

Abstract
Genome-wide expression microarray studies have revealed that the biological and clinical heterogeneity of breast cancer can be partly explained by information embedded within a complex but ordered transcriptional architecture. Comprising this architecture are gene expression networks, or signatures, reflecting biochemical and behavioral properties of tumors that might be harnessed to improve disease subtyping, patient prognosis and prediction of therapeutic response. Emerging 'hypothesis-driven' strategies that incorporate knowledge of pathways and other biological phenomena in the signature discovery process are linking prognosis and therapy prediction with transcriptional readouts of tumorigenic mechanisms that better inform therapeutic options.
Introduction
DNA microarrays are tools for assessing the functional dynamics of genes and genomes in a highly parallel fashion. Historically defined as ordered collections of DNA probes for the specific detection of complementary DNA targets, microarrays enable genome-wide surveys of the relative abundance of mRNA transcripts, the high-resolution mapping of genomic copy number alterations, the identification of binding sites of nucleic acid-binding proteins, and the comprehensive analysis of single-nucleotide polymorphisms (SNPs). Although microarray technology and its applications have evolved considerably over the years to meet a growing range of genomic challenges [1], the classical format for microarrays in interrogating the transcriptome (that is, expression microarrays) has been a key technology for discovery in functional and medical genomics.
Since the mid-1990s, expression microarrays have been extensively applied to the study of cancer, and no cancer type has seen as much genomic attention as breast cancer. The most prolific area of breast cancer genomics has been the elucidation and interpretation of gene expression patterns that underlie biological and clinical properties of tumors. In a seminal study that analyzed expression profiles of primary breast tumors, Perou and colleagues [2] showed that the vast and complex transcriptional data generated by microarrays contained discernible patterns of gene expression that related to tumor biology and behavior. Through hierarchical cluster analysis, numerous 'gene clusters' could be recognized as biologically distinct networks reflecting the phenotypic wiring of individual tumors. These 'molecular portraits' revealed information on multiple biological tiers – from broad tumorigenic properties to discrete biochemical pathways to intra-tumor tissue heterogeneity – and led to the discovery of an 'intrinsic' gene subset that could distinguish between multiple new cancer subtypes on the basis of fundamental tumor properties associated with cell-type origin. These subtypes, termed Luminal A/ER+, Luminal B/ER+, Normal Breast-like, ERBB2+, and Basal-like (that is, the Perou–Sorlie subtypes), were subsequently shown to be stable and reproducible classes observable in different patient populations, and correlated significantly with tumor recurrence and patient survival [3,4].
Together, these studies provided early evidence that the transcriptional circuitry of breast cancer, as revealed by microarrays, could not only provide novel insights into the biology of cancer but could also accurately identify certain previously discernible clinical phenotypes (for example estrogen receptor (ER) status, HER2/neu expression, and proliferation rate) and robustly define new molecularly informed classifications that delineate novel disease entities associated with patient outcomes.
More recently, new investigative techniques have begun to refine our understanding of the breast cancer onco-transcriptome and how it relates to tumor biology and behavior. From this vantage point, the intersections between pathological mechanisms and clinical endpoints are being explored with new vigor. Traditional microarray methods for uncovering prognostic expression signatures, based primarily on empirical associations not requiring plausible biological relevance of the markers used, are now sharing the stage with mechanistically motivated strategies driven by knowledge of oncogenic pathways and processes. More commonly, experimental approaches show that patho-biological simulations performed in vitro reveal transcriptional configurations predictive of tumor biology in vivo. Together, these functional genomics strategies are changing the scientific process of breast cancer biomarker discovery, towards one that incorporates mechanistic knowledge.
Patient prognosis
The work by Perou, Sorlie and colleagues demonstrated the power of expression genomics to stratify patients clinically on the basis of the complex molecular configurations of their tumors. Questions remained, however, about the practical utility of the Perou–Sorlie subtypes in prognosis, and whether other genomic strategies might provide greater prognostic resolution in certain clinically challenging patient subpopulations.
Van 't Veer and colleagues [5] and Wang and colleagues [6] both focused on the identification of gene expression 'signatures' (rather than tumor subtypes) that could predict outcome in patients with early-stage breast cancer (N0, T1/T2), the majority of whom would unnecessarily receive systemic adjuvant therapy according to conventional guidelines. Working with primary tumor material from patients who did not receive adjuvant systemic therapy, each group identified and validated a prognostic signature capable of predicting 5-year disease recurrence [5-7]. The signature by van 't Veer and colleagues (otherwise known as the Amsterdam signature) consisted of 70 genes, whereas the predictor of Wang and colleagues (otherwise known as the Rotterdam signature) was composed of 76 genes: 60 for prognosis of patients with ER-positive tumors, and 16 for prognosis of those with ER-negative disease. In each case, the prognostic power of the signature was independent of, and even superior to, conventional risk factors (such as tumor size, histologic grade, and patient age), and, in comparison with the St Gallen's and National Institutes of Health consensus guidelines for establishing patient eligibility for adjuvant chemotherapy, the signatures were better at predicting which patients should not receive adjuvant therapy (and similar at predicting who should receive adjuvant therapy), potentially sparing a significant fraction of 'cured' patients from overtreatment.
Both the Amsterdam and Rotterdam signatures have now been further validated in large multicenter investigations that confirm the prognostic advantages of the expression signatures over conventional guidelines for selecting patients for adjuvant systemic therapy [8,9]. The Amsterdam signature has now been marketed for clinical use through the Amsterdam-based diagnostics company, Agendia, founded in 2003 by the Netherlands Cancer Institute (NKI).
An interesting aspect of these two studies is that although the two gene lists were derived from the same basic scientific question and using similar patient cohorts, only three genes were found in common to both signatures [6]. Various technical differences have been proposed to account for this discrepancy, but others have noted that if one looks beyond the genes to the pathways they represent, multiple pathways can be found in common between the signatures, indicating that the signatures and their predictive powers may converge on the same underlying biology [6]. Although the endpoints of these two investigations were clinical in nature, a compelling biological interpretation of the results has emerged: that early primary tumors may already possess the hardwiring necessary for future metastasis, thus countering the view that metastatic potential is an acquired trait that develops later in the course of tumorigenesis and in a rare subpopulation of cells.
Tailored treatment
If early-stage primary breast tumors are already hardwired for metastatic potential, might their propensity for therapeutic response also be molecularly ingrained, and measurable via a transcriptional readout? Valuable evidence supporting this hypothesis was first demonstrated in the context of diffuse large-B-cell lymphoma (DLBCL). Alizadeh and colleagues [10] used expression microarrays to elucidate transcriptional patterns that could dichotomize DLBCL samples into at least two distinct classes reflecting different aspects of normal B-cell physiology. One class showed expression of genes commonly induced in germinal-center B cells (the GCB-like class), whereas the other was characterized by expression of genes associated with mitogenic stimulation of blood B cells (termed the activated B-cell (ABC)-like class). Importantly, these two classes showed distinct clinical behaviors after chemotherapy; patients with GCB-like disease had twice the 5-year survival rate of those with ABC-like disease [10-12].
