

Abstract
The stepwise movements generated by myosin, dynein, and kinesin were observed in living cells in an attempt to understand the molecular mechanisms of movement within cells. First, the sequential process of the transport of vesicles, including human epidermal factor 2 receptor, after endocytosis was observed for long periods in three dimensions using quantum dots (QDs) and a three-dimensional confocal microscope. QD vesicles, after being endocytosed into the cells, moved along the membrane by transferring actin filaments and were then rapidly transported toward the nucleus along microtubules. Second, the position of vesicles was detected with a precision up to 1.9 nm and 330 μs using a new two-dimensional tracking method. The movement of the QDs transported by myosin VI lying just beneath the cell membrane consisted of 29- and 15-nm steps with a transition phase between these two steps. QD vesicles were then transported toward the nucleus or away from the nucleus toward the cell membrane with successive 8-nm steps. The stepwise movements of these motor proteins in cells were observed using new imaging methods that allowed the molecular mechanisms underlying traffic to and from the membrane to be determined.
INTRODUCTION
Motor proteins such as myosin, kinesin, and dynein play roles in cell motility, such as vesicle transport, mitosis, and muscle contraction (1,2). To understand the molecular mechanisms of this motility, an in vitro motility assay employing motor proteins and methods for detecting single molecules of purified motor proteins was developed using optical tweezers and fluorescence molecules (3–8). Single kinesin-1 moves processively using 8-nm steps at a maximum velocity of ∼900 nm/s at room temperature and produces forces of ∼7 pN (7). Dynein also proceeds using 8-nm steps and produces forces of ∼7 pN (9,10). Myosin V and VI move processively with step sizes of ∼36 and ∼30 nm, respectively, at a maximum velocity of 300–600 nm/s and produce forces of 2–3 pN (11–14). The movements of kinesin, dynein, and myosin V and VI have been explained by a “hand-over-hand” model (8,10,15–18).
It is not known if these detailed molecular functions operate in living cells. Physiological conditions in the cell environment are very different from those in in vitro assays where the experiments are performed; for example, the ion composition is different in the cells and the assays cells have adaptor proteins and cytoskeletal networks that are not present in the assays. Therefore, it is crucial to measure the molecular functions of motor proteins in cells to understand the molecular mechanisms of vesicle transport by motor proteins. The movement of kinesin in cells can be detected with nanometer accuracy using a cross correlation analysis of the differential interference contrast of the image (19). Recently, the transport of peroxisomes in cells has been detected with nanometer accuracy using GFP-labeled peroxisomes in Drosophila S2 cells (20). The 8-nm steps of presumably dynein during inward transport have been detected using “fluorescence imaging with one-nanometer accuracy” (FIONA) methods (8,20). Green fluorescent protein (GFP) is a very useful protein to investigate molecular mechanisms in cells. However, the bleaching of the fluorescence signal of single GFP molecules in cells is rapid, making it unsuitable for long-term observations with FIONA (16,17,21,22). The bright and stable fluorescence particles, quantum dots (QDs), are useful for detecting the position of the proteins with a high precision for long periods of time because QDs take longer to photobleach than GFP (10,18,23–26).
Vesicle transport is an excellent model for exploring motor function in cells, because both actin- and microtubule-based motors are involved in the transport. For example, endocytosis of a membrane protein, such as human epidermal growth factor receptor 1 (HER1), is a multistep process involving several different motor proteins. The endocytosis is initiated by the clustering of clathrin, dynamin, and many other proteins triggered by a ligand binding to HER1 (27). The vesicle then removes the clathrin coat, and is transported via an actin-based motor, myosin VI (28,29). The vesicle is then switched to the microtubule-based motor and transported toward the nucleus by dynein and away from the nucleus by kinesin (1,30). Some endocytic vesicles reattach to the membrane in what is known as the recycling circuit (31–33). The recycling vesicles could be transported via myosin V (34,35).
