

Summary
We have investigated the function of the p110δ catalytic subunit of phosphoinositide 3-kinase (PI 3-kinase) in platelets using p110δ knockout (p110δ-/-) mice and p110δ knock-in (p110δD910A/D910A) mice, which express a catalytically inactive form of the enzyme. Aggregation to threshold concentrations of the GPVI-specific agonist, CRP, was partially reduced in p110δ-/- and p110δD910A/D910A platelets. This inhibition was overcome by higher concentrations of CRP. The degree of inhibition was considerably weaker than that induced by LY294002 and wortmannin, which inhibit all isoforms of PI 3-kinase. p110δ-/- platelets showed decreased spreading on fibrinogen- or von Willebrand factor (VWF)-coated surfaces under static conditions, whereas they spread normally on collagen. LY294002 had a more pronounced inhibitory effect on spreading on all three surfaces. Adhesion and aggregate formation of p110δ-/- platelets to collagen or fibrinogen/VWF at intermediate/high rates of shear were normal. This study demonstrates a minor role for the p110δ catalytic subunit in mediating platelet activation by the collagen receptor GPVI and integrin αIIbβ3. The more pronounced inhibitory effect of LY294002 and wortmannin indicates that other isoforms of PI 3-kinase play a more significant role in signalling by the two platelet glycoprotein receptors.
Keywords: PI 3-kinase, p110δ, platelets, signalling, flow adhesion
Introduction
The primary physiological function of platelets is to stop bleeding from sites of vascular injury. They do this by adhering to exposed subendothelial components such as collagen, fibronectin, fibrinogen and VWF, becoming activated and forming aggregates, which plug the damaged blood vessel. The platelet aggregates become covered with fibrinogen, which is cleaved to fibrin polymers by thrombin and acts to consolidate the primary haemostatic plug.
Three of the main receptors present on the platelet surface required for tethering, activation and aggregation are the GPIb-IX-V complex, which binds VWF; GPVI/FcR γ-chain complex, which binds collagen; and αIIbβ3, which binds fibrinogen and other extracellular matrix proteins (ECM), including VWF. All three of these receptors utilize many of the same signalling molecules to achieve their respective responses. Some of the common signalling molecules shared by these receptors include Src family tyrosine kinases (1-3), the tyrosine kinase Syk (4-6), phospholipase (PLC) γ2 (7-10) and phosphoinositide 3-kinases (PI 3-kinases) (2, 11, 12). GPVI and αIIbβ3 also share the adapters SLP-76 and adhesion- and degranulation-promoting adapter protein (ADAP) (13, 14), and Vav GDP/GTP exchange factors (14-16). Targeted disruption or pharmacological inhibition of many of these proteins diminishes the ability of platelets to adhere and respond to ECM.
PI 3-kinases are a family of lipid kinases that are activated in response to growth factors, hormones, and ECM proteins. They have been implicated in a variety of cellular processes including growth, proliferation, survival, migration, and secretion. PI 3-kinases catalyze the conversion of the plasma membrane lipid phosphatidylinositol-4,5-bisphosphate [PI(4,5)P2] to phosphatidylinositol-3,4,5-trisphosphate [PI(3,4,5)P3] (17). PI 3,4,5-P3 is a potent second messenger that recruits pleckstrin homology (PH) domain-containing effector molecules to the cytoplasmic surface of the plasma membrane. Signalling proteins that accumulate at these sites include the serine-threonine kinases Akt (also referred to as protein kinase B [PKB]), phosphoinositide-dependent kinase 1 (PDK1), Tec family tyrosine kinases, and exchange factors that regulate heterotrimeric guanosine triphosphate (GTP)-binding proteins (G proteins) such as Vav, and PLC (18).
PI 3-kinases are categorized into three classes (I-III) (18). Class I PI 3-kinases are further subdivided into classes IA and IB depending on whether they are activated downstream of tyrosine kinase-linked receptors (class IA) or G protein-coupled receptors (class IB). There is also evidence that class IA enzymes can be regulated by G protein βγ subunits. Class IA PI 3-kinases are heterodimeric proteins consisting of a p110 catalytic subunit and a p85 regulatory subunit. Three types of class IA p110 catalytic subunits (p110α, p110β, and p110δ) encoded by 3 distinct genes have been identified. p110α and p110β are expressed in variety of cell types, whereas p110δ is primarily haematopoietic-specific (19, 20). Five class IA regulatory subunits (p85α, p55α, p50α, p85β, and p55γ) derived from three different genes have been identified, p55α and p50α being splice variants of p85α (21). The regulatory subunit acts to localize and activate the catalytic subunit. The class IB PI 3-kinase consists of one catalytic subunit, p110γ, associated with the regulatory subunit, p101. The enzyme is activated downstream of G protein-coupled receptors via G protein βγ subunits.
Most of the studies aimed at elucidating the role of PI 3-kinases in platelets have relied on the structurally distinct inhibitors LY294002 and wortmannin, which do not distinguish between the different forms of PI 3-kinases. These studies have demonstrated that PI 3-kinases are critical for regulating different aspects of platelet activation, including cytoskeletal rearrangements associated with spreading and activation of PLCγ2 through the major platelelet glycoprotein receptors, namely GPIb-IX-V, GPVI and αIIbβ3. The presence of all class IA and IB catalytic PI 3-kinases in platelets has been reported (22, 23), although this is controversial for the p110δ catalytic subunit (19, 24-27). Almost nothing is known, however, about the role of each of these catalytic isoforms in mediating signalling pathways by the major platelet glycoprotein receptors. The class IB PI 3-kinase catalytic subunit, p110γ, is activated in response to stimulation of platelets by G protein-coupled receptors (22, 28, 29), and has been shown to support activation downstream of the P2Y12 ADP receptor (25).
Studies have yet to be performed using p110α- and p110β-deficient platelets as deletion of either protein is embryonic lethal (30, 31). Simultaneous deletion of p85α, p55α and p50α regulatory subunits also causes perinatal lethality, whereas deletion of p85α alone does not (32, 33). A recent study of platelets from p85α-/- mice showed severe impairment of CRP-induced platelet aggregation, secretion, integrin activation, lamellipodia formation and tyrosine phosphorylation of effector molecules (24). These mice also exhibited a concomitant reduction in levels of p110α, p110β and p110δ, which is consistent with the instability of the p110 proteins in the absence of adaptor subunits. Since p85α is more abundant than p55α and p50α PI 3-kinase regulatory subunits in platelets and it heterodimerizes with all three class IA PI 3-kinase catalytic subunits, this report demonstrates the importance of class IA PI 3-kinases in signalling by GPVI.
In this study, we utilized human platelets, human megakaryoctytic cell lines DAMI and HEL, and two different mouse models, p110δ knockout (p110δ-/-) mice, which completely lack p110δ expression, and p110δ knockin (p110δD910A/D910A) mice, which express catalytically inactive p110δ, to address the role of the PI 3-kinase isoform p110δ in platelet activation. We demonstrate that platelets express low levels of p110δ and that it is involved in regulating spreading on fibrinogen and in mediating activation through GPVI. Studies with wortmannin and LY294002 suggest that it is not the major catalytic isoform that fulfills these roles or that other catalytic subunits are able to compensate for its absence.