Because NF-κB activity is critical for the development and survival of normal B cells and is known to be important in several cancer types, Davis and colleagues [13] investigated the possibility that the NF-κB pathway might be differentially activated between GCB-like and ABC-like forms of DLBCL. Indeed, the authors identified, in the microarray data, a handful of NF-κB target genes that were significantly differentially expressed between the two groups, with higher expression in the ABC-like class. Using cell lines representative of the two classes, the authors showed that constitutive NF-κB activity was required for survival of the ABC-like class, but not the GCB-like class. That NF-κB can protect cells from death induced by certain chemo-therapeutics may partly explain the poor survival outcomes observed in the ABC-like class. Moreover, the results suggest that patients of the poor-outcome ABC-like class, as defined by gene expression profiling, may derive benefit from treatment with NF-κB inhibitors that are known to work in synergy with chemotherapy to enhance cell death. This hypothesis is currently under investigation in a phase II clinical trial at the National Cancer Institute, Rockville, MD, USA.
In the context of breast cancer, several expression profiling studies have provided preliminary evidence for the existence of therapy-predictive signatures. These studies have relied primarily on empirical approaches that assess, either directly or indirectly, tumor sensitivity to drugs. The direct approach is prospective, involving expression analysis of preoperative tumor biopsies taken in the neoadjuvant setting, and subsequent 'supervised' class prediction to determine whether a multigene predictor can distinguish tumors that will show complete pathologic response (pCR) from those that will exhibit residual or progressive disease. So far, this approach has been used in several contexts to elucidate therapy-predictive signatures for treatments such as docetaxel [14,15], T/FAC (paclitaxel, 5-fluorouracil, adriamycin, and cyclophosphamide) [16,17], AC (adriamycin and cyclophosphamide) [18], and AT (adriamycin and paclitaxel) [19]. Although each study has reported the discovery of predictive genes with some promising classification accuracy, in most cases little or no independent validation has yet been reported. In the largest and most validated of these studies, Hess and colleagues [17] discovered a 30-probe predictor of pCR after T/FAC therapy that, in validation, showed high sensitivity for identifying pCR cases (92%) and a high negative predictive value for predicting cases that exhibited residual disease (96%). In comparison with the predictive power of conventional variables, this result could be viewed as a marginal, but valuable, prognostic improvement, but it will require further validation in larger cohorts to demonstrate significant clinical value.
A more indirect approach to identifying therapy-predictive genes involves the retrospective analysis of historical samples in which patient outcome data can be used as an approximate measure of therapeutic response. An advantage of this approach is that it uses a long-term measurement of therapeutic efficacy, such as whether or not the cancer returns over time, rather than a short-term pathologic response that does not always correlate with future outcome. However, a drawback is that the line between therapy prediction and patient prognosis is blurred. Whereas a relapsing cancer can be viewed as a therapy failure, one that does not return may have been successfully treated at surgery and may thus have no bearing on the effectiveness of adjuvant therapy. Nevertheless, prediction of therapy failure can indicate the need for a more aggressive treatment strategy. Studies pursuing this line of investigation have described a 2-gene test [20] and a 21-gene test [21] both for tamoxifen failure, that, when validated, outperformed conventional predictors of recurrence. Not found in these studies, however, was direct evidence that alternative therapies would provide benefit for these patients. In a follow-up to the latter study, Paik and colleagues [22] showed a significant interaction between the 21-gene test and combined tamoxifen and chemotherapy (cyclophosphamide, methotrexate and fluorouracil or methotrexate and fluorouracil), suggesting that women predicted to fail tamoxifen treatment could potentially benefit from additional chemotherapy.
Ultimately, prognostic signatures resulting from empirical methods that group tumors into biologically uncharacterized classes (such as 'responders' and 'nonresponders') may be performance limited. The molecular heterogeneity of breast cancer suggests that the biological programs driving tumor progression are both numerous and diverse, and these programs, operating independently or in aggregate, may dictate how a tumor or subgroup of tumors will progress clinically or will respond to certain drugs. The ability to define these circuitries biologically, parse them out at the transcriptional level, and assess their prognostic associations will allow the identification of tumor subtypes based on pathway activities that not only predict for tumor behaviors but also explain them.
Surrogate signatures
In breast cancer, several clinicopathological markers are frequently used alone or in combination to assess patient risk. For example, lymph node stage, tumor size, and histologic grade are important elements of the major prognostic indices, whereas ER status is widely regarded as the primary predictor of response to hormonal (antiestrogen) therapy. Microarray data sets from large studies of breast cancer have provided unique opportunities to investigate the relationships between gene expression patterns and these clinical/laboratory parameters. These studies have revealed several underlying signatures associated with the primary physiology of the tumor with important prognostic and predictive implications, and suggest that the sum of multiple gene-expression measurements may provide greater diagnostic precision than the biochemical or morphological marker on which they are based.
Perhaps the most apparent and widely observed of these expression signatures is the one that reflects ER status. Composed of hundreds of genes that include known direct and indirect targets of the ER, this signature is strongly correlated with clinical measurements of ER (for example by immunohistochemistry, ligand-binding assay, and enzyme immunoassay) and faithfully partitions tumors into ER-positive and ER-negative classes with reproducible accuracy [2,5,23]. This close link between the signature and ER status is further demonstrated by the observation that the relative levels of the ER signature genes are predictive of ER protein levels as measured by enzyme immunoassay in a panel of human breast tumors [24]. Even the expression of the ER gene itself (as measured by microarray) is highly correlated with ER status [2], leading some groups, for data analysis purposes, to substitute microarray-based ER expression levels for clinical measures of ER status in the absence of clinical data [5,7]. Because the ER transcript is itself a central figure in the ER signature, together with a number of known ER target genes, it is plausible that the transcriptional activity of ER drives expression of the ER signature genes. In this context, the signature could be viewed as a functional readout of ER activity. Recently, the ER signature was analyzed in a cohort of ER-positive tumors and found to be prognostic of disease-free survival in patients receiving adjuvant tamoxifen mono-therapy [25], suggesting that a gene-expression-based readout of ER functionality may be a greater predictor of antiestrogen response than a measure based on ER protein level alone.