Endocytosis of human epidermal growth factor receptor 2 (HER2), one of the HER family, simplifies the experimental setup because HER2 does not have a ligand binding step (36,37). Therefore, the fluorescence labeled antibody delivers the vesicle and/or switching to the motor proteins in a living cell without using a ligand (24,25). Furthermore, there is special antibody against HER2 (Trastuzumab), which inhibits the recycling circuit (38,39). The signaling pathway can be greatly simplified if the recycling of the endosome is excluded. This allows the actual steps involved in the transport processes to be separated from the fission process. In this study, the movements of motor proteins in a cell need to be visualized for extended periods of time. To achieve this, stable and intense QDs were bound to anti-HER2 antibody, which were then endocytosed into breast cancer cells in which HER2 had been overexpressed on the cell membrane (31,39,40). The QDs containing vesicle could be detected for periods of time up to 330 μs and at a spatial precision of ∼2 nm using a newly developed analytical method with a higher temporal resolution without a reduction in the observation area. The stepwise movements and velocities of myosin VI, dynein, and kinesin reported in purified proteins were confirmed in this cell line. An unexpected transition between the small and large step sizes produced by myosin VI in vesicle transport in a living cell was observed only in these cells.
MATERIALS AND METHODS
Preparation of anti-HER2-QDs
The fluctuation of the fluorescent intensity of a single QD caused by the blinking was obvious (Supplementary Fig. S1 A, Supplementary Material). We could not analyze the position of a single QD when the QD was in the nonfluorescent state. The number of nonfluorescent single QDs was minimized by condensing multiple QD into a single one similar to the previous study (10) (Fig. S1, B–D). Multiple QDs were cross-linked by ethylene-diamine-carbodiimide (EDC) using anti-HER2 antibody. The procedure to make the condensed QD via anti-HER2 antibody (anti-HER2-QD) is described as follows: 50 μl of 2 μM QD solution (emission 605 nm, Evitag; Evidot, Evident Technologies, Troy, NY), 450 μl of 50 mM MES (pH 5.0) and 500 μl of 20 mg/ml EDC (Pierce, Rockwell, IL) in 50 mM MES (pH 5.0) were mixed and incubated for 5 min at 25°C. After centrifugation (330,000 × g, 20 min, 25°C), the pellet was suspended in 160 μl of a 250-mM HEPES (pH 8.0) solution. Then, 40 μl of a 1-mg/ml solution of anti-HER2 (Trastuzumab; Genetics, Chugai Pharmaceuticals, Tokyo, Japan) was added to the solution, and incubated for 30 min at 25°C. After the addition of 200 μl of 50 mg/ml bovine serum albumin (BSA), the solution was cooled to 4°C. To remove the free anti-HER2, the QDs were rinsed three times in 200 μl of 100 mM HEPES and 5 mg/ml BSA using centrifugation (330,000 × g, 20 min, 4°C). The anti-HER2 coated QDs were resuspended in 100 mM HEPES (pH 8.0). The supernatant was used after centrifuging at 40,000 × g for 3 min at 4°C.
Culturing of KPL4 cells and drug treatment
Human breast cancer cells, KPL-4, in which HER2 was overexpressed on the cell membrane, were cultured in Dulbecco's modified eagle's medium (DMEM) (GIBCO, Carlsbad, CA) medium with 5% fetal bovine serum (FBS) (GIBCO) at 37°C in 5% CO2. One hour before the QD experiment, the culture medium was exchanged for DMEM without FBS. Cells were then incubated for 2 h in DMEM medium containing 0.2 nM anti-HER2-QD. After washing the free anti-HER2-QDs from the medium, the cell was incubated for 2–12 h in DMEM medium containing 10% FBS. The QD experiments were performed at 30°C or 20°C on the microscope. To depolymerize the microtubules, the cells were incubated in a medium containing 40 μM nocodazole for 10 min just before the experiment. To depolymerize the actin filaments, the cells were incubated in a medium containing 10 μM cytochalasin D for 10 min before the experiments. Nocodazole or cytochalasin D was present in the medium throughout the experiments.
Immunofluorescence microscopy
KPL-4 cells were stained and fixed using a previously described method (41). Cells that had been fixed were treated with phalloidine-rhodamine (Molecular Probe, Eugene, OR), anti-tubulin (Molecular Probe A11126), and anti-rabbit immunoglobulin G-fluorescein isothiocyanate (IgG-FITC) (Sigma F7512, St. Louis, MO). Immunofluorescence images were taken under a confocal microscope (Pascal, Zeiss). Cells which endocytosed anti-HER2-QDs were fixed and then reacted with anti-myosin VI (Sigma M5187) and anti-rabbit IgG-FITC. Cells were observed under an objective-type total internal reflection fluorescent (TIRF) microscope (5).