Materials and methods
Reagents
Collagen (Horm) was purchased from Nycomed (Munich, Germany). Collagen related peptide (CRP) was prepared as previously described (34, 35). Heparin (25,000 U/ml; monoparin) was obtained from CP Pharmaceuticals (Wrexham, UK). Rhodamine-conjugated phalloidin, anti-actin monoclonal antibody, adenosine diphosphate (ADP), bovine thrombin, phorbol 12-myristate 13-acetate PMA, wortmannin, fatty acid free bovine serum albumin (BSA) were obtained from Sigma-Aldrich (Poole, UK). Human fibrinogen depleted of plasminogen, VWF and fibronectin was obtained from Enzyme Research Laboratories (Swansea, UK). LY294002 was obtained from Merck Biosciences Ltd. (Nottingham, UK). DAMI and HEL human megakaryocytic cell lines were obtained from the American Type Culture Collection (Manassas, VA, USA). RPMI, fetal bovine serum, glutamine, penicillin, and streptomycin were obtained from Gibco-Invitrogen Corporation (Paisley, UK). The αIIbβ3 antagonist lotrafiban was a gift from GlaxoSmithKline (King of Prussia, PA, USA). Anti-p110β rabbit polyclonal antibody was obtained from Santa Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). Anti-p110α and anti-p110δ rabbit polyclonal antibodies, recombinant bovine p110α, human p110β and human p110δ originated from the BV laboratory (Ludwig Institute for Cancer Research, London, UK) (20, 36). Anti-phosphotyrosine monoclonal antibody (4G10) was obtained from Upstate Biotechnology Incorporated (Lake Placid, NY, USA). Anti-PLCγ2 and anti-Btk polyclonal antibodies were kindly supplied by Dr. Mike Tomlinson (formerly of DNAX, Palo Alto, CA, USA; presently at The Medical School, University of Birmingham, Birmingham, UK). Other reagents were from previously described sources (4, 37).
Mice
p110δ-/- (mixed 129Sv/C57BL6J strain) and p110δD910A/D910A (backcrossed onto C57BL/6 strain for 10 generations) mice were generated as previously described (38, 39). All mice used in this study were 8-12 weeks of age. Age and sex matched wild-type (wt) mice were used as controls in all experiments. First generation (F1) mice from C57BL/6 × CBA/Ca breedings were used as controls with p110δ-/- mice. Wild-type littermate mice from p110δD910A/+ × p110δD910A/+ breedings were used as controls with the p110δD910A/D910A mice.
Blood collection and preparation of platelets
Blood was collected from mice by cardiac puncture following carbon dioxide asphyxiation and platelets were prepared as previously described (40). Platelets were resuspended in modified Tyrode’s-HEPES buffer pH 7.3 (134 nM NaCl, 2.9 mM KCl, 20 mM HEPES, 12 mM NaHCO3, 1 mM MgCl2, 5 mM glucose) to a final concentration of 2 × 108/mL for aggregation studies and 5 × 108/mL in modified Tyrode’s-HEPES buffer containing 1 mM ethyleneglycotetraacetic acid (EGTA) and 10 μM indomethacin for biochemical analysis.
Aggregation analysis
Stimulation of platelets with various concentrations of CRP, 1 μM ADP and 30 nM PMA was performed in an Born optical aggregometer (Chrono-Log, Havertown, PA, USA) with continuous stirring at 1200 rpm at 37°C. For inhibitor studies, platelets were treated with LY294002 for 10 min or 10 μM lotrafiban for 5 min at room temperature prior to stimulation with CRP.
Cell culture and preparation of soluble cell lysates
Both DAMI and HEL cells were cultured in RPMI containing 10% fetal bovine serum, 2 mM glutamine, 100 U/ml penicillin and 10 μg/ml streptomycin in a humidified incubator at 37°C and 5% CO2. Cells were maintained at an exponential phase of growth. Cells in suspension were resuspended to 1 × 106/ml in Tyrode’s-HEPES buffer pH 7.3 and lysed with an equal volume of ice-cold lysis buffer (2% NP-40, 300 mM NaCl, 20 mM Tris, 10 mM ethylenediaminetetraacetic acid [EDTA], 2 mM Na3V04, 1 mM AEBSF, 10 μg/mL leupeptin, 10 μg/ml aprotinin, and 1 μg/ml pepstatin A, pH 7.4).
Immunoprecipitations and immunoblotting
Platelets were stimulated with 10 μg/ml CRP for 2 min at 37°C with stirring at 1200 rpm. For inhibitor studies, platelets were treated with 25 μM LY294002 for 10 min at room temperature prior to stimulation with CRP. Platelets were then lysed with an equal volume of ice-cold lysis buffer. Cell lysates were precleared with protein A-Sepharose for 30 min at 4°C prior to immunoprecipitations. Sepharose beads and cell debris were removed by centrifugation for 5 min at 13,000 × g at 4°C. Supernatants were collected and PLCγ2 and Btk were immunoprecipitated with the appropriate antibodies. Soluble cell lysates and immunoprecipitates were resolved on 10% sodium dodecylsulfate-polyacrylamide gel electrophoresis (SDS-PAGE) gels, transferred to polyvinylidene difluoride (PVDF) membranes using a semi-dry transfer system (Trans-blot SD, BioRad Laboratories, Hertfordshire, UK) and immunoblotted. Proteins were detected by incubating membranes with enhanced chemiluminescence reagents (Amersham Biosciences, Bucks, UK) or SuperSignal West Femto maximum sensitivity substrate (Pierce, Rockford, IL) followed by exposure to hyperfilm MP (Amersham Biosciences, Buckinghamshire, UK).
Platelet adhesion and spreading under static conditions
Platelets (3 × 107/mL) were allowed to sediment on to glass coverslips coated with either 200 μg/ml fibrinogen or 100 μg/ml collagen and blocked with 1% fatty acid free BSA for 1 h at room temperature. Platelets were allowed to spread on the different surfaces for 30 min at 37°C before fixing with 3.7% paraformaldehyde and permeabilized with 0.2% Triton X-100 in phosphate buffered saline (PBS) (140 mM NaCl, 3 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4 pH 7.4). Following 2 washes with PBS, the fixed platelets were stained for F-actin with rhodamin-conjugated phalloidin for 1 h and then washed 3 more times with PBS before mounting with coverslips and analyzed. Fluorescence was visualized using a Zeiss Axiovert S100 microscope (Zeiss, Oberkochen, Germany) with a monochromatic light source and a charge-coupled device (CCD) camera as previously described. Openlab version 3.0 software (Improvision Ltd, Coventry, UK) was used for image capture and subsequent analysis.
Platelet adhesion under flow conditions
Heparinized whole blood (10 U/mL heparin final concentration) was drawn through 1 × 0.1 mm microslides (Camlab, Cambridge, UK) coated in the presence of either 300 μg/mL collagen, 200 μg/mL fibrinogen, or a combination of 200 μg/mL fibrinogen and 200 μg/mL VWF and blocked with 2% BSA in PBS. Shear rates of 800 s-1 and 1500 s-1 with corresponding flow rates of 0.08 mL/min and 0.15 mL/min were generated by a syringe pump (Harvard Apparatus, Southnatick, MA, USA). After 2 min perfusion with whole blood, modified Tyrodes-HEPES buffer was drawn through the microslides for 8 min at the same shear rate as the blood. Platelet thrombi that had formed on the different surfaces were visualised with an inverted stage videomicroscope system (DM IRB, Leica). Percent surface coverage was quantified using Image-Pro Plus software (Media Cybernetics, Silver Spring, MD, USA). Subsequently, adherent platelets were lysed with ice-cold lysis buffer (as described above) and total protein was quantified with a BioRad DC protein kit (Hertfordshire, UK). Cell lysates were also resolved on 12% SDS-PAGE gels, transferred to PVDF membranes, and immunoblotted with anti-actin monoclonal antibody.