In a study aimed at understanding the relevance of p53 status in breast cancer prognosis, we recently identified a 32-gene signature capable of distinguishing p53 mutant and p53 wildtype breast tumors with moderate (85%) accuracy [26]. Subsequent analysis of the misclassified tumors, however, shed light on the reason for classification failure. Misclassified wildtype tumors (that is, with the mutant-like signature; n = 26) showed highly significant underexpression of several known direct target genes of p53, as well as the p53 gene itself, whereas p53 mutant tumors with the wildtype-like signature (n = 12) showed significantly higher expression of the p53 target genes than other mutant tumors.
Furthermore, in an independent study of p53 activity, over half of the p53 signature genes identified in the breast tumors were found to be significantly modulated by p53 activation in HCT116 colorectal cancer cells [27]. These observations suggest that the signature, as a gauge of p53 transcriptional endpoints, may be more tuned to p53 function than mutational status as ascertained by the gold standard for mutational analysis, direct sequencing. Moreover, survival analysis of patients with p53 wildtype tumors showed that those with the mutant-like signature had a significantly shorter interval to disease-specific death than those with the wildtype-like profile. In several independent breast cancer cohorts, this signature of p53 deficiency was highly correlated with metastatic recurrence and therapeutic failure, regardless of treatment type, and remained a significant prognostic predictor in multivariate analyses with conventional risk factors, whereas p53 mutational status alone did not. Together, these observations suggest that an expression signature derived from the molecular differences between p53 mutant and wildtype tumors may provide a more comprehensive and clinically useful readout of p53 functionality than mutational status alone.
In a similar vein, we and others have recently investigated the clinical utility of gene expression patterns associated with the histologic grade of breast cancer. Although histologic grade is widely regarded as a strong indicator of disease recurrence, its acceptance as a routine prognostic variable has been limited by the subjective nature of the grading process and its history of inter-observer variability. Recently, a 5-gene genetic grade signature [28] and a 97-gene genomic grade index [29] have been identified, both capable of discriminating grade I and grade III tumors with high accuracy, and partitioning intermediate grade II tumors into grade I-like and grade III-like classes with enhanced prognostic resolution. Patients with grade II disease classified as grade I-like and grade III-like showed significantly different 10-year survival curves – similar to those of patients with histologic grade I and grade III tumors, respectively. Moreover, in multivariate analyses with conventional prognostic variables, we found that the genetic grade signature remained highly significant, even outperforming lymph node status and tumor size in most cohorts analyzed [28]. That most of these signature genes have known functions in cell-cycle-related processes and are significantly correlated with tumor mitotic index and Ki67 scores (A. Ivshina, personal communication) suggests that these grade-associated signatures are also markers of proliferation.
Thus, multigene predictors that objectively capture the prognostic essence of histologic grade and cellular proliferation have surprising precision in assessing risk of recurrence, particularly for women with grade II disease. Indeed, from a purely prognostic perspective, these studies suggest that there is no grade II, only shades of low and high grade. Furthermore, from a biological perspective, these findings offer insight into the pathobiological nature of breast cancer, suggesting that tumors of low and high grade may reflect independent biological entities rather than a continuum through which cancer progresses.
Parsing pathways
The expression signatures derived from ER status, p53 mutation, and histologic grade are products of 'bottom-up' analytical strategies [30] that are biologically motivated rather than empirically derived. These strategies first define relationships between a physiologic or biochemical phenomenon and patterns of gene expression, then use the expression patterns to predict the relative contribution of the phenomenon or pathway to clinical tumor behavior. In contrast to 'top-down' strategies that identify predictive signatures in the absence of biological input, the bottom-up approach has several advantages. First, by defining the downstream genes, insights into the molecular underpinnings of a discrete pathophysiologic phenomenon (such as an oncogenic pathway) are obtained. Second, the transcript levels of the genes themselves can be used to predict the extent of pathway activation in individual tumors, with the potential to select patients for pathway-targeted therapies. Third, such signatures can be assessed singly or in parallel to study the individual and combinatorial effects of distinct pathways on tumor aggressiveness, patient outcome or therapeutic response, in contrast to the dilution of individual pathway contributions that occurs in signatures derived from empirically based methods.
Desai and colleagues at the National Cancer Institute (USA) were among the first to investigate the global transcriptional outputs of multiple oncogenic pathways and their discriminatory powers [31]. Profiling breast tumors of transgenic mice harboring different mammary-gland-specific oncotransgenes (MMTV-Ha-ras, MMTV-neu, MMTV-myc, MMTV-polyoma middle T antigen, C3T-SV40 large T antigen and WAP-SV40 large T antigen), the authors identified expression cassettes unique to the different transgenes, indicating that transcriptional fingerprints of the earliest initiating oncogenic events could be identified within primary tumors.
Building on this concept, Joseph Nevins and colleagues at Duke University have recently published a series of reports that illustrate a systematic approach to the discovery and clinical application of pathway-specific and drug-specific signatures. Using primary mouse embryo fibroblasts [32] and human mammary epithelial cells [33] transfected with oncogenes such as HRAS, MYC, E2F and SRC, the authors identified expression signatures that distinguished oncogene-activated cells from controls. These signatures, representing transcriptional readouts of pathway activity derived in vitro, were then tested for their ability to assess pathway activation states in vivo with the use of mouse and human primary tumors previously characterized for aberrations in these pathways. The relative probability of pathway activation (or deregulation) was then estimated by comparing the configuration of the tumor profiles with that of the (in vitro) pathway-activated signatures. In this manner, the authors demonstrated that, on a probability scale, pathway activity could be predicted in vivo with significant accuracy. When applied to data sets of breast, ovarian and lung tumors, hierarchical clustering of the relative probabilities of pathway activation (as measured for multiple pathway signatures) could distinguish between patient subgroups with significantly different survival rates, demonstrating a strong association between multimodal pathway deregulation and clinical tumor behavior [33]. Moreover, when applied to a panel of cancer cell lines with known sensitivities to pathway-specific compounds (for example, for Ras and Src), the signatures were found to be significantly correlated with drug response [33].
These results demonstrate that expression signatures anchored to pathway activation states may aid in our biological understanding of tumor behavior and potentiate a means for selecting patients who will respond to pathway-specific therapies. Furthermore, where traditional classification methods have involved assigning patients (or tumors) to classes with definitive boundaries, assessing the likelihood that a tumor or patient will exhibit a certain trait (such as pathway deregulation or survival), as demonstrated in these studies, translates class prediction to a probability scale whereby sensitivity relative to specificity may be adjusted according to clinical need.