Three-dimensional single particle tracking using a confocal microscope
The optical system for the three-dimensional (3D) observation of the vesicles consisted of an epi-fluorescent microscope (IX-71, Olympus, Tokyo, Japan), a Nipkow lens type confocal unit (CSU10, Yokokawa, Tokyo, Japan), and an electron multiplier type charge-coupled device (CCD) camera (EM-CCD, Ixon DV887, Andor Technology, Belfast, Ireland) (Supplementary Fig. S2 A). The objective lens (60× PlanApo, 1.45 NA, oil, Olympus) was moved by a Piezo actuator (FL301, Nanocontrol, Tokyo, Japan) with a feedback loop (NS202, Nanocontrol). A computer controlled the Piezo actuator that was used to synchronize the image acquisitions. This ensured that the object lens stayed within the exposure time of the CCD camera (Supplementary Fig. S2 B). An area ∼30 × 30 μm2 was illuminated by a green laser (532 nm, ∼0.01 mW/μm2 for a specimen, Crystalaser, Reno, NV).
The FIONA method was modified so as to observe fluorescent particles in a living cell (8). Fig. 1 A shows the fluorescence images of a single QD fixed on the glass surface at various focal positions. The intensity of the image decreased independently of the distance of the just-in-focus position. The distribution of the intensity along the focal axis followed a Gaussian distribution (Fig. 1 B). Therefore, the three-dimensional position of the fluorophore was calculated by fitting the fluorescent images at various focal positions with a three-dimensional Gaussian function according to the equation,
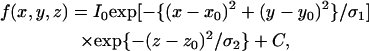 |
where I0 is the peak intensity, (x0, y0, z0) are the positions of the fluorophore, and C is the background intensity.
FIGURE 1.

Three-dimensional single particle tracking. (A) Image set of an optical section of a QD at various focal positions. Upper panels are the fluorescence images of the QD at the various focal positions. Lower panels show the 3D expressions of the intensity profiles of the upper panels. (B) Relationship between the focal (Z) position and the fluorescence intensity. Black line is a single Gaussian curve fitted to the intensities. The peak occurred at 0.1 μm. (C) Spatial precision of 3D single particle tracking. Standard deviations of tracking using 3D single particle tracking were 6 nm (XY-plane) and 45 nm (Z axis). (D) Precision of the Z-direction determined using 3D single particle tracking. QDs bound on a coverslip were moved 100 nm along the focal axis by a Piezo actuator. The movement of the QDs was tracked using 3D single particle tracking in three dimensions. Only the Z-position of the QD is shown here.
The standard deviations of the position of single QD, tracked by 3D single particle tracking, were 6 nm for the XY-plane and 45 nm for the Z axis (Fig. 1 C). The vibration of the objective and/or the glass surface during scanning caused a reduction in the precision of the Z axis. Artificial 100-nm steps of the QD along the Z axis could be clearly observed with 3D single particle tracking (Fig. 1 D).
Binning FIONA
The optical system for two-dimensional (2D) observations consisted of an epi-fluorescent microscope (IX-71, Olympus) and an electron microscopy (EM) camera (Ixon DV860, Andor Technology). An area ∼10 × 10 μm2 was illuminated by a green laser (532 nm, ∼0.1 mW/μm2 for each specimen, Bigskylaser, Bozeman, MT) using a total internal reflection irradiation or diagonal irradiation. The fluorescence of the QDs was filtered (transmission wavelength of the filter was >580 nm; Omega Optics, Austin, TX).
QDs near the surface of cells were observed under an epi-fluorescence microscope using the illumination produced from total internal reflection (5) and imaged on EM-CCD. QDs inside cells were also observed by diagonal illumination. The temporal resolution was limited by the acquisition time of the EM-CCD camera. To shorten the acquisition time, the volume of information was reduced by binning the 16 pixels on Y axis on the CCD without reducing the total area of observation (Fig. 2 A). The binning operation was carried out on the software attached with the EM-CCD (Andor Technology). This improved the acquisition time up to 0.33 ms. The fluorescence image of the fluorophore was projected onto the two bins on the Y axis (Fig. 2 B). The X-position of the fluorescent spot was calculated by fitting the profile of the fluorescence intensity with a Gaussian curve according to the equation,
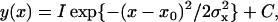 |
where x0, σx, and C are the position of a centroid, the width of the Gaussian curve, and the background intensity, respectively (Fig. 2 B). The Y-position was calculated from the difference of the fluorescent intensity of the adjoining bins. This is similar to the nanometry used for a quadrant photodiode (7). To obtain the Y-position at high precision, the difference in the intensity was fitted to a theoretical 2D Gaussian curve in which the width in the Y-direction was assumed to be the same as σx (Fig. 2 C).