Analysis of data
All experiments were performed 3-5 times and data shown are means ± standard error of the mean (SEM). Statistical analysis was conducted using Student’s unpaired t test. A P value < 0.05 defined significant differences between test groups.
Results
p110δ is expressed in human and mouse platelets
There are contrasting reports on the presence of p110δ catalytic subunit in human and mouse platelets (19, 20, 24-26). To resolve this issue, we investigated the presence of the PI 3-kinase isoform in human and mouse platelets, and in human megakaryocyte cell lines by Western blotting. Recombinant protein and mouse spleen lysates from wt and p110δ-/- platelets were used as controls. A protein band of the correct molecular weight was observed in both human and wt mouse platelet samples (Figure 1A) and, significantly, was absent from platelets and splenocytes from p110δ-/- mice (Figure 1B). As a precaution against the band being due to leukocyte contamination, platelets were isolated from only the top third of human platelet rich plasma, while mouse platelets were prepared from both the top and the bottom thirds of the platelet rich plasma, with similar results being observed in both cases. A similar size protein was also present at low level in the human megakaryocytic cell lines DAMI and HEL. The doublet observed in the human platelet lysate (Figure 1A, lane 1) may represent a splice variant of p110δ, a differentially post-translationally modified form of p110δ, or a degradation product. The most likely explanation is that it is a degradation fragment as p110δ is a notoriously unstable protein (26). These results show that p110δ is expressed in both human and mouse platelets. We also detected a weak p110α signal in human and mouse platelets, whereas a strong band signal was seen for p110β (Figure 1C), confirming that p110β is a prominent PI 3-kinase catalytic subunit present in platelets.
Figure 1.
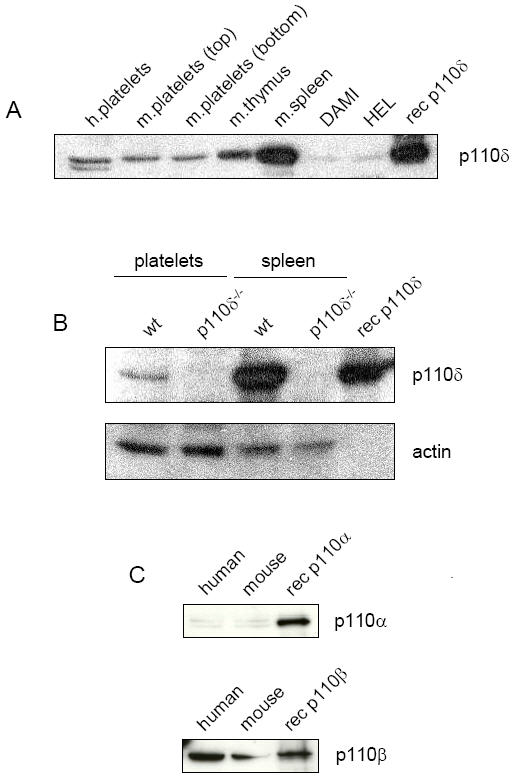
p110δ is expressed in human megakaryocytic cell lines and human and mouse platelets. Soluble cell lysates were prepared of human and mouse platelets, and DAMI and HEL human megakaryocytic cell lines as outlined in Materials and Methods. Lysates were resolved on 10% SDS-PAGE gels then Western blotted as indicated. (A) Human platelets were prepared from the top third of the platelet rich plasma (PRP). Mouse platelets (m.platelets) were prepared from the top third (top) and bottom (bottom) third of the PRP. This was done to demonstrate that the signal was not the result of leukocyte contamination from the buffy coat. Lysates were Western blotted with an anti-p110δ rabbit polyclonal antibody (p110δ) directed against the C-terminal end of the protein. Recombinant human p110δ (rec p110δ) was run in the outside lane as a positive control. (B) Lysates of washed platelets and splenocytes prepared from wt and p110δ-deficient (p110δ-/-) mice were Western blotted with anti-p110δ rabbit polyclonal antibody. The membrane was stripped and reprobed with an anti-actin monoclonal antibody (actin). (C) Human and mouse platelet lysates were Western blotted with either anti-p110α (p110α) or anti-p110β (p110β) rabbit polyclonal antibodies. Recombinant human p110α (rec p110α) and p110β (rec p110β) were included as positive controls. Results are representative of three experiments.
p110δ regulates aggregation to CRP, but not to ADP, thrombin or PMA
Mice lacking p110δ or expressing a catalytically inactive point mutated p110δ (p110δD910A/D910A) do not exhibit any overt bleeding disorder and have normal platelet counts (data not shown). Furthermore, platelets from both mice aggregate normally in response to low and intermediate concentrations of the G protein-coupled receptor agonist ADP and to phorbol ester, which activate protein kinase C (Figure 2A). In contrast, both p110δ-/- and p110δD910A/D910A platelets exhibited slower shape change and decreased rate and amplitude of aggregation in response to a submaximal concentration (0.3 μg/ml) of the GPVI-specific agonist CRP as exemplified in Figure 2B. A similar set of observations were made in six experiments performed alongside aged-matched controls (P < 0.05). Importantly, there were no differences in aggregation between the two sets of wild type controls used in this study. The defect in onset and rate of aggregation in the p110δ-/- mice was also seen with intermediate and high concentrations of CRP, but was less pronounced (Figure 2B). To examine whether the increase in shape change observed in the absence of functional p110δ was due to the reduction in rate of aggregation, studies were performed in the presence of the αIIbβ3 antagonist, lotrafiban. Under these conditions, the response to CRP was reduced, confirming a direct role for p110δ in mediating shape change (Figure 2C). Aggregation induced by collagen was slightly reduced in the absence of functional p110δ, although this effect was lower in magnitude and not statistically significant (data not shown). The absence of an effect on collagen may reflect a greater reliance on ADP and thromboxanes, whose response is not affected by the absence of p110δ, as well as the fact that collagen also binds to the integrin α2β1 which modulates GPVI signalling (41). Maximally-effective concentrations of the general PI 3-kinase inhibitors LY294002 and wortmannin had a considerably more powerfully inhibitory action in murine platelets, as illustrated by the blockade of aggregation to 3 μg/ml CRP (Figure 2D).
Figure 2.