Taking these concepts further, Potti and colleagues [34] combined microarray data from the NCI-60 cell lines with historical pharmacologic data generated from the NCI-60 panel at the National Cancer Institute to define expression signatures capable of discriminating between cell lines that are sensitive to various drugs and those that are resistant. In this manner, drug response signatures were obtained for compounds such as docetaxel, topotecan, adriamycin, paclitaxel, 5-fluorouracil, and cyclophosphamide. The predictive capacity of these signatures was then validated by using two types of independent data set: first, those composed of cell line expression profiles generated in independent pharmacologic studies, and second, those composed of primary tumor profiles taken in the context of neoadjuvant therapy. Remarkably, with the latter validation approach, these predictors derived in vitro achieved more than 80% accuracy in each of five independent neoadjuvant studies involving breast and ovarian cancer patients treated with docetaxel, topotecan, adriamycin, or paclitaxel. However, it should be noted that the separation of patients into predicted response groups (sensitive versus resistant) was based on a 'best-fit' line; nevertheless in each case this line fell close to the 50% probability score, thus introducing only a small bias into the reported accuracies. Furthermore, the authors showed that multiple drug response signatures could be combined to predict sensitivity to multidrug regimens such as T/FAC) and FAC (5-fluorouracil, adriamycin, and cyclophosphamide), again with more than 80% accuracy.
Finally, the authors superimposed predictions based on the two types of signature: for drug response and for oncogenic pathways. In one example they found a significant association between predicted activation of the phosphoinositide 3-kinase (PI3-kinase) pathway and predicted docetaxel resistance in the NCI-60 data set. In a separate group of lung cancer cell lines, this association not only remained significant but the cells predicted to be PI3-kinase activated were significantly sensitive to a PI3-kinase inhibitor. This demonstrates that the drug response and pathway activation signatures can not only be used individually to predict treatment outcomes, but can also be combined for insight into the mechanisms modulating drug sensitivity. Together, these studies present a rational knowledge-based approach to individualized treatment, whereby the combinatorial analysis of biologically and experimentally defined expression signatures might one day guide therapeutic decisions that are truly tailored to the unique molecular anatomy of an individual's tumor.
Moving forward with in vitro-based models for building genomic predictors, several important considerations regarding system design and prediction accuracy must be addressed. What is the optimal number of models (namely cell lines, pathway targets, and so on), and how much biological diversity should be included in the system? What phenotypic endpoints should be used (IC50? LC50? a specific time point?) and how do these relate to tumor pharmacokinetics or pathway activation states? How do different classification strategies compare with respect to the robustness and accuracy of the genomic predictors they generate?
Mining mechanisms
The vast quantities of data generated from large-scale expression profiling studies provide a rich ground for exploring the complex and conditional relationships that exist between genes, their expression patterns, and tumor phenotypes. These relationships, although complex, exhibit a natural order governed by biological rules. This order is manifested in the hierarchical structure of gene–gene correlations from which the various prognostic expression signatures have been mined. Although bottom-up investigations have elucidated the biology underlying several of these signatures, most multigene expression patterns associated with prognosis remain biologically anonymous. Understanding this biology, and the transcriptional mechanisms regulating these signatures, may lead to the discovery of new oncogenic pathways and therapeutic targets.
To explore the diversity of gene correlations that underlie the clinical behavior of cancer, we have analyzed large microarray data sets of primary breast tumors for genes that are both coordinately expressed (in clusters) and individually related to clinical outcomes, and have discovered numerous distinct expression cassettes that may signify clinically relevant pathways in breast carcinogenesis (Figure 1). However, a biological definition of these pathways and the mechanisms that regulate them requires more than simple inference, but rather the integration of multiple forms of information (for example biological, clinical, and genomic) coupled with statistical and experimental validation methods.
Figure 1.
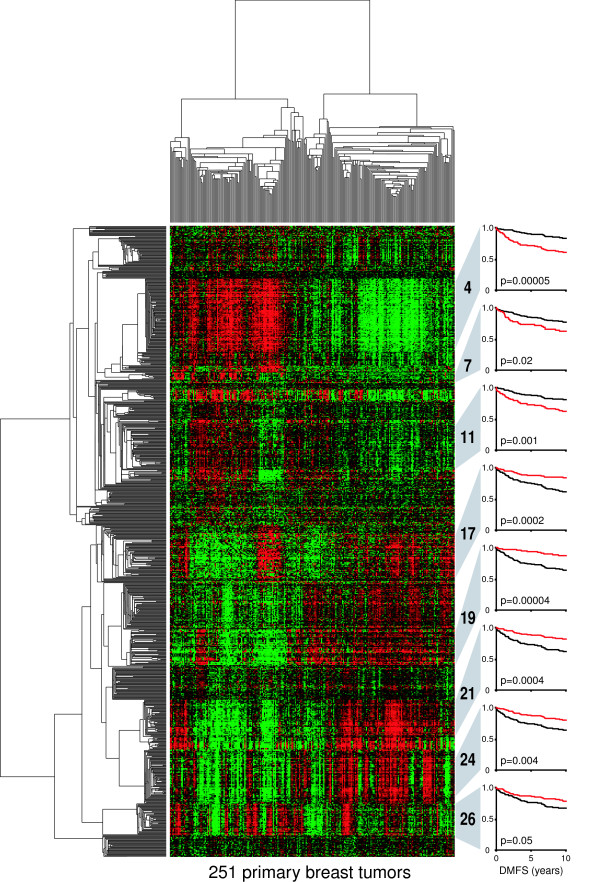
Clustergram of diverse gene expression signatures prognostic of breast cancer recurrence. Tumors (n = 251; columns) and gene probe sets (n = 816; rows) of the Uppsala cohort (GEO ID GSE3494) [26] were hierarchically clustered by using Pearson correlation and average linkage analysis. Probe-set values were natural-log-transformed and mean centered before clustering. Initially, all 44,928 probe sets (on the Affymetrix U133A and U133B arrays) were assessed for survival correlations as follows. The expression value for each gene was used to dichotomize patients into below-mean and above-mean expression groups. The two groups were then assessed for differences in distant metastasis-free survival (DMFS) by Cox regression analysis. Probe sets significantly associated with DMFS (that is, with likelihood-ratio test P values of less than 0.05) were hierarchically clustered as described above, and clusters with average correlations of more than 0.5 were selected for inclusion in the figure. Probe sets within clusters were then averaged for each tumor, and cluster survival associations were determined as described above. Kaplan-Meier plots for selected numbered clusters are shown at the right. The red survival curves indicate patients with above-mean cluster expression. Cluster 7 is composed of five genes, all mapping to chromosome 17q12 (ORMDL3, PSMD3, CRKRS, PERLD1, and C17ORF37) with an average expression correlation of 0.64. Cluster 11, with an average correlation of 0.65, consists of 31 distinct genes, 18 of which map to chromosome 16p13 (PPP4C, PARN, ATP6V0C, C16orf14, GBL, HAGH, ITFG3, MGC13114, MRPS34, NDUFB10, NMRAL1, NTHL1, NUBP2, POLR3K, RNPS1, STUB1, TBL3, and USP7).