FIGURE 2.

Procedure of binning FIONA. (A) Binning of fluorescent image of QD. Left panel is a part (32 × 32 pixels) of full image (128 × 128 pixels) and the right panel is a binning image with 16 pixels in the bin. The information volume decreased 16-fold and the acquisition time of EM-CCD improved from 2 to 0.33 ms. (B) Fluorescent profiles of two bins after the binning process. Ia and Ib indicate the fluorescent intensities of a and b in right side of panel A. (C) Calibration table for the Y axis. Calibration curve depends on the width of the point spread function (σx). (D and E) QD fixed on a coverslip was moved by 10-nm steps using a Piezo actuator. The position of the QDs was determined with a temporal resolution of 330 μs, and showed 10-nm steps with noise of 1.9 nm (X axis, D) and 2.0 nm (Y axis, E).
The spatial precision of binning FIONA was determined by moving a single QD on the glass surface by 10 nm with a Piezo actuator (Fig. 2, D and E). The spatial precision of the X-position was estimated to be 1.9 nm (Fig. 2 D). Although only two bins were applied to the calculation of the Y-position, the spatial precision of Y-position was 2.0 nm, which was the same as that for the X-position (Fig. 2 E).
Data analysis
The steps involved in the movement were automatically detected using computer programming. In this algorithm, the following three parameters were recognized at each step (Fig. 3).
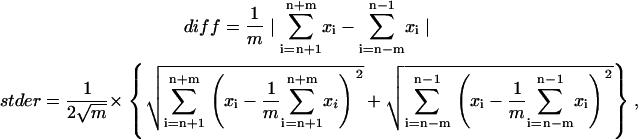 |
where xn is the position at the n-th point, and m is the number of data points calculated. The parameter of diff indicates the difference between the mean positions before and after the n-th point over m data points, eliminating n-th point itself (Fig. 3 A, upper). The parameter of stder is an average of the mean ± SE of the data before and after the n-th point (Fig. 3 A, lower). If motor proteins generated steps, the difference, diff, should be larger than stder. To minimize the probability of misjudgment, a step is deemed to be a step if it fell within a step area of diff > 3stder (Fig. 3 B). The probability of misjudgments was only <0.4% assuming a Gaussian distribution of the noise and m ≥ 4. When the number of points within one step area was <4, the stepping area was omitted to minimize the misjudgment further. Therefore, the steps that were detected were within an interval of 4 or more points. The algorithm recognized the stepping point after the step area had been determined where the standard deviation at the n-th point over m data points (stdev) is the maximum amount among the step area (Fig. 3 C). The parameter of stdev was defined as:
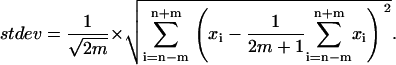 |
FIGURE 3.

Determination of steps and step size. These panels show a procedure of a step detection algorithm used in this study. (A) The differences between the averages of the data points in the forward and backward over n-th data (diff) have been compared to the degree of fluctuation in same data (stder). (B) When diff was larger than 3stder the area was considered to be a step area (orange). (C and D) The algorithm defined the data that had the maximum standard deviation (stdev, C) in one step area as a stepping point (cyan circle, D). (E) Dwell displacement was calculated by averaging adjoining stepping points. Left panel shows the definition of each of the parameters, diff, stder, and stdev. A detail explanation is described in Materials and Methods.
Finally, the data was averaged between the stepping point and the next stepping point (Fig. 3 D).
RESULTS
3D movements of QD vesicles in cells
Human breast cancer cells of KPL-4, overexpressing HER2, were cultured on coverslips in DMEM containing 5% FBS (40). Just before the experiment, FBS was removed for 1 h to starve the cells. Anti-HER2 (Trastuzumab) was used as a vesicle marker and was bound to multiple particles of QD to avoid the blinking effect induced by single QDs (Supplementary Fig. S1). Anti-HER2-QDs were added to the cells on the coverslip. Unbound anti-HER2-QDs were removed 2 h after their addition by washing with DMEM. Endocytosis was triggered by the addition of 10% FBS.