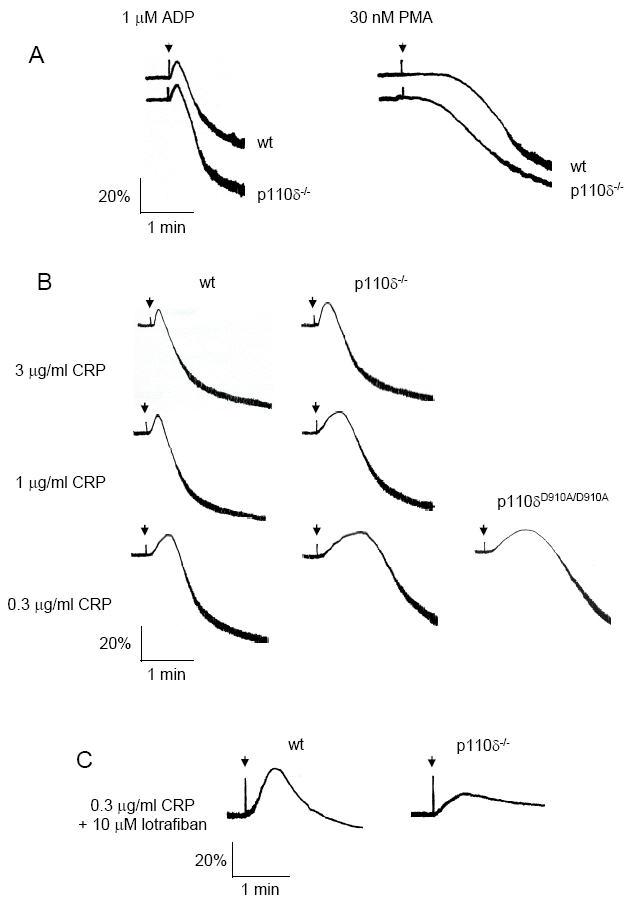
Platelet aggregation responses to various agonists. (A) Washed wt and p110δ-/- platelets (2 × 108/ml) were induced to aggregate with either 1 μM ADP or 30 nM PMA. The extent of aggregation (percent aggregation) was measured as a change in optical density as a function of time. (B) Platelets from wt, p110δ-/- and p110δD910A/D910A mice were stimulated with different concentrations of collagen related peptide (CRP) and platelet aggregation was recorded. (C) Platelets from wt and p110δ-/- mice were pretreated with 10 μM of the αIIbβ3 antagonist lotrafiban prior to stimulation with 0.3 μg/ml CRP. (D) Mouse platelets were pretreated with either 0.1% DMSO (control), 25 μM LY294002 or 100 nM wortmannin prior to stimulation with 3 μg/ml CRP. Wild-type platelets were from F1 offspring from C57BL/6 × CBA/Ca breedings. Results are representative of six experiments.
These results demonstrate that p110δ plays a partial role in signalling by GPVI/FcR γ-chain complex, but not by G protein-coupled receptor agonists. The more pronounced inhibitory action of LY294002 and wortmannin, however, indicates that it is not the major catalytic isoform regulating the formation of 3-phosphorylated inositides.
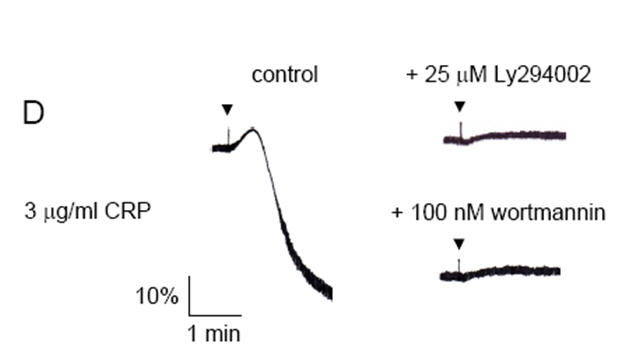
Phosphorylation of PLCγ2 and Btk are not reduced in p110δ-deficient platelets
To investigate the molecular basis of the aggregation defect observed in p110δ-/- platelets, we analyzed the phosphorylation status of several of the main signalling molecules downstream of GPVI/FcR γ-chain. Soluble cell lysates prepared from wt or p110δ-/- platelets stimulated with CRP in the presence of LY294002 and analyzed for tyrosine phosphorylation upon stimulation showed the same qualitative and quantitative band patterns by Western blotting (Figure 3A). These results demonstrate that complete inhibition of PI 3-kinase does not result in a qualitative change in the overall tyrosine kinase activity in CRP-induced platelets. Furthermore, immunoprecipitation and densitometric analysis did not reveal decreases in phosphorylation of PLCγ2 and Btk in p110δ-/- platelets in response to CRP (Figure 3B,C and data not shown). However, a 65% reduction in the tyrosine phosphorylation status of PLCγ2 and a 90% reduction in the tyrosine phosphorylation status of Btk were observed in CRP-induced wild-type platelets in the presence of LY294002 (Figure 3B,C). These reductions in phosphorylation in the presence of LY294002 are consistent with the model in which PI(3,4,5)P3 is required for recruitment of PLCγ2 and Btk to the plasma membrane and that this precedes phosphorylation. The absence of a significant decrease in tyrosine phosphorylation of these PI 3-kinase effector molecules in p110δ deficient platelets in response to CRP suggests that p110δ plays only a minor role in activation of PLCγ2 and Btk in platelets.
Figure 3.
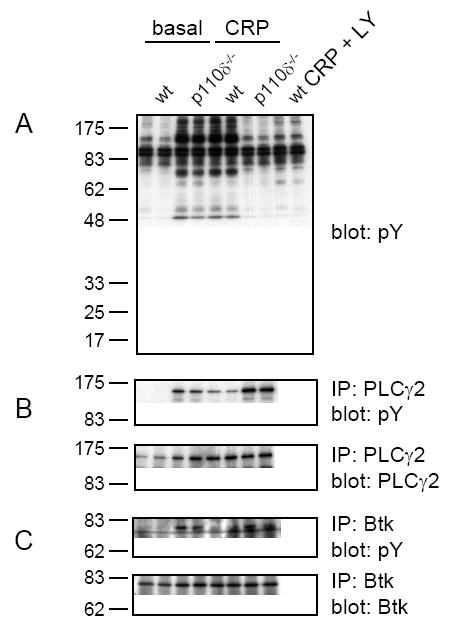
Tyrosine phosphorylation of PLCγ2 and Btk is not reduced in CRP stimulated p110δ-/- platelets. Washed platelets (5 × 108/ml) from wt and p110δ-/- mice were treated with either 0.1% DMSO (basal) or 25 μM LY294002 prior to stimulation with 10 μg/ml CRP for 2 min at 37°C under constant mixing. (A) Soluble cell lysates were resolved on 10% SDS-PAGE gels under reducing conditions then Western blotted (blot) with anti-phosphotyrosine (pY) monoclonal antibody. (B,C) PLCγ2 and Btk were immunoprecipitated (IP) and Western blotted with pY. Membranes were stripped and reprobed with either anti-PLCγ2 (PLCγ2) or anti-Btk (Btk) rabbit polyclonal antibodies. Results are representative of three experiments.
Impaired spreading of p110δ-/- platelets on fibrinogen
In order to investigate the role of p110δ in signalling by fibrinogen, we monitored spreading on a fibrinogen-coated surface and compared this with spreading on collagen. Platelets were allowed to adhere and spread for 30 min then fixed and stained with the actin-specific marker phalloidin labeled with rhodamine. Murine platelets undergo limited spreading on fibrinogen, forming filopodia and limited lamellipodia. In the presence of ADP, more extensive spreading is seen together with formation of stress fibers. Spreading of p110δ-/- platelets on fibrinogen was markedly inhibited, although this was restored in the presence of ADP (Figure 4A and Table 1). In comparison, LY294002 abolished spreading of wt platelets on fibrinogen both in the absence or presence of ADP, confirming previous reports that PI 3-kinase is essential for this response (42). p110δ-/- platelets also exhibited decreased spreading on VWF in the presence of botrocetin, suggesting that p110δ may also play a role in signalling downstream of the GPIb-IX-V complex (data not shown). However, this observation could also be explained by disruption of αIIbβ3 signalling, which is a second receptor for VWF and is essential for this response (43, 44). p110δ-/- platelets spread normally on collagen in the absence and presence of ADP (Figure 4B and Table 1), whereas spreading on collagen was inhibited in murine platelets deficient in the p85α regulatory subunit of PI 3-kinase or in the presence of LY294002 or wortmannin (24).