Early microarray studies involving breast cancer cell lines identified a large cluster of coordinately expressed genes associated with cell proliferation rates [35]. Later dubbed the proliferation signature, these genes have since been linked to various aspects of tumorigenesis in breast and other cancer types including neoplastic transformation [36], histologic grade [28,29,37], and poor patient survival [38,39]. (Cluster 4 in Figure 1 represents this signature.) For statistical support of the notion that this signature reflects cellular proliferation in primary breast tumors, we analyzed various subsets of these signature genes for correlations with different forms of biological and clinicopathological information. Gene ontology analysis of the signature genes consistently resulted in the significant enrichment of proliferative processes such as mitosis, cytokinesis, chromosomal segregation, chromatin packaging and remodeling, and DNA metabolism and replication (LDM and ETL, unpublished results). Using clinical tumor annotations, we found significant correlations between expression of the signature genes and pathologic markers of proliferation including Ki67, S-phase fraction and mitotic index (LDM and ETL, unpublished results), further supporting the link between gene expression and tumor cell proliferation. Furthermore, a significant fraction of these signature genes have been observed in cell synchronization experiments involving HeLa cells (cervical carcinoma) as being expressed periodically at specific phases of the cell cycle [40]. Thus, as illustrated in this simple example, the integrative analysis of functional, clinical, and experimental information can provide substantial support for the hypothesis that an expression signature reflects a specific biological phenomenon – in this case, the proliferative capacity of tumor cells.
Integration of additional forms of data, such as genomic sequence, location, and copy number alterations, can potentially expose the transcriptional mechanisms that regulate the expression of these correlated genes. For example, Gasch and Eisen [41], exploring mechanisms of gene co-regulation in yeast, demonstrated that promoter analysis of coordinately expressed genes could reveal significant enrichments of binding motifs specific for the transcription factor(s) responsible for the observed coordinate expression. However, despite the success of this approach in identifying gene regulatory mechanisms in organisms of lower complexity [42], it has so far shown little success in elucidating transcriptional mechanisms in cancer, perhaps owing in part to the greater complexity and lack of spatial compactness of human gene promoters. In a recent study by Kristensen and colleagues [43], the impact of genetic variation on breast cancer gene expression was examined. Using a panel of 50 primary human tumors with matched patient blood samples, the authors found that selected germline SNPs at putative regulatory loci in 115 of 203 candidate genes (of the reactive oxygen species pathway) showed highly significant associations with microarray expression patterns, indicative of both cis-acting and trans-acting effects. In some instances, transcripts associated with SNPs in trans showed significant enrichment for certain gene ontology terms and pathways, suggesting linkages between SNPs and the activity of biological programs. This work indicates that the coordinate expression of genes in breast cancer may be markedly influenced by genetic variation at gene regulatory loci, and opens up a new avenue for the discovery of transcriptional regulatory mechanisms and genetic biomarkers in breast cancer.
Alterations in chromosomal copy number are also manifested in the gene expression patterns of breast cancer. In Figure 1, for example, clusters 7 and 11 are significantly enriched for genes mapping to cytobands 17q12 and 16p13, respectively (see Figure 1 legend). Both loci are frequently amplified in breast cancer, suggesting that the correlated expression of these genes may be explained, in large part, by the transcriptional consequences of genomic amplification. This hypothesis is supported by the work of Pollack and colleagues [44], who first examined the intersection between expression array and array comparative genomic hybridization (CGH) data from breast cancer cell lines and primary breast tumors, and observed that more than 60% of high-level copy-number gains coincided with the coordinate overexpression of involved genes, producing, in effect, a residual expression footprint of a genomic amplicon. The integrative analysis of high-resolution array CGH and microarray expression data is now frequently applied to investigations of the mechanistic context of genomic aberrations. In breast cancer, focused studies on 17q12 and 8p11 have revealed new oncogene candidates in which amplification and overexpression are highly correlated [45,46]. Genes identified by this strategy, such as LSM1, BAG4, and C8orf4 on 8p11, have subsequently been shown to drive neoplastic transformation in vitro, and when expressed in combination can induce growth that is independent of both growth factors and anchorage to substrate [47].
The intersection between gene amplification and over-expression has also been exploited to uncover transcriptional regulators of a prognostic expression signature in breast cancer. In a series of work, Howard Chang and colleagues explored the relationship between wound healing and cancer progression [30,48,49]. Initial microarray analysis defined an expression signature of serum response in fibroblasts that, when applied to breast and other epithelial cancer data sets, seemed indicative of tumors exhibiting an active wound response [48]. This wound response signature was subsequently found to be prognostic of survival for patients with breast, lung, and gastric cancers [30,48].
To uncover the transcriptional mechanisms driving expression of the wound response genes, Adler and colleagues [49] used a genetic linkage approach (stepwise linkage analysis of microarray signatures (SLAMS)) involving the integration of gene expression and array CGH data. Considering the possibility that the origin of the wound response signature may be rooted in chromosomal alterations, the authors identified genes with patterns of copy number gain or loss that significantly distinguished breast tumors positive and negative for the wound signature. They observed an enrichment of genes localized to 8q and amplified in tumors with the activated wound response. Analysis of the distributions of 8q-amplified genes within tumor groups led the authors to deduce the possibility of a regulatory interaction between components of 8q24 and 8q13. Closer examination of the expression patterns of the amplified genes revealed that the MYC gene on 8q24 was the one most highly induced by fibroblasts upon serum stimulation, and the CSN5 gene on 8q13 was the one most highly correlated with the wound signature, suggesting a synergistic role for these two proteins in modulating the expression of the wound signature genes. MYC encodes an oncogenic transcription factor frequently amplified in breast cancer, and CSN5 encodes the catalytic subunit of the COP9 signalosome, a multifunctional activator of cullin-based ubiquitin ligases.
To test for a functional interaction, Adler and colleagues over-expressed MYC and CSN5 in noncancerous MCF10A breast epithelial cells. Co-expression of MYC and CSN5, but not the expression of a green fluorescent protein control or of either gene alone, resulted in the induction of more than 75% of the 255 genes overexpressed in the activated wound signature, as well as significant increases in cellular proliferation and invasion through Matrigel that were consistent with the association between the activated wound response and more aggressive disease. Thus, from in silico prediction to experimental validation, Adler and colleagues demonstrate a methodology of integrative genomic analysis that can facilitate the discovery of complex transcriptional mechanisms regulating gene expression signatures. The increased complexity is that the expression phenotype is manifested only with the activation of two cooperating gene products: a synthetic or conditional effect.
