD is always located between putative translocation regions and catalytic or adherence regions, and are thus appropriately positioned to release activity domains from large bacterial toxins after translocation (Figure 10A). Therefore, GTP-stimulated autoprocessing may be a newly recognized common mechanism by which large bacterial toxins deliver catalytic domains to the cytosol of host eukaryotic cells.
In addition to the CPD-mediated cleavage representing a new mechanism for bacterial toxin delivery, the CPDs are apparently a novel cysteine protease family. The characterization of the CPD as a cysteine protease was surprising since alignment programs, including a search of the MEROPS peptidase database (Rawlings et al, 2006), did not reveal sequence similarity to known cysteine proteases. The closest relative of the CPD may be NS2 from Hepatitis C virus, a cysteine protease that remains unclassified in the MEROPS database (Rawlings and Barrett, 1997). Both CPD and NS2 catalyze autocleavage of their respective proteins. Indeed, NS2 also cleaves between a Leu-Ala pair although the site of cleavage is to the C-terminal side of NS2 (Grakoui et al, 1993).
Comparison of CPD and NS2 revealed that the orientation and particularly the spacing of the catalytic residues are very similar suggesting a similar mechanism of catalysis. Although there are similarities between these two proteins, a mutation of Glu972 between the catalytic His952 and Cys993 within NS2 had a defect in processing of the HCV polyprotein suggesting that this residue may be required to form a catalytic triad (Grakoui et al, 1993; Hijikata et al, 1993; Wu et al, 1998). Our studies determined that His3519 and Cys3568 comprise a catalytic dyad of the CPD but mutation of the putative third catalytic Glu3543 residue of RTX did not disrupt CPD activity.
Additionally, activation of CPD was achieved by addition of GTP. By contrast, the NS2/3 cleavage required ATP for in trans processing although ATP hydrolysis is more likely needed for activity of host cofactor Hsp90 and not for stimulation of protease activity (Waxman et al, 2001). Hsp90 is not likely involved in autoprocessing of the CPD, since addition of Hsp90 inhibitor geldanamycin had no effect on in vitro CPD processing stimulated by cytosol (data not shown) and previous experiments have also shown that geldanamycin did not inhibit TcdB activity (Schirmer and Aktories, 2004). In total, the inability to assign CPD to any known family and the requirement of GTP binding for CPD activity indicates that this is a novel cysteine protease that could share a mechanism of catalysis with the unclassified NS2 cysteine proteases.
Overall, in this study, we have identified a mechanism for the processing of the V. cholerae RTX toxin by a novel cysteine protease that is conserved in other large bacterial proteins. Hence, our proposed model for delivery of activity domains by this bacterial toxin could contribute to our understanding of the mechanism of translocation of other virulence factors. Future studies investigating these other organisms will determine if this is indeed a common mechanism and will provide a greater understanding of the contribution of the cysteine protease autocleavage in bacterial pathogenesis.
Materials and methods
Cell lines, bacterial strains, and reagents
COS-7 cells were cultured at 37°C with 5% CO2 in DMEM containing 50 U/ml penicillin, 50 μg/ml streptomycin, and 10% FBS (Invitrogen, Carlsbad, CA). V. cholerae strains KFV119, KFV92 (Sheahan et al, 2004), and KSV10 (see below) were grown at 30°C in Luria broth (LB) containing 100 μg/ml streptomycin. All restriction enzymes were obtained from New England Biolabs (Beverly, MA). All chemicals were purchased from Sigma (St Louis, MO) except NEM, which was from Pierce (Rockford, IL), Calpeptin, E64, and Pepstatin were from Calbiochem (La Jolla, CA), IPTG was from Denville Scientific (Metuchen, NJ), and mant-GTP and BODIPY-GTPγS were from Invitrogen (Carlsbad, CA). Oligonucleotides listed in Supplementary Table 2 were obtained from either Sigma or IDT (Coralville, IA).