Figure 4.
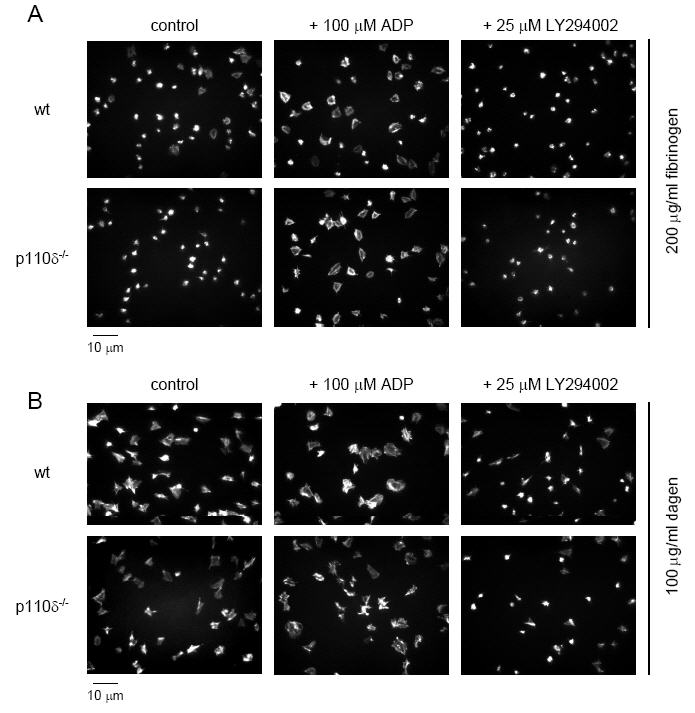
Static adhesion and spreading of platelets on fibrinogen and collagen. Washed wt and p110δ-/- platelets (3 × 107/ml) were placed on slides coated with either 200 μg/ml fibrinogen (A) or 100 μg/ml fibrillar collagen (B) for 30 min at 37°C under static conditions. Some platelets were activated with 100 μM ADP prior to placement on the surfaces, whereas others were treated with 25 μM LY294002. Platelets were fixed, permeabilized, and stained for F-actin with rhodamine-phalloidin. Results are representative of 3-5 experiments.
These results demonstrate a partial role for p110δ in mediating spreading by the integrin αIIbβ3 and possibly also by the GPIb-IX-V complex. However, this role is overcome in the presence of ADP, which activates the p110γ catalytic isoform. On the other hand, a role of p110δ in mediating spreading on collagen was not observed, possibly because activation of PI 3-kinase is not rate limiting in response to high concentrations of ligand (see Discussion).
Adhesion and aggregate formation of p110δ-/- platelets at an intermediate rate of shear
The ability of p110δ-/- platelets in whole blood to adhere to different surfaces at an intermediate rate of shear was investigated. The results were compared to the effect of the general inhibitor of PI 3-kinase, LY294002. In these experiments, a three fold higher concentration of LY294002 was used relative to studies in washed platelets, because of extensive binding of the inhibitor to plasma proteins. Aggregation studies in plasma demonstrated that a three fold higher concentration of LY294002 caused the same blockade of aggregation to CRP as seen in washed platelets.
Heparinized whole blood was collected from mice and flowed through microslides coated with collagen, fibrinogen, or a combination of VWF and fibrinogen for 2 min at 800 s-1 or 1500 s-1. Blood was rinsed from microslides and adherent platelet aggregates were visualized by phase-contrast microscopy. Adhesion and aggregate formation in p110δ-/- platelets on collagen was similar to that seen in wild type cells, both in terms of aggregate morphology and volume (Figure 5A). This was confirmed by analysis of surface coverage (not shown), protein tyrosine phosphorylation and quantitation of protein levels by blotting for actin (Figure 5B). In comparison, aggregate formation on a collagen-coated surface was significantly inhibited in the presence of LY294002, demonstrating that PI 3-kinase is necessary for platelet aggregation at an intermediate rate of flow (Figure 5C), as previously shown (45). Platelet adhesion on surfaces coated with VWF, or VWF and fibrinogen was unaltered for p110δ-/- platelets or in the presence of LY294002. Neither of these surfaces supported aggregate formation under the conditions used. The present results demonstrate that p110δ plays a negligible role in supporting adhesion and aggregate formation at an intermediate/high rate of flow on a variety of surfaces.
Figure 5.
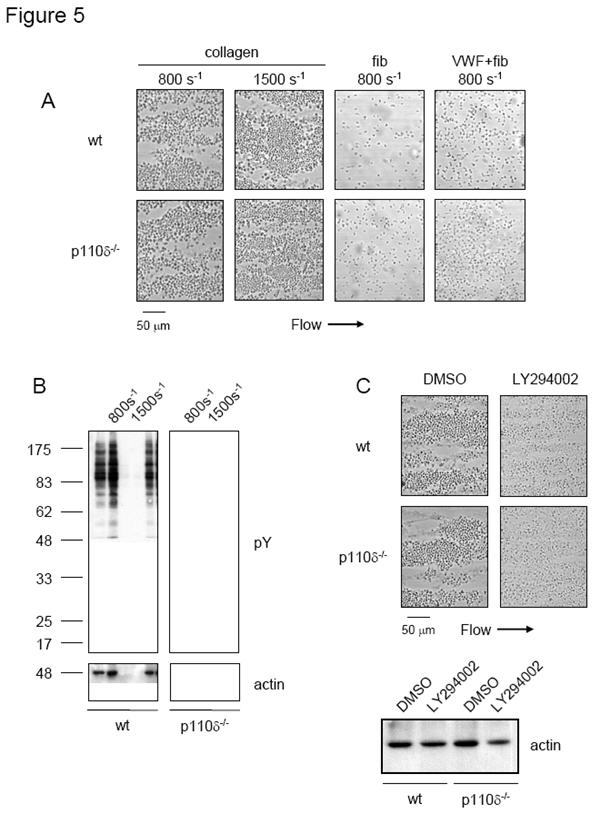
p110δ-/- platelets adhere and aggregate normally to collagen, fibrinogen, and VWF under intermediate flow conditions. (A) Heparinized blood from wt and p110δ-/- mice was flowed through microslides coated overnight in the presence of 300 μg/ml fibrillar collagen (col), 200 μg/ml fibrinogen (fib), or a mixture of 200 μg/ml VWF and 200 μg/ml fibrinogen (VWF + fib) at 800 s-1 and 1500 s-1 for 2 minutes. (B) Adherent platelets from (A) were lysed with 1% NP-40; proteins were resolved on 10% SDS-PAGE gels under reducing conditions and Western blotted with anti-phosphotyrosine (pY) monoclonal antibody. Membranes were stripped and reprobed with anti-actin (actin) monoclonal antibody. (C) Heparinized blood from wt mice was pretreated with 75 μM LY294002 for 5 min prior to being flowed through microslides coated with 300 μg/ml fibrillar collagen at 800 s-1 for 2 minutes. Adherent platelets were lysed and Western blotted with actin. Results are reprentative of three experiments.