Future challenges
Expression arrays initially began simply as a method of multiplexing single gene discovery, akin to running several thousand quantitative RNA dot-blots. From this one-dimensional approach evolved the current state of the art: expression profiling to uncover pathway regulation of gene expression and to define molecular classes on the basis of integration of the total signals experienced by the cancer cell. Fundamental to this transition has been the ability to analyze and model complex systems made possible by mathematical algorithms coupled with computational capacity. It is in this realm of complexity analysis that the future of array-based expression genomics will lie. One can clearly see some of the more immediate areas of expansion.
First, data content can increase. Other characteristics of the transcriptome such as exon usage and noncoding RNAs (including microRNAs) are not well covered by the existing array technologies and their inclusion would inevitably result in greater precision and comprehensiveness. Exon junctions could conceivably be included in the battery of tests yet to be applied. Of course, this will require greater array capacity in terms of encompassing more probes in smaller spaces. Given the advances in microelectronics, those possibilities are currently available but are perhaps not cost-effective for broad biological experimentation.
Second, the analytical systems can be more informed. Although the output of individual probes can be viewed as events that are independent from that of any other probe, biologically, the degrees of freedom of transcriptional systems are already constrained by biochemical and even evolutionary reality. Thus, gene X is always coordinately expressed with gene Y, or gene A is always upstream of gene B, or proteins C, E, and F are always in a complex and function only as a unit, never alone. These genetic, biochemical, or physiologic relationships validated by other means can be incorporated as 'priors' as we seek higher orders of interaction.
Last, metadata sets will emerge that will markedly expand the ability to validate and to model transcriptional networks of biological and clinical significance. This is already taking place with Oncomine [36], and follows the success of other genomic databases. As a result of standardization, the availability of large numbers of data sets describing the transcriptional behavior of breast cancers has permitted the validation of local observations in silico. In the context of prognosis, the performance of expression signatures can now be validated in and compared across numerous independent cohorts [4,26,28,50], and analyzed in combination for synergistic interactions [30]. At some point, the content of the expression metadata sets for breast cancer will be large enough to sustain continuous activity in data mining, hypothesis generation, and validation. This requires the inclusion of detailed clinical information. In some medical research communities, this metadata set approach is more advanced. Comparative and evolutional geneticists use the growing number of complete genomes in publicly available databases as their primary substrate for investigation. In molecular epidemiology, whole-genome SNP databases with linked clinical data are being made available to qualified researchers for analysis and data mining.
These trends will have a great impact on breast cancer research. The advantage will be the ability to be comprehensive and yet precise at the same time, and the speed of discovery will be breathtaking. The challenge, however, will shift to organizational issues. How fast can we validate new marker sets? What kind of incentives can we use to encourage groups to share primary data? How can we sustain teams of computer scientists, basic molecular biologists, molecular pathologists, and oncologists to meet these challenges?
Abbreviations
ABC = activated B cell; CGH = comparative genomic hybridization; DLBCL = diffuse large-B-cell lymphoma; ER = estrogen receptor; GCB = germinal-center B cell; NF = nuclear factor; pCR = complete pathologic response; PI3-kinase = phosphoinositide 3-kinase; SNP = single-nucleotide polymorphism; T/FAC = paclitaxel, 5-fluorouracil, adriamycin and cyclophosphamide).
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
Acknowledgements
The authors are supported by the Singapore Agency for Science, Technology and Research (A*STAR). The authors apologize to colleagues whose work could not be cited because of space limitations.
Contributor Information
Lance D Miller, Email: millerL@gis.a-star.edu.
Edison T Liu, Email: liue@gis.a-star.edu.sg.
References
- Gershon D. DNA microarrays: more than gene expression. Nature. 2005;437:1195–1198. doi: 10.1038/4371195a. [DOI] [PubMed] [Google Scholar]
- Perou CM, Sorlie T, Eisen MB, van de Rijn M, Jeffrey SS, Rees CA, Pollack JR, Ross DT, Johnsen H, Akslen LA, et al. Molecular portraits of human breast tumours. Nature. 2000;406:747–752. doi: 10.1038/35021093. [DOI] [PubMed] [Google Scholar]
- Sorlie T, Perou CM, Tibshirani R, Aas T, Geisler S, Johnsen H, Hastie T, Eisen MB, van de Rijn M, Jeffrey SS, et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc Natl Acad Sci USA. 2001;98:10869–10874. doi: 10.1073/pnas.191367098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, Deng S, Johnsen H, Pesich R, Geisler S, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA. 2003;100:8418–8423. doi: 10.1073/pnas.0932692100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van 't Veer LJ, Dai H, van de Vijver MJ, He YD, Hart AA, Mao M, Peterse HL, van der Kooy K, Marton MJ, Witteveen AT, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–536. doi: 10.1038/415530a. [DOI] [PubMed] [Google Scholar]
- Wang Y, Klijn JG, Zhang Y, Sieuwerts AM, Look MP, Yang F, Talantov D, Timmermans M, Meijer-van Gelder ME, Yu J, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. Lancet. 2005;365:671–679. doi: 10.1016/S0140-6736(05)17947-1. [DOI] [PubMed] [Google Scholar]
- van de Vijver MJ, He YD, van't Veer LJ, Dai H, Hart AA, Voskuil DW, Schreiber GJ, Peterse JL, Roberts C, Marton MJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347:1999–2009. doi: 10.1056/NEJMoa021967. [DOI] [PubMed] [Google Scholar]