Cloning of the CPD
Primers CPDc-BglII and CPD-EcoRI were used to amplify the DNA corresponding to amino acids 3376–3637 of RTX with flanking BglII and EcoRI from N16961 genomic DNA. To construct pxlCPDc and pCPDn, PCR products generated using primers xlCPDc-BglII and CPDn-BglII in the forward direction and CPD-EcoRI in the reverse direction were cloned into the Invitrogen pCR-BluntII-TOPO vector, then subcloned into either pEGFP-N3 or pEGFP-C3 (Clontech, Mountain View, CA) as BglII-EcoRI fragments to create in-frame fusions to egfp.
Point mutations within pCPDc were engineered using the Quickchange Mutagenesis kit (Stratagene, La Jolla, CA). Cys3568 was mutated to Ser using the primers C3568S-sense and C3568S-antisense incorporating a BamHI site to aid in screening. The restriction enzyme ApaI was used to excise a fragment from pCPDc C-S containing the C3568S point mutation for religation into similarly digested pxlCPDc and pCPDn to construct pxlCPDc C-S and pCPDn C-S. The His3519A and Glu3543A mutations were generated with primers H3519A-sense, H3519A-antisense, E3543A-sense, and E2543A-antisense. Plasmid DNA prepared using the Qiaprep Spin Miniprep kit (Qiagen, Valencia, CA) was sequenced to confirm gene sequence and egfp fusions.
Construction of a point mutation on the rtxA gene of V. cholerae
A point mutation within the rtxA gene on the V. cholerae chromosome was created by double homologous recombination using the sacB-lethality counterselection method as previously described (Fullner and Mekalanos, 1999). Briefly, a 2097 bp fragment corresponding to RTX codons for amino acids 3201–3899 was amplified from N16961 genomic DNA and cloned into pCR-BluntII-TOPO. The Cys3568S point mutation was generated as described above. After mutagenesis the fragment was moved into the sacB-counterselection vector pWM91 (Metcalf et al, 1996). The resulting plasmid was transformed into E. coli SM10λpir and mated into the recipient strains KFV119. Colonies containing the cointegrated plasmid were subjected to counterselection on sucrose. The colonies obtained after selection were screened by digestion of PCR products with BamHI to detect the incorporation of the point mutation and by DNA sequencing.
Recombinant CPD cloning, expression, and purification
Primers CPD-LICF and CPD-LICR were used to amplify the CPD from the V. cholerae strains KFV119 and KSV10 and then ligation independent cloning was used to construct overexpression plasmids pHisCPD and pHisCPDC-S, respectively, according to the manufacturer's protocol for pET30-XA-LIC cloning (Novagen, Madison, WI) using modified vector pMCSG7 (Stols et al, 2002).
CPD and CPD C-S were expressed in E. coli strain BL21(λDE3). Overnight cultures were diluted 1:500 into LB containing 100 μg/ml ampicillin and grown at 37°C to OD600=0.6. Protein expression was induced with 0.2 mM IPTG for 2 h. Bacterial pellets were resuspended in 20 mM Tris, 500 mM NaCl, and 5 mM imidazole pH 8.0, 1 mg/ml lysozyme and incubated on ice for 30 min. Triton X-100 (1%) was added and lysates were sonicated with a Bronson Digital Sonifier 450 at 40% amplitude for 2 min. Insoluble debris was pelleted at 27 000 g and the 6 × His tagged proteins were purified by affinity chromatography using a HisTrap HP column on the ÄKTA purifier (GE Healthcare, Piscataway, NJ). The column was washed with 20 mM Tris, 500 mM NaCl, and 60 mM imidazole pH 8.0 and eluted with 250 mM imidazole. Eluted proteins were dialyzed into 20 mM Tris, 500 mM NaCl pH 7.5 and then adjusted to 10% glycerol and stored at −80°C in aliquots. Protein purity was assessed on Coomassie blue R250-stained SDS–PAGE gel and concentration was determined by using a BCA acid protein assay kit (Pierce, Rockford, IL) or by A280 using extinction coefficient 40 ,450 deduced using the PROTPARAM software at http://ca.expasy.org/tools/protparam.html (Gasteiger et al, 2005). For binding studies, proteins were diluted with dialysis buffer to 5% glycerol and concentrated to 14–22 mg/ml by centrifugation using a Millipore Microcon YM-10 filter device.