Discussion
The primary aim of this study was to investigate the role of the PI 3-kinase catalytic subunit p110δ in platelet function. p110δ was shown to be expressed in human megakaryocytic cell lines, and in human and mouse platelets. Functionally, p110δ plays a minor role in signalling downstream of the GPVI/FcR γ-chain complex, αIIbβ3 and possibly GPIb-IX-V. In contrast, wt platelets pretreated with the general PI 3-kinase inhibitor LY294002 and p85α-deficient platelets show more severe activation defects (24). These results demonstrate that one of the other isoforms of Class IA catalytic subunits of PI 3-kinase, most notably p110β, which is expressed in high levels in platelets, appear to be more important in regulating platelet function than p110δ. Confirmation of this is hampered by the fact that targeted ablation of either p110α or p110β catalytic subunits results in embryonic lethality (30, 31).
Binding of collagen or CRP to GPVI activates a tyrosine kinase-based signalling cascade that can be considered as a “hybrid” of the B and T cell receptor (BCR and TCR, respectively) signalling pathways (46). ITAM domains present in the cytoplasmic tails of all three receptors become tyrosine phoshorylated by Src family kinases and act as docking sites for the tyrosine kinase Syk, which goes on to phosphorylate a variety of effector molecules. Two critical events common to signalling downstream of BCR, TCR, and GPVI receptors are activation of PLCγ and class IA PI 3-kinases. Platelets lacking PLCγ2 show decreased Ca2+ mobilization, secretion, aggregation, adhesion, and aggregate formation under flow in response to collagen or CRP (9). PLCγ2-/- platelets also exhibited impaired spreading on fibrinogen (10). Inhibition of PI 3-kinases with pharmacologic inhibitors or through deletion of the p85α class IA regulatory subunit causes similar, but less severe decreases in responsiveness to those observed in the absence of PLCγ2, consistent with the observation that PI 3-kinases function lies upstream of PLCγ2 (11, 24, 47, 48).
The p110δ isoform of PI 3-kinase has recently been shown to play a non-redundant role in B cell development and function (38, 39, 49). Knockout mouse models of p110δ generated by two separate groups using different gene-targeting strategies exhibited severely impaired BCR signalling function (38, 49). p110δ function was shown to be essential for BCR-mediated Ca2+ flux, and activation of PLCγ2, Btk, and PKB. The bulk of PI 3,4,5P3 production downstream of the BCR was due to p110δ activity rather than p110α and p110β, which were expressed at normal levels in the absence of p110δ.
Reduced spreading of p110δ-/- platelets on fibrinogen suggests that p110δ is also involved in outside-in signalling from αIIbβ3. Vav family of GDP/GTP exchange factors and PLCγ2 are regulated by PI 3-kinase and are implicated in mediating spreading by αIIbβ3 as discussed in the introduction (10, 15, 16, 50). The observation that spreading of p110δ-/- platelets on collagen was unaltered, however, was unexpected, in view of the defect in spreading in p85α-/- platelets (24) and the impairment in aggregation observed in p110δ-/- platelets. This suggests that activation of p110δ is not rate-limiting for this response. This possibility is supported by the observation that the spreading defect displayed by p110δ-/- platelets on fibrinogen was overcome by pretreating platelets with ADP, which activates the class IB PI 3-kinase catalytic subunit p110γ via the Gi-coupled receptor, P2Y12 (25). This indicates that under conditions of strong PI 3-kinase activation, the isoform of PI 3-kinase, which provides 3-phosphorylated lipids is not critical. The lack of a bleeding defect in the p110δ-/- mice and normal aggregate formation ex vivo on various surfaces is probably due to activation of p110γ and p110β isoforms by ECM proteins and by release of ADP, which activates p110γ.
The present observations suggest a minor role for p110δ in supporting mouse platelet activation in comparison to the more profound reduction seen in the presence of LY294002 or wortmannin in CRP-stimulated human platelets. It is unclear whether the relatively minor role of p110δ is due to a low level of expression relative to other isoforms, notably p110β, or because of redundancy between isoforms. In this context, it is of future interest to compare the present results to those induced by ablation of the other two catalytic subunits, p110α and p110β, as well as in platelets deficient in any two or all three of these isoforms. In this context, it is noteworthy, for example, to compare the minor phenotype of Tec-deficient platelets (40), with the more pronounced role that is seen in the absence of Btk, or the negligible phenotypes of Vav1 and Vav3 deficient platelets with that seen in the absence of both GDP/GTP exchange factors (16).
In conclusion, we have demonstrated expression of p110δ in human megakaryocytic cell lines and in both human and mouse platelets. We have also shown that p110δ plays a partial role in platelet aggregation and spreading. The small contribution of p110δ to platelet function may be due to a low level of expression in platelets relative to other isoforms, notably p110β, although redundancy may also be an important factor.
Acknowledgments
We would like to thank Dr. Owen McCarty for assisting with the image analysis.
Footnotes
Support: This work is supported by the British Heart Foundation (BHF) and the Wellcome Trust. YAS holds a BHF Research Fellowship. JMA holds a BHF Studentship. SPW holds a BHF Professorship. Research in the laboratory of BV is supported by the Ludwig Institute for Cancer Research, the Biotechnology and Biological Sciences Research Council and the European Union Fifth Framework Programme QLG1-2001-02171.