The moving vesicles containing QDs were observed under a Nipkow-type confocal microscope (Supplementary Fig. S2) 2 h after the FBS had been added. Many QD vesicles moved in a straight line, indicating that the QD vesicles were transported on either the actin filaments or microtubules (Fig. 4 A and Supplementary Movie S1). To observe the sequence of vesicular transport in three dimensions, one set of the nine confocal 3D images of the QDs were taken every 330 ms with 1.0-μm step movements of the objective (Fig. 4 B; Supplementary Fig. S2 and Movie S2). The 3D position of the fluorophore was calculated by fitting the fluorescence images at various focal planes with 3D Gaussian functions (3D single particle tracking) (Figs. 1 and 4 C). The Z axis in Fig. 4 C is directed from the membrane to the nucleus perpendicular to the cell membrane. Anti-HER2-QDs moved in continuous lines at a velocity of 300–800 nm/s at 30°C just beneath the cell membrane (red symbols in Fig. 4, C and D) after endocytosis (Supplementary Movie S3). Occasionally the QDs paused (0.5 s on average) during their movements when moving along the same lines or when changing the direction of their movement. When the QDs were transported toward the nucleus (inward movement), their speed of movement increased (0.8–1.5 μm/s) (blue symbols in Fig. 4, C and D). Once close to the nucleus, the speed decreased dramatically. In the areas in the vicinity of the nucleus, anti-HER2-QDs moved straight for short distances (within a range of 0.5 μm) and exhibited changes in direction (green symbols in Fig. 4, C and D).
FIGURE 4.

Movements of anti-HER2-QDs in living cells. (A) Fluorescence confocal image of anti-HER2-QDs in KPL4 cells. Bar indicates 10 μm. White spots are individual vesicles labeled with anti-HER2-QD. Cyan lines are the trajectories of the movements of individual anti-HER2-QDs for a 10-s period. (B) Three-dimensional movement of the anti-HER2-QD. The confocal images at various focus positions are arranged vertically. The red spot is an example of a moving QD. (C) Three-dimensional movement of the transport of HER2 from the membrane to the nucleus after endocytosis. Z axis is the axis from the membrane to the nucleus, and XY-plane is parallel with the membrane. Red, blue, and green circles indicate movement along the membrane, transport toward the nucleus, and drift around the nucleus, respectively. (D) The time course of the velocity of the movement in panel C averaged for a 3-s period. Red circles indicate the movements near the membrane, and blue circles indicate movement toward the nucleus. Arrowheads indicate the cessation of movement. The colors correspond, respectively, to those explained in C. (E) Colocalization of myosin VI and anti-HER2-QD observed under the TIRF microscope. Myosin VI was immunolabeled with anti-myosin VI and FITC-anti-IgG (green spots). Anti-HER2-QDs bound to HER2 on the membrane, and some of the QDs internalized into the cytoplasm after endocytosis (red spots). White arrows indicate the QD spots that overlapped with those of myosin VI. Scale bar is 5 μm. Lower panel is an enlargement of the region depicted in cyan in the upper panel.
To determine whether QD vesicles were transported by actin- or microtubule-based motor proteins, actin filaments and microtubules in the chemically fixed cells were stained with phalloidin-rhodamine and FITC antibody against anti-tubulin, respectively. Images taken with a confocal microscope indicated that the actin filaments were localized directly underneath the cell membrane, whereas the microtubules were positioned ∼1 μm away from the membrane (Supplementary Fig. S3).
To confirm the relationship between the movements of anti-HER2-QDs in the cytoplasm and the localization of the actin and microtubules, either the actin filament or the microtubule were depolymerized by adding cytochalasin D or nocodazole, respectively (Supplementary Fig. S4 A) (42,43). Treating the QDs with cytochalasin D or nocodazole affected their movement. The numbers of moving QDs beneath the membrane, in the cytosols, and near the nucleus were counted (Supplementary Fig. S4 B). QDs that moved slowly, <100 nm/s with random direction, were not included in the count. After the addition of 10 μM cytochalasin D, the number of the QDs moving beneath the membrane decreased to 33% of that before the treatment (22 cells). In contrast, the population of QDs transported toward the nucleus increased up to 189% of that before the treatment. After the addition of 40 μM nocodazole, the transport of anti-HER2-QDs toward and away from the nucleus was inhibited. The movement of anti-HER2-QDs beneath the cell membrane was not inhibited, but rather, increased to 122%. These results indicate that the movements of anti-HER2-QDs beneath the membrane involved the actin filaments and such motor proteins as myosin VI or myosin V (2). Transports toward and away from the nucleus are most likely performed by microtubule-based motors, such as dynein or kinesin, respectively (1).