Transient transfection
COS-7 cells were plated at approximately 50% confluency in either six-well dishes or on coverslips 24 h before transfection. Cells were transfected using the Fugene 6 reagent (Roche Applied Sciences, Indianapolis, IN) mixed with plasmid DNA at a 3:1 reagent:DNA ratio in DMEM. This mixture was preincubated for 45 min before addition to cultured cells followed by incubation at 37°C with 5% CO2 for 24 h.
Microscopy
Cells grown on coverslips were washed with phosphate-buffered saline (PBS), fixed with 4% paraformaldehyde and stained with 0.5 μg/ml Hoechst 33342 24 h after transfection. The coverslips were mounted using Anti-Fade mounting media (Molecular Probes, Eugene, OR). Transfected cells were observed for expression of EGFP at × 630 magnification under fluorescence microscopy at 550–575 and 440–470 nm to visualize nuclei. Images were captured using an inverted Leica DMIRE2 microscope with a C4742-95-12ERG digital charge-coupled device (CCD) camera (Hamamatsu Photonics, Tokyo) in conjunction with the OPENLAB software (Improvision, Coventry, UK).
In vitro cleavage assay
Cell lysate was prepared from approximately 107 COS-7 cells that were washed with PBS, collected and suspended in 1 ml of homogenization buffer (250 mM sucrose, 3 mM imidazole pH 7.5). The cells were lysed by three pulses of 10 s using a Sonicel disrupter and then were centrifuged at 2000 g for 5 min at 4°C. For subcellular fractionation, the lysate was fractionated by centrifugation at 100 000 g for 30 min at 4°C. The supernatant (cytosolic fraction) was removed and the insoluble pellet (membrane fraction) was resuspended in an equivalent volume of homogenization buffer containing 1% Triton X-100. The in vitro cleavage assay was performed by first diluting 2 μg rCPD or rCPD C-S in a buffer of 20 mM Tris, 60 mM NaCl, 250 mM sucrose, 3 mM imidazole, pH 7.5. Reactions were initiated by addition of cell lysate or chemical compounds at concentrations indicated and incubated at 37°C for the time indicated. Samples were heated at 95°C in SDS–PAGE loading buffer for 5 min and proteins were separated by SDS–PAGE (15% gel) and staining with Coomassie blue R250.
Western blotting
Transiently transfected cells were collected 24 h after transfection, whereas cells exposed to KFV92, KFV119, and KSV10 were collected 90 and 180 min after addition of bacteria. All cells were washed with PBS, collected by scraping, centrifuged at 4500 g, and resuspended in SDS–PAGE sample buffer. Proteins in boiled cell lysates were separated on SDS–PAGE and transferred to Hybond-C nitrocellulose (GE Healthcare, Piscataway, NJ). EGFP expression was detected using a monoclonal anti-GFP antibody at 1:1000 dilution (Santa Cruz Biotechnology, Santa Cruz, CA) followed by the anti-mouse IgG HRP secondary antibody at 1:5000 dilution (Sigma, St Louis, MO). Actin crosslinking was detected by Western blotting with a polyclonal anti-actin antibody at 1:1000 dilution (Sigma, St Louis, MO) and a 1:5000 dilution of anti-rabbit IgG HRP (Jackson Immuno Research, West Grove, PA).