References
- 1.Quek LS, Pasquet JM, Hers I, Cornall R, Knight G, Barnes M, et al. Fyn and Lyn phosphorylate the Fc receptor gamma chain downstream of glycoprotein VI in murine platelets, and Lyn regulates a novel feedback pathway. Blood. 2000;96(13):4246–53. [PubMed] [Google Scholar]
- 2.Jackson SP, Schoenwaelder SM, Yuan Y, Rabinowitz I, Salem HH, Mitchell CA. Adhesion receptor activation of phosphatidylinositol 3-kinase. von Willebrand factor stimulates the cytoskeletal association and activation of phosphatidylinositol 3-kinase and pp60c-src in human platelets. J Biol Chem. 1994;269(43):27093–9. [PubMed] [Google Scholar]
- 3.Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci U S A. 2003;100(23):13298–302. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Poole A, Gibbins JM, Turner M, van Vugt MJ, van de Winkel JG, Saito T, et al. The Fc receptor gamma-chain and the tyrosine kinase Syk are essential for activation of mouse platelets by collagen. Embo J. 1997;16(9):2333–41. doi: 10.1093/emboj/16.9.2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suzuki-Inoue K, Wilde JI, Andrews RK, Auger JM, Siraganian RP, Sekiya F, et al. Glycoproteins VI and Ib-IX-V stimulate tyrosine phosphorylation of tyrosine kinase Syk and phospholipase Cgamma2 at distinct sites. Biochem J. 2004;378(Pt 3):1023–9. doi: 10.1042/BJ20031430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Woodside DG, Obergfell A, Leng L, Wilsbacher JL, Miranti CK, Brugge JS, et al. Activation of Syk protein tyrosine kinase through interaction with integrin beta cytoplasmic domains. Curr Biol. 2001;11(22):1799–804. doi: 10.1016/s0960-9822(01)00565-6. [DOI] [PubMed] [Google Scholar]
- 7.Blake RA, Schieven GL, Watson SP. Collagen stimulates tyrosine phosphorylation of phospholipase C-gamma 2 but not phospholipase C-gamma 1 in human platelets. FEBS Lett. 1994;353(2):212–6. doi: 10.1016/0014-5793(94)01037-4. [DOI] [PubMed] [Google Scholar]
- 8.Mangin P, Yuan Y, Goncalves I, Eckly A, Freund M, Cazenave JP, et al. Signaling role for phospholipase C gamma 2 in platelet glycoprotein Ib alpha calcium flux and cytoskeletal reorganization. Involvement of a pathway distinct from FcR gamma chain and Fc gamma RIIA. J Biol Chem. 2003;278(35):32880–91. doi: 10.1074/jbc.M302333200. [DOI] [PubMed] [Google Scholar]
- 9.Suzuki-Inoue K, Inoue O, Frampton J, Watson SP. Murine GPVI stimulates weak integrin activation in PLCgamma2-/-platelets: involvement of PLCgamma1 and PI3-kinase. Blood. 2003;102(4):1367–73. doi: 10.1182/blood-2003-01-0029. [DOI] [PubMed] [Google Scholar]
- 10.Wonerow P, Pearce AC, Vaux DJ, Watson SP. A critical role for phospholipase Cgamma2 in alphaIIbbeta3-mediated platelet spreading. J Biol Chem. 2003;278(39):37520–9. doi: 10.1074/jbc.M305077200. [DOI] [PubMed] [Google Scholar]
- 11.Pasquet JM, Bobe R, Gross B, Gratacap MP, Tomlinson MG, Payrastre B, et al. A collagen-related peptide regulates phospholipase Cgamma2 via phosphatidylinositol 3-kinase in human platelets. Biochem J. 1999;342(Pt 1):171–7. [PMC free article] [PubMed] [Google Scholar]
- 12.Ji P, Haimovich B. Integrin alpha IIb beta 3-mediated pp125FAK phosphorylation and platelet spreading on fibrinogen are regulated by PI 3-kinase. Biochim Biophys Acta. 1999;1448(3):543–52. doi: 10.1016/s0167-4889(98)00160-8. [DOI] [PubMed] [Google Scholar]
- 13.Asazuma N, Wilde JI, Berlanga O, Leduc M, Leo A, Schweighoffer E, et al. Interaction of linker for activation of T cells with multiple adapter proteins in platelets activated by the glycoprotein VI-selective ligand, convulxin. J Biol Chem. 2000;275(43):33427–34. doi: 10.1074/jbc.M001439200. [DOI] [PubMed] [Google Scholar]
- 14.Obergfell A, Judd BA, del Pozo MA, Schwartz MA, Koretzky GA, Shattil SJ. The molecular adapter SLP-76 relays signals from platelet integrin alphaIIbbeta3 to the actin cytoskeleton. J Biol Chem. 2001;276(8):5916–23. doi: 10.1074/jbc.M010639200. [DOI] [PubMed] [Google Scholar]
- 15.Pearce AC, Wilde JI, Doody GM, Best D, Inoue O, Vigorito E, et al. Vav1, but not Vav2, contributes to platelet aggregation by CRP and thrombin, but neither is required for regulation of phospholipase C. Blood. 2002;100(10):3561–9. doi: 10.1182/blood.V100.10.3561. [DOI] [PubMed] [Google Scholar]
- 16.Pearce AC, Vigorito E, Senis YA, Billadeau DD, Watson SP, Turner M. Impaired platelet activation by GPVI in mice deficient in both Vav1 and Vav3. Blood. 2003;102(11):208a. [Google Scholar]
- 17.Vanhaesebroeck B, Leevers SJ, Ahmadi K, Timms J, Katso R, Driscoll PC, et al. Synthesis and function of 3-phosphorylated inositol lipids. Annu Rev Biochem. 2001;70:535–602. doi: 10.1146/annurev.biochem.70.1.535. [DOI] [PubMed] [Google Scholar]
- 18.Vanhaesebroeck B, Waterfield MD. Signaling by distinct classes of phosphoinositide 3-kinases. Exp Cell Res. 1999;253(1):239–54. doi: 10.1006/excr.1999.4701. [DOI] [PubMed] [Google Scholar]
- 19.Chantry D, Vojtek A, Kashishian A, Holtzman DA, Wood C, Gray PW, et al. p110delta, a novel phosphatidylinositol 3-kinase catalytic subunit that associates with p85 and is expressed predominantly in leukocytes. J Biol Chem. 1997;272(31):19236–41. doi: 10.1074/jbc.272.31.19236. [DOI] [PubMed] [Google Scholar]
- 20.Vanhaesebroeck B, Welham MJ, Kotani K, Stein R, Warne PH, Zvelebil MJ, et al. P110delta, a novel phosphoinositide 3-kinase in leukocytes. Proc Natl Acad Sci U S A. 1997;94(9):4330–5. doi: 10.1073/pnas.94.9.4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Inukai K, Anai M, Van Breda E, Hosaka T, Katagiri H, Funaki M, et al. A novel 55-kDa regulatory subunit for phosphatidylinositol 3-kinase structurally similar to p55PIK Is generated by alternative splicing of the p85alpha gene. J Biol Chem. 1996;271(10):5317–20. doi: 10.1074/jbc.271.10.5317. [DOI] [PubMed] [Google Scholar]
- 22.Zhang J, Shattil SJ, Cunningham MC, Rittenhouse SE. Phosphoinositide 3-kinase gamma and p85/phosphoinositide 3-kinase in platelets. Relative activation by thrombin receptor or beta-phorbol myristate acetate and roles in promoting the ligand-binding function of alphaIIbbeta3 integrin. J Biol Chem. 1996;271(11):6265–72. doi: 10.1074/jbc.271.11.6265. [DOI] [PubMed] [Google Scholar]
- 23.Tang X, Downes CP. Purification and characterization of Gbetagamma-responsive phosphoinositide 3-kinases from pig platelet cytosol. J Biol Chem. 1997;272(22):14193–9. doi: 10.1074/jbc.272.22.14193. [DOI] [PubMed] [Google Scholar]
- 24.Watanabe N, Nakajima H, Suzuki H, Oda A, Matsubara Y, Moroi M, et al. Functional phenotype of phosphoinositide 3-kinase p85alpha-null platelets characterized by an impaired response to GP VI stimulation. Blood. 2003;102(2):541–8. doi: 10.1182/blood-2002-11-3327. [DOI] [PubMed] [Google Scholar]