The transport of the clathrin-mediated vesicle that had been endocytosed including the EGFR (HER1) family has been shown to be related to myosin VI (28,29,44). To explore this further, we investigated whether myosin VI and anti-HER2-QD colocalized beneath the membrane using an immunofluorescence method and TIRF microscopy (5). The QDs were colocalized with myosin VI (Fig. 4 E). The probability of colocalization of QD (red) and myosin VI (green) was 9.9 ± 3.1% (10 cells measured) of all QDs located on the membrane. The probability that the fluorescent spots of QDs overlapped randomly with those of myosin VI within a distance of 100 nm was estimated as: (the number of myosin VI spots) × (the area of myosin VI spot) / (the area of cell body) / (the area of QD spots) × 0.12 (μm2) × π = 98.7 × 0.31 (μm2) / 585.4 (μm2) / 0.38 (pixels2) × 0.12 (μm2) × π = 0.004.
This value of 0.4% was significantly smaller than that obtained from the overlapped spots of 9.9% in Fig. 4 E. This is consistent with the finding of <9.9% of the anti-HER2 antibody being internalized and transported into the cells within 2 h of the incubation previously reported (45). Myosin V is thought to not participate in the transport of vesicles containing QD antibody, since the anti-HER2 antibody inhibited the recycling of the endosome including HER2, which is transported by myosin V (34,35,38,39). The movement along the actin filament in this study, therefore, could be produced most likely by myosin VI similar to the HER1 endosome. These results suggest that QD vesicles were transported along actin filaments by myosin VI soon after endocytosis.
Movement of QD vesicles on actin filaments
To observe the movement of QD vesicles transported beneath the cell membrane with high temporal and spatial precision, 2D images at a fixed focal plane were taken under TIRF microscopy equipped with fast EM-CCD. Only the fluorescent spots emitting constant intensities were analyzed when they moved parallel to the coverslip and near the cell membrane, presumably on the actin filaments (Supplementary Movie S4 and S5). Positions of the QD spots were determined using FIONA with a spatial resolution of 1.8 nm with an imaging cycle time of 2 ms (Fig. 5 A). Stepwise movements were detected using computer analysis (see Materials and Methods). The steps detected varied greatly in size ranging from 8 and 50 nm. To investigate if step size was correlated with time, the step sizes were plotted as a function of time (Fig. 5 B and Supplementary Fig. S5). Stepwise changes in step size were detected by the same method as that used for the displacement. It was assumed that the distribution of the step size could be well fitted to a Gaussian curve similar to that for the noise of the QD position. The average step sizes until the step size changing were plotted in a histogram (Fig. 5 C). The histogram showed two peaks at 17.2 and 30.9 nm fitted by two Gaussian curves. This leads to the question of whether these periods where the step sizes are different play any significant role in cell transport. The traces were separated into the large step period where mean step size was >23 nm and the small step period where steps were <23 nm. The value of 23 nm was used because this is the middle value of the two Gaussian distribution shown in Fig. 5 C. The step size and the dwell time, i.e., the time between adjacent steps, of the small and large step period have been analyzed separately (Fig. 5, D–G). The distribution of the step sizes also had peaks at 15 nm for the small step period and 29 nm for the long step period (Fig. 5, D and E). The distribution and mean time (∼30 ms) of the dwell times for the small step periods was similar to that for long periods (Fig. 5, F and G).
FIGURE 5.

Stepwise movements of the transport of the vesicles beneath the membrane. (A) Typical traces obtained for the transport beneath the membrane at 30°C. Red lines indicate the individual steps. Values are the sizes of each step detected by the computer programming. (B) Switching of the step size. Red lines indicate the stepwise changes of the step size. Yellow and cyan indicate the periods where the steps were small and large, respectively. (C) Histogram of the average step size until the step size changed. Data was fitted with the Gaussian distribution with peaks at 30.9 and 17.2 nm (n = 25). (D and E) Histogram of the step sizes for periods with small (D) and large steps (E). The peak of the Gaussian distribution fitted to the data for periods where the steps were small was 15 nm with a halfwidth of 5.6 nm (n = 202). The peak for the period where the steps were large occurred at 29 nm with a halfwidth of 10 nm (n = 188). (F and G) Dwell time of period where the steps were small (F) and large (G). The mean dwell time of period where the steps were small and large were 29 ms (n = 202) and 32 nm (n = 188), respectively.