Bioinformatics
Proteins with CPDs were identified by searching GenBank and the nonredundant protein database through the National Center for Biotechnology Information (NCBI) (http://www.ncbi.nlm.nih.gov) and San Diego Supercomputing Center (SDSC) (http://www.sdsc.edu) using PSI-BLAST (Altschul et al, 1997) and MEME/MAST (Bailey and Elkan, 1994; Bailey and Gribskov, 1998), respectively. Protein sequences identical to YPTB3219 and FhaL from other related species were excluded from analysis. V. cholerae RTX CPD was further searched against the MEROPS peptidase database (http://merops.sanger.ac.uk) (Rawlings et al, 2006). CLUSTALW alignments were prepared using the identity weight matrix followed by preparation of PHYLIP rooted phylogenetic tree in DRAWGRAM and alignment in TeXshade using the Biology WorkBench V. 3.2 program at SDSC (Felsenstein, 1989; Thompson et al, 1994; Beitz, 2000). Primary amino acids sequences used were from NCBI GenBank except Xenorhabdus sequences were acquired by FTP from the Donald Danforth Plant Science Center. Accession numbers are listed in Supplementary Table 1.
GTP binding assay
Fluorescence binding assays were performed with rCPD and rCPD C-S in 20 mM Tris, 150 mM NaCl, 1.5% glycerol, 0.2% octaethylene glycol monododecyl ether, pH 7.5 with 0.5 μM mant-GTP or 0.25 μM BODIPY FL-GTPγS. The assay mixtures were incubated in 96-well dishes at 37°C for 30 min. For BODIPY experiments, fluorescence emission for each sample was collected every 5 nm from 502 to 602 nm after excitation at λ=488 nm using the Safire 2 spectrofluorometer (Tecan, Durham, NC). For mant experiments, excitation was at λ=350 nm and peak emission at 440 nm was measured. Results are reported as fluorescence intensity (I) or fractional intensity change (FIC) calculated as (Iobs−Ifree)/Ifree. The Kd was determined from a plot of FIC vs total enzyme concentration using the one site binding equation Y=Bmax × X/Kd+X using the Graphpad Prism 4 Software package. The Ki was determined using the same software by plotting FIC vs log[GTPγS] using the one site competition equation Y=Bottom+(Top−Bottom)/(1+10^(X−LogEC50).
Fourier transform mass spectrometry
FT-MS was performed at Chicago Biomedical Consortium Proteomic Facility at the University of Illinois at Chicago. 100 pmol rCPD cleaved by addition of nucleotide for 24 h was separated on an Agilent Zorbax C-8 (300A pore size, 3.5 μm particle size) 2.1 × 50 mm id column run at a flow rate of 200 μl/min in 90% 0.1% formic acid:water/10% acetonitrile to 50:50 in 10 min. Eluted samples were run on a Thermo LTQ-FT mass spectrometer using FTMS+p ESI Full MS scanning mode, mass range 450–2000, FT resolution 100 000, scan speed approximately 0.9 s/scan. The mw of eluted peptides was determined by deconvolving multiply charge peaks to the proteins effective mass using the charge state determination afforded by the instrument high resolution capability. The measured mass was compared to the predicted mw of possible peptide cleavage fragments determined from the primary sequence of rCPD using PROTPARAM at http://ca.expasy.org/tools/protparam.html (Gasteiger et al, 2005).
Supplementary Material
Supplementary Figure 1
Supplementary Figure 2
Supplementary Legends
Supplementary Figure 3
Supplementary Table 1
Supplementary Table 2
Supplementary Figure 4
Acknowledgments
We thank H Howell and A Bonebrake for technical assistance, D Freymann for assistance with the nucleotide-binding studies, and A Schilling for technical advice on FT-MS. Proteomics and informatics services were provided by the CBC-UIC Research Resources Center Proteomics and Informatics Services Facility, which was established by a grant from The Searle Funds at the Chicago Community Trust to the Chicago Biomedical Consortium. This work was supported by PHS grant AI051490, a subcontract from the GLRCE grant U54AI057153, and a Burroughs Wellcome Fund Investigators in Pathogenesis of Infectious Disease Award (to KJFS). KLS was supported by predoctoral NRSA Fellowship T32-AI07476.