- Buyse M, Loi S, van't Veer L, Viale G, Delorenzi M, Glas AM, d'Assignies MS, Bergh J, Lidereau R, Ellis P, et al. Validation and clinical utility of a 70-gene prognostic signature for women with node-negative breast cancer. J Natl Cancer Inst. 2006;98:1183–1192. doi: 10.1093/jnci/djj329. [DOI] [PubMed] [Google Scholar]
- Foekens JA, Atkins D, Zhang Y, Sweep FC, Harbeck N, Paradiso A, Cufer T, Sieuwerts AM, Talantov D, Span PN, et al. Multicenter validation of a gene expression-based prognostic signature in lymph node-negative primary breast cancer. J Clin Oncol. 2006;24:1665–1671. doi: 10.1200/JCO.2005.03.9115. [DOI] [PubMed] [Google Scholar]
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature. 2000;403:503–511. doi: 10.1038/35000501. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med. 2002;346:1937–1947. doi: 10.1056/NEJMoa012914. [DOI] [PubMed] [Google Scholar]
- Wright G, Tan B, Rosenwald A, Hurt EH, Wiestner A, Staudt LM. A gene expression-based method to diagnose clinically distinct subgroups of diffuse large B cell lymphoma. Proc Natl Acad Sci USA. 2003;100:9991–9996. doi: 10.1073/pnas.1732008100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis RE, Brown KD, Siebenlist U, Staudt LM. Constitutive nuclear factor κB activity is required for survival of activated B cell-like diffuse large B cell lymphoma cells. J Exp Med. 2001;194:1861–1874. doi: 10.1084/jem.194.12.1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JC, Wooten EC, Tsimelzon A, Hilsenbeck SG, Gutierrez MC, Elledge R, Mohsin S, Osborne CK, Chamness GC, Allred DC, et al. Gene expression profiling for the prediction of therapeutic response to docetaxel in patients with breast cancer. Lancet. 2003;362:362–369. doi: 10.1016/S0140-6736(03)14023-8. [DOI] [PubMed] [Google Scholar]
- Iwao-Koizumi K, Matoba R, Ueno N, Kim SJ, Ando A, Miyoshi Y, Maeda E, Noguchi S, Kato K. Prediction of docetaxel response in human breast cancer by gene expression profiling. J Clin Oncol. 2005;23:422–431. doi: 10.1200/JCO.2005.09.078. [DOI] [PubMed] [Google Scholar]
- Ayers M, Symmans WF, Stec J, Damokosh AI, Clark E, Hess K, Lecocke M, Metivier J, Booser D, Ibrahim N, et al. Gene expression profiles predict complete pathologic response to neoadjuvant paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide chemotherapy in breast cancer. J Clin Oncol. 2004;22:2284–2293. doi: 10.1200/JCO.2004.05.166. [DOI] [PubMed] [Google Scholar]
- Hess KR, Anderson K, Symmans WF, Valero V, Ibrahim N, Mejia JA, Booser D, Theriault RL, Buzdar AU, Dempsey PJ, et al. Pharmacogenomic predictor of sensitivity to preoperative chemotherapy with paclitaxel and fluorouracil, doxorubicin, and cyclophosphamide in breast cancer. J Clin Oncol. 2006;24:4236–4244. doi: 10.1200/JCO.2006.05.6861. [DOI] [PubMed] [Google Scholar]
- Folgueira MA, Carraro DM, Brentani H, Patrao DF, Barbosa EM, Netto MM, Caldeira JR, Katayama ML, Soares FA, Oliveira CT, et al. Gene expression profile associated with response to doxorubicin-based therapy in breast cancer. Clin Cancer Res. 2005;11:7434–7443. doi: 10.1158/1078-0432.CCR-04-0548. [DOI] [PubMed] [Google Scholar]
- Dressman HK, Hans C, Bild A, Olson JA, Rosen E, Marcom PK, Liotcheva VB, Jones EL, Vujaskovic Z, Marks J, et al. Gene expression profiles of multiple breast cancer phenotypes and response to neoadjuvant chemotherapy. Clin Cancer Res. 2006;12:819–826. doi: 10.1158/1078-0432.CCR-05-1447. [DOI] [PubMed] [Google Scholar]
- Ma XJ, Wang Z, Ryan PD, Isakoff SJ, Barmettler A, Fuller A, Muir B, Mohapatra G, Salunga R, Tuggle JT, et al. A two-gene expression ratio predicts clinical outcome in breast cancer patients treated with tamoxifen. Cancer Cell. 2004;5:607–616. doi: 10.1016/j.ccr.2004.05.015. [DOI] [PubMed] [Google Scholar]
- Paik S, Shak S, Tang G, Kim C, Baker J, Cronin M, Baehner FL, Walker MG, Watson D, Park T, et al. A multigene assay to predict recurrence of tamoxifen-treated, node-negative breast cancer. N Engl J Med. 2004;351:2817–2826. doi: 10.1056/NEJMoa041588. [DOI] [PubMed] [Google Scholar]
- Paik S, Tang G, Shak S, Kim C, Baker J, Kim W, Cronin M, Baehner FL, Watson D, Bryant J, et al. Gene expression and benefit of chemotherapy in women with node-negative, estrogen receptor-positive breast cancer. J Clin Oncol. 2006;24:3726–3734. doi: 10.1200/JCO.2005.04.7985. [DOI] [PubMed] [Google Scholar]
- Gruvberger S, Ringner M, Chen Y, Panavally S, Saal LH, Borg A, Ferno M, Peterson C, Meltzer PS. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001;61:5979–5984. [PubMed] [Google Scholar]
- Gruvberger-Saal SK, Eden P, Ringner M, Baldetorp B, Chebil G, Borg A, Ferno M, Peterson C, Meltzer PS. Predicting continuous values of prognostic markers in breast cancer from microarray gene expression profiles. Mol Cancer Ther. 2004;3:161–168. [PubMed] [Google Scholar]
- Symmans WF, Sotiriou C, Anderson SK, Hatzis C, Valero V, Booser DJ, Larsimont D, Desmedt C, Gold DL, Sneige N, et al. Measurements of estrogen receptor and reporter genes from microarrays determine receptor status and time to recurrence following adjuvant tamoxifen therapy. Breast Cancer Res Treat. 2005;94(Suppl 1):308a. [Google Scholar]
- Miller LD, Smeds J, George J, Vega VB, Vergara L, Ploner A, Pawitan Y, Hall P, Klaar S, Liu ET, et al. An expression signature for p53 status in human breast cancer predicts mutation status, transcriptional effects, and patient survival. Proc Natl Acad Sci USA. 2005;102:13550–13555. doi: 10.1073/pnas.0506230102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, Liu ET, Yu Q. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem. 2004;279:21183–21192. doi: 10.1074/jbc.M311912200. [DOI] [PubMed] [Google Scholar]
- Ivshina AV, George J, Senko O, Mow B, Putti TC, Smeds J, Lindahl T, Pawitan Y, Hall P, Nordgren H, et al. Genetic reclassification of histologic grade delineates new clinical subtypes of breast cancer. Cancer Res. 2006;66:10292–10301. doi: 10.1158/0008-5472.CAN-05-4414. [DOI] [PubMed] [Google Scholar]
- Sotiriou C, Wirapati P, Loi S, Harris A, Fox S, Smeds J, Nordgren H, Farmer P, Praz V, Haibe-Kains B, et al. Gene expression profiling in breast cancer: understanding the molecular basis of histologic grade to improve prognosis. J Natl Cancer Inst. 2006;98:262–272. doi: 10.1093/jnci/djj052. [DOI] [PubMed] [Google Scholar]