- 25.Hirsch E, Bosco O, Tropel P, Laffargue M, Calvez R, Altruda F, et al. Resistance to thromboembolism in PI3Kgamma-deficient mice. Faseb J. 2001;15(11):2019–21. doi: 10.1096/fj.00-0810fje. [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Vanhaesebroeck B, Rittenhouse SE. Human platelets contain p110delta phosphoinositide 3-kinase. Biochem Biophys Res Commun. 2002;296(1):178–81. doi: 10.1016/s0006-291x(02)00744-1. [DOI] [PubMed] [Google Scholar]
- 27.Vanhaesebroeck B, Leevers SJ, Panayotou G, Waterfield MD. Phosphoinositide 3-kinases: a conserved family of signal transducers. Trends Biochem Sci. 1997;22(7):267–72. doi: 10.1016/s0968-0004(97)01061-x. [DOI] [PubMed] [Google Scholar]
- 28.Zhang J, Benovic JL, Sugai M, Wetzker R, Gout I, Rittenhouse SE. Sequestration of a G-protein beta gamma subunit or ADP-ribosylation of Rho can inhibit thrombin-induced activation of platelet phosphoinositide 3-kinases. J Biol Chem. 1995;270(12):6589–94. doi: 10.1074/jbc.270.12.6589. [DOI] [PubMed] [Google Scholar]
- 29.Stephens LR, Eguinoa A, Erdjument-Bromage H, Lui M, Cooke F, Coadwell J, et al. The G beta gamma sensitivity of a PI3K is dependent upon a tightly associated adaptor, p101. Cell. 1997;89(1):105–14. doi: 10.1016/s0092-8674(00)80187-7. [DOI] [PubMed] [Google Scholar]
- 30.Bi L, Okabe I, Bernard DJ, Wynshaw-Boris A, Nussbaum RL. Proliferative defect and embryonic lethality in mice homozygous for a deletion in the p110alpha subunit of phosphoinositide 3-kinase. J Biol Chem. 1999;274(16):10963–8. doi: 10.1074/jbc.274.16.10963. [DOI] [PubMed] [Google Scholar]
- 31.Bi L, Okabe I, Bernard DJ, Nussbaum RL. Early embryonic lethality in mice deficient in the p110beta catalytic subunit of PI 3-kinase. Mamm Genome. 2002;13(3):169–72. doi: 10.1007/BF02684023. [DOI] [PubMed] [Google Scholar]
- 32.Fruman DA, Snapper SB, Yballe CM, Davidson L, Yu JY, Alt FW, et al. Impaired B cell development and proliferation in absence of phosphoinositide 3-kinase p85alpha. Science. 1999;283(5400):393–7. doi: 10.1126/science.283.5400.393. [DOI] [PubMed] [Google Scholar]
- 33.Terauchi Y, Tsuji Y, Satoh S, Minoura H, Murakami K, Okuno A, et al. Increased insulin sensitivity and hypoglycaemia in mice lacking the p85 alpha subunit of phosphoinositide 3-kinase. Nat Genet. 1999;21(2):230–5. doi: 10.1038/6023. [DOI] [PubMed] [Google Scholar]
- 34.Morton LF, Hargreaves PG, Farndale RW, Young RD, Barnes MJ. Integrin alpha 2 beta 1-independent activation of platelets by simple collagen-like peptides: collagen tertiary (triple-helical) and quaternary (polymeric) structures are sufficient alone for alpha 2 beta 1-independent platelet reactivity. Biochem J. 1995;306(Pt 2):337–44. doi: 10.1042/bj3060337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Asselin J, Knight CG, Farndale RW, Barnes MJ, Watson SP. Monomeric (glycine-proline-hydroxyproline)10 repeat sequence is a partial agonist of the platelet collagen receptor glycoprotein VI. Biochem J. 1999;339(Pt 2):413–8. [PMC free article] [PubMed] [Google Scholar]
- 36.Vanhaesebroeck B, Jones GE, Allen WE, Zicha D, Hooshmand-Rad R, Sawyer C, et al. Distinct PI(3)Ks mediate mitogenic signalling and cell migration in macrophages. Nat Cell Biol. 1999;1(1):69–71. doi: 10.1038/9045. [DOI] [PubMed] [Google Scholar]
- 37.Pasquet JM, Quek L, Stevens C, Bobe R, Huber M, Duronio V, et al. Phosphatidylinositol 3,4,5-trisphosphate regulates Ca(2+) entry via btk in platelets and megakaryocytes without increasing phospholipase C activity. Embo J. 2000;19(12):2793–802. doi: 10.1093/emboj/19.12.2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Clayton E, Bardi G, Bell SE, Chantry D, Downes CP, Gray A, et al. A crucial role for the p110delta subunit of phosphatidylinositol 3-kinase in B cell development and activation. J Exp Med. 2002;196(6):753–63. doi: 10.1084/jem.20020805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Okkenhaug K, Bilancio A, Farjot G, Priddle H, Sancho S, Peskett E, et al. Impaired B and T cell antigen receptor signaling in p110delta PI 3-kinase mutant mice. Science. 2002;297(5583):1031–4. doi: 10.1126/science.1073560. [DOI] [PubMed] [Google Scholar]
- 40.Atkinson BT, Ellmeier W, Watson SP. Tec regulates platelet activation by GPVI in the absence of Btk. Blood. 2003;102(10):3592–9. doi: 10.1182/blood-2003-04-1142. [DOI] [PubMed] [Google Scholar]
- 41.Inoue O, Suzuki-Inoue K, Dean WL, Frampton J, Watson SP. Integrin alpha2beta1 mediates outside-in regulation of platelet spreading on collagen through activation of Src kinases and PLCgamma2. J Cell Biol. 2003;160(5):769–80. doi: 10.1083/jcb.200208043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Heraud JM, Racaud-Sultan C, Gironcel D, Albiges-Rizo C, Giacomini T, Roques S, et al. Lipid products of phosphoinositide 3-kinase and phosphatidylinositol 4′,5′-bisphosphate are both required for ADP-dependent platelet spreading. J Biol Chem. 1998;273(28):17817–23. doi: 10.1074/jbc.273.28.17817. [DOI] [PubMed] [Google Scholar]
- 43.Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate-receptor interactions in platelet thrombus formation under flow. Cell. 1998;94(5):657–66. doi: 10.1016/s0092-8674(00)81607-4. [DOI] [PubMed] [Google Scholar]
- 44.Mekrache M, Legendre P, Kieffer N, Baruch D. Activation of integrin alphaIIbbeta3 expressed in Chinese hamster ovary cells is required for interaction with solid-phase von Willebrand factor. Br J Haematol. 2002;119(4):1024–32. doi: 10.1046/j.1365-2141.2002.03960.x. [DOI] [PubMed] [Google Scholar]
- 45.Yap CL, Anderson KE, Hughan SC, Dopheide SM, Salem HH, Jackson SP. Essential role for phosphoinositide 3-kinase in shear-dependent signaling between platelet glycoprotein Ib/V/IX and integrin alpha(IIb)beta(3) Blood. 2002;99(1):151–8. doi: 10.1182/blood.v99.1.151. [DOI] [PubMed] [Google Scholar]
- 46.Watson SP, Asazuma N, Atkinson B, Berlanga O, Best D, Bobe R, et al. The role of ITAM- and ITIM-coupled receptors in platelet activation by collagen. Thromb Haemost. 2001;86(1):276–88. [PubMed] [Google Scholar]
- 47.Falasca M, Logan SK, Lehto VP, Baccante G, Lemmon MA, Schlessinger J. Activation of phospholipase C gamma by PI 3-kinase-induced PH domain-mediated membrane targeting. Embo J. 1998;17(2):414–22. doi: 10.1093/emboj/17.2.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Gratacap MP, Payrastre B, Viala C, Mauco G, Plantavid M, Chap H. Phosphatidylinositol 3,4,5-trisphosphate-dependent stimulation of phospholipase C-gamma2 is an early key event in FcgammaRIIA-mediated activation of human platelets. J Biol Chem. 1998;273(38):24314–21. doi: 10.1074/jbc.273.38.24314. [DOI] [PubMed] [Google Scholar]
- 49.Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Wang D, et al. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22(24):8580–91. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Obergfell A, Eto K, Mocsai A, Buensuceso C, Moores SL, Brugge JS, et al. Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol. 2002;157(2):265–75. doi: 10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]