References
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson BM, Vasini EC (1970) Nonpolar effects in reactions of the sulfhydryl group of papain. Biochemistry 9: 3348–3352 [DOI] [PubMed] [Google Scholar]
- Bailey TL, Elkan C (1994) Fitting a mixture model by expectation maximization to discover motifs in bioploymers. In Proceedings of the Second International Conference on Intelligent Systems for Molecular Biology, pp 28–36. Menlo Park, CA: AAAI Press [PubMed]
- Bailey TL, Gribskov M (1998) Combining evidence using p-values: application to sequence homology searches. Bioinformatics 14: 48–54 [DOI] [PubMed] [Google Scholar]
- Barroso LA, Moncrief JS, Lyerly DM, Wilkins TD (1994) Mutagenesis of the Clostridium difficile toxin B gene and effect on cytotoxic activity. Microb Pathog 16: 297–303 [DOI] [PubMed] [Google Scholar]
- Beitz E (2000) TEXshade: shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics 16: 135–139 [DOI] [PubMed] [Google Scholar]
- Boardman BK, Satchell KJ (2004) Vibrio cholerae strains with mutations in an atypical Type I secretion apparatus accumulate RTX toxin intracellularly. J Bacteriol 186: 8137–8143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordero CL, Kudryashov DS, Reisler E Satchell KJ (2006) The actin cross-linking domain of the Vibrio cholerae RTX toxin directly catalyzes the covalent cross-linking of actin. J Biol Chem 281: 32366–32374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falnes PO, Sandvig K (2000) Penetration of protein toxins into cells. Curr Opin Cell Biol 12: 407–413 [DOI] [PubMed] [Google Scholar]
- Felsenstein J (1989) PHYLIP—Phylogeny Inference Package (v3.2). Cladistics 5: 164–166 [Google Scholar]
- Fullner KJ, Mekalanos JJ (1999) Genetic characterization of a new type IV-A pilus gene cluster found in both classical and El Tor biotypes of Vibrio cholerae. Infect Immun 67: 1393–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fullner KJ, Mekalanos JJ (2000) In vivo covalent cross-linking of cellular actin by the Vibrio cholerae RTX toxin. EMBO J 19: 5315–5323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasteiger E, Hoogland C, Gattiker A, Duvaud S, Wilkins MR, Appel RD, Bairoch A (2005) Protein identification and analysis tools on the ExPASy server. In The Proteomics Protocols Handbook, Walker JM (ed) pp 571–607. Totowa, NJ: Humana Press [Google Scholar]
- Grakoui A, McCourt DW, Wychowski C, Feinstone SM, Rice CM (1993) A second hepatitis C virus-encoded proteinase. Proc Natl Acad Sci USA 90: 10583–10587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada K, Tamai M, Yamagishi M, Ohmura S, Sawada J, Tanaka I (1978) Isolation and chracterization of E-64, a new thiol protease inhibitor. Agric Biol Chem 42: 523–528 [Google Scholar]
- Henderson IR, Navarro-Garcia F, Desvaux M, Fernandez RC, Ala'Aldeen D (2004) Type V protein secretion pathway: the autotransporter story. Microbiol Mol Biol Rev 68: 692–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hijikata M, Mizushima H, Akagi T, Mori S, Kakiuchi N, Kato N, Tanaka T, Kimura K, Shimotohno K (1993) Two distinct proteinase activities required for the processing of a putative nonstructural precursor protein of hepatitis C virus. J Virol 67: 4665–4675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimple RJ, De Vries L, Tronchere H, Behe CI, Morris RA, Garquhar MG, Siderovski DP (2001) RGS12 and RGS14 GoLoco motifs are Gαi interaction sites with guanine nucleotide dissociation inhibitor activity. J Biol Chem 276: 29275–29281 [DOI] [PubMed] [Google Scholar]