- Chang HY, Nuyten DS, Sneddon JB, Hastie T, Tibshirani R, Sorlie T, Dai H, He YD, van't Veer LJ, Bartelink H, et al. Robustness, scalability, and integration of a wound-response gene expression signature in predicting breast cancer survival. Proc Natl Acad Sci USA. 2005;102:3738–3743. doi: 10.1073/pnas.0409462102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai KV, Xiao N, Wang W, Gangi L, Greene J, Powell JI, Dickson R, Furth P, Hunter K, Kucherlapati R, et al. Initiating oncogenic event determines gene-expression patterns of human breast cancer models. Proc Natl Acad Sci USA. 2002;99:6967–6972. doi: 10.1073/pnas.102172399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang E, Ishida S, Pittman J, Dressman H, Bild A, Kloos M, D'Amico M, Pestell RG, West M, Nevins JR. Gene expression phenotypic models that predict the activity of oncogenic pathways. Nat Genet. 2003;34:226–230. doi: 10.1038/ng1167. [DOI] [PubMed] [Google Scholar]
- Bild AH, Yao G, Chang JT, Wang Q, Potti A, Chasse D, Joshi MB, Harpole D, Lancaster JM, Berchuck A, et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature. 2006;439:353–357. doi: 10.1038/nature04296. [DOI] [PubMed] [Google Scholar]
- Potti A, Dressman HK, Bild A, Riedel RF, Chan G, Sayer R, Cragun J, Cottrill H, Kelley MJ, Petersen R, et al. Genomic signatures to guide the use of chemotherapeutics. Nat Med. 2006;12:1294–1300. doi: 10.1038/nm1491. [DOI] [PubMed] [Google Scholar]
- Ross DT, Scherf U, Eisen MB, Perou CM, Rees C, Spellman P, Iyer V, Jeffrey SS, Van de Rijn M, Waltham M, et al. Systematic variation in gene expression patterns in human cancer cell lines. Nat Genet. 2000;24:227–235. doi: 10.1038/73432. [DOI] [PubMed] [Google Scholar]
- Rhodes DR, Yu J, Shanker K, Deshpande N, Varambally R, Ghosh D, Barrette T, Pandey A, Chinnaiyan AM. Large-scale meta-analysis of cancer microarray data identifies common transcriptional profiles of neoplastic transformation and progression. Proc Natl Acad Sci USA. 2004;101:9309–9314. doi: 10.1073/pnas.0401994101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XJ, Salunga R, Tuggle JT, Gaudet J, Enright E, McQuary P, Payette T, Pistone M, Stecker K, Zhang BM, et al. Gene expression profiles of human breast cancer progression. Proc Natl Acad Sci USA. 2003;100:5974–5979. doi: 10.1073/pnas.0931261100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Wiestner A, Chan WC, Connors JM, Campo E, Gascoyne RD, Grogan TM, Muller-Hermelink HK, Smeland EB, et al. The proliferation gene expression signature is a quantitative integrator of oncogenic events that predicts survival in mantle cell lymphoma. Cancer Cell. 2003;3:185–197. doi: 10.1016/S1535-6108(03)00028-X. [DOI] [PubMed] [Google Scholar]
- Dai H, van't Veer L, Lamb J, He YD, Mao M, Fine BM, Bernards R, van de Vijver M, Deutsch P, Sachs A, et al. A cell proliferation signature is a marker of extremely poor outcome in a subpopulation of breast cancer patients. Cancer Res. 2005;65:4059–4066. doi: 10.1158/0008-5472.CAN-04-3953. [DOI] [PubMed] [Google Scholar]
- Whitfield ML, Sherlock G, Saldanha AJ, Murray JI, Ball CA, Alexander KE, Matese JC, Perou CM, Hurt MM, Brown PO, et al. Identification of genes periodically expressed in the human cell cycle and their expression in tumors. Mol Biol Cell. 2002;13:1977–2000. doi: 10.1091/mbc.02-02-0030.. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasch AP, Eisen MB. Exploring the conditional coregulation of yeast gene expression through fuzzy k-means clustering. Genome Biol. 2002;3:RESEARCH0059. doi: 10.1186/gb-2002-3-11-research0059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Karuturi RK, Miller LD, Lin K, Jia Y, Kondu P, Wang L, Wong LS, Liu ET, Balasubramanian MK, et al. Identification of cell cycle-regulated genes in fission yeast. Mol Biol Cell. 2005;16:1026–1042. doi: 10.1091/mbc.E04-04-0299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristensen VN, Edvardsen H, Tsalenko A, Nordgard SH, Sorlie T, Sharan R, Vailaya A, Ben-Dor A, Lonning PE, Lien S, et al. Genetic variation in putative regulatory loci controlling gene expression in breast cancer. Proc Natl Acad Sci USA. 2006;103:7735–7740. doi: 10.1073/pnas.0601893103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollack JR, Sorlie T, Perou CM, Rees CA, Jeffrey SS, Lonning PE, Tibshirani R, Botstein D, Borresen-Dale AL, Brown PO. Microarray analysis reveals a major direct role of DNA copy number alteration in the transcriptional program of human breast tumors. Proc Natl Acad Sci USA. 2002;99:12963–12968. doi: 10.1073/pnas.162471999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauraniemi P, Barlund M, Monni O, Kallioniemi A. New amplified and highly expressed genes discovered in the ERBB2 amplicon in breast cancer by cDNA microarrays. Cancer Res. 2001;61:8235–8240. [PubMed] [Google Scholar]
- Ray ME, Yang ZQ, Albertson D, Kleer CG, Washburn JG, Macoska JA, Ethier SP. Genomic and expression analysis of the 8p11-12 amplicon in human breast cancer cell lines. Cancer Res. 2004;64:40–47. doi: 10.1158/0008-5472.CAN-03-1022. [DOI] [PubMed] [Google Scholar]
- Yang ZQ, Streicher KL, Ray ME, Abrams J, Ethier SP. Multiple interacting oncogenes on the 8p11-p12 amplicon in human breast cancer. Cancer Res. 2006;66:11632–11643. doi: 10.1158/0008-5472.CAN-06-2946. [DOI] [PubMed] [Google Scholar]
- Chang HY, Sneddon JB, Alizadeh AA, Sood R, West RB, Montgomery K, Chi JT, van de Rijn M, Botstein D, Brown PO. Gene expression signature of fibroblast serum response predicts human cancer progression: similarities between tumors and wounds. PLoS Biol. 2004;2:E7. doi: 10.1371/journal.pbio.0020007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adler AS, Lin M, Horlings H, Nuyten DS, van de Vijver MJ, Chang HY. Genetic regulators of large-scale transcriptional signatures in cancer. Nat Genet. 2006;38:421–430. doi: 10.1038/ng1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan C, Oh DS, Wessels L, Weigelt B, Nuyten DS, Nobel AB, van't Veer LJ, Perou CM. Concordance among gene-expression-based predictors for breast cancer. N Engl J Med. 2006;355:560–569. doi: 10.1056/NEJMoa052933. [DOI] [PubMed] [Google Scholar]