- Lin W, Fullner KJ, Clayton R, Sexton JA, Rogers MB, Calia KE, Calderwood SB, Fraser C, Mekalanos JJ (1999) Identification of a Vibrio cholerae RTX toxin gene cluster that is tightly linked to the cholera toxin prophage. Proc Natl Acad Sci USA 96: 1071–1076 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metcalf WW, Jiang W, Daniels LL, Kim SK, Haldimann A, Wanner BL (1996) Conditionally replicative and conjugative plasmids carrying lacZα for cloning, mutagenesis, and allele replacement in bacteria. Plasmid 35: 1–13 [DOI] [PubMed] [Google Scholar]
- Pfeifer G, Schirmer J, Leemhuis J, Busch C, Meyer DK, Aktories K, Barth H (2003) Cellular uptake of Clostridium difficile toxin B. Translocation of the N-terminal catalytic domain into the cytosol of eukaryotic cells. J Biol Chem 278: 44535–44541 [DOI] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ (1994) Families of cysteine peptidases. Methods Enzymol 244: 461–486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings ND, Barrett AJ (1997) Classification of peptidases by comparisons of primary and secondary structures. In Proteolysis in Cell Functions, Hopsuhavu VK, Jarvinen M, Kirschke H (eds), pp 13–21. Amsterdam: IOS Press [Google Scholar]
- Rawlings ND, Morton FR, Barrett AJ (2006) MEROPS: the peptidase database. Nucleic Acids Res 34: D270–272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogel A, Hanski E (1992) Distinct steps in the penetration of adenylate cyclase toxin of Bordetella pertussis into sheep erythrocytes. Translocation of the toxin across the membrane. J Biol Chem 267: 22599–22605 [PubMed] [Google Scholar]
- Rupnik M, Pabst S, von Eichel-Streiber C, Urlaub H, Soling HD (2005) Characterization of the cleavage site and function of resulting cleavage fragments after limited proteolysis of Clostridium difficile toxin B (TcdB) by host cells. Microbiology 151: 199–208 [DOI] [PubMed] [Google Scholar]
- Sasaki T, Kishi M, Saito M, Tanaka T, Higuchi N, Kominami E, Katunuma N, Murachi T (1990) Inhibitory effect of di- and tripeptidyl aldehydes on calpains and cathepsins. J Enzyme Inhib 3: 195–201 [DOI] [PubMed] [Google Scholar]
- Schirmer J, Aktories K (2004) Large clostridial cytotoxins: cellular biology of Rho/Ras-glucosylating toxins. Biochim Biophys Acta 1673: 66–74 [DOI] [PubMed] [Google Scholar]
- Sheahan KL, Cordero CL, Satchell KJ (2004) Identification of a domain within the multifunctional Vibrio cholerae RTX toxin that covalently cross-links actin. Proc Natl Acad Sci USA 101: 9798–9803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheahan KL, Satchell KJ (2007) Inactivation of Rho family GTPases by the RTX toxin of Vibrio cholerae. Cell Microbiol 9: 1324–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stols L, Gu M, Dieckman L, Raffen R, Collart FR, Donnelly MI (2002) A new vector for high-throughput, ligation-independent cloning encoding a tobacco etch virus protease cleavage site. Protein Expr Purif 25: 8–15 [DOI] [PubMed] [Google Scholar]
- Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22: 4673–4680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waxman L, Whitney M, Pollok BA, Kuo LC, Darke PL (2001) Host cell factor requirement for hepatitis C virus enzyme maturation. Proc Natl Acad Sci USA 98: 13931–13935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whitaker JR, Perez-Villase nor J (1968) Chemical modification of papain. I. Reaction with the chloromethyl ketones of phenylalanine and lysine and with phenylmethyl-sulfonyl fluoride. Arch Biochem Biophys 124: 70–78 [DOI] [PubMed] [Google Scholar]
- Wu Z, Yao N, Le HV, Weber PC (1998) Mechanism of autoproteolysis at the NS2-NS3 junction of the hepatitis C virus polyprotein. Trends Biochem Sci 23: 92–94 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1
Supplementary Figure 2
Supplementary Legends
Supplementary Figure 3
Supplementary Table 1
Supplementary Table 2
Supplementary Figure 4