To determine whether the stepwise change was a random event as a part of noise or due to a biological process, step sizes were compared with a result of a simulation assuming a random event. The step sizes obtained in the experiment were arranged at random. The number of step size changes was counted by the method described in the Fig. 5 B. The number of stepwise changes in the simulated step traces was 0.7 ± 0.5 per 2 s (mean ± SD, n = 1400). This is considerably smaller than the average number of stepwise changes (2.4 times/2 s; n = 5) obtained in the experiment (Fig. 5 B and Supplementary Fig. S5), suggesting that most of the stepwise change was developed not by random events, but by actual meaningful biological events. Furthermore, to determine whether there was any correlation of step size between successive steps in Fig. 5 B and Supplementary Fig. S5, we had another type of an analysis that is zero crossing analysis (Supplementary Fig. S6). The average step size of all step sizes (24.3 nm) was defined as the zero, and we counted the data points between two successive zero upcrossings (Supplementary Fig. S6 A). Assuming the step size was randomly distributed, the dwell during zero crossing followed to the single exponential function. The experimental data did not show the single exponential distribution that the data simulated as random events showed (Supplementary Fig. S6 B). This analytical result also indicates the step size change happened due to some correlation, not randomly.
Movement of QD vesicles on microtubules analyzed by the binning FIONA
The transport of QD vesicles rapidly toward the nucleus on the microtubules (inward movement), which was presumably produced by dynein, was observed a few micrometers from the cell membrane (Fig. 6). The inward transport could be observed by illuminating the inside of the cell on a diagonal, avoiding the visualization of the actin-based transport. The microtubules are congregated near the nucleus, thus a region was selected far enough from the nucleus to show the microtubules lying roughly parallel to the microscope stage (Fig. 6 A, in the red square). When the vesicle transport could be observed at a temporal resolution of 2 ms similar to what occurs on actin filaments, clear steps of the vesicle could not be clearly detected. It was expected that steps <10 nm would be generated by dynein because, from the velocity of the QD of 0.8–1.5 μm/s in cells (Fig. 4 D), dynein will take only 4–8 ms or 2–4 sampling points calculated to move from the cell membrane to the nucleus. We developed a new analytical method to determine the position of QDs at a higher temporal resolution (binning FIONA). The temporal resolution of the captured images has been improved to 330 μs by binning 16 pixels on the Y axis of CCD without reducing the observation area (Fig. 6 A, middle, and Supplementary Movie S6). The spatial precision or standard deviation of the noise produced by the binning process was 1.9 nm along the X axis and 2.0 nm along the Y axis per 330 μs. This is very similar to the value of 1.8 nm that was obtained when no binning was performed (Fig. 2, D and E). When 6 points with the 330-μs period were averaged for a total of 2 ms, the noise in the X and Y axis could be reduced to 0.8 and 0.9 nm, respectively. These values are considerably better than those obtained without the binning process. Each step in the inward movement toward the nucleus could be clearly detected by binning FIONA (Fig. 6 B and Supplementary Fig. S7). The inward movement at 30°C was composed of successive 6–10 nm steps with occasional steps of 12–20 nm and steps in the backward direction. To confirm that the successive 8-nm steps were not produced as a result of the binning processes, experiments were also conducted at a lower temperature (20°C, Fig. 6 C). At the lower temperature, 8-nm steps were observed with a temporal resolution of 2 ms in the absence of binning and the successive large (>20 nm) steps were not observed in the cytosol. The velocity was three times slower (250–500 nm/s) than that recorded at 30°C (Fig. 6 C). The majority of the steps were also 8 nm in size inside cells, which is consistent with those steps measured at 30°C using binning FIONA.
FIGURE 6.

Stepwise movements of the transport of the vesicles toward the nucleus (inward movement). (A) An incident-light image and fluorescence image obtained using binning FIONA. To confirm the direction of the movement of vesicles, the incident-light image of the cell was observed when the fluorescence image of QD vesicles was overlapping (left). The 16 pixels on the Y axis of the fluorescent image were binned into one bar (middle). The 8-nm step was clearly observed by using binning FIONA (right). (B) A typical trace of the movement toward the nucleus at 30°C. Gray line is the raw data, and the black line is the average line for the three data sets. (C) Typical trace of the movement toward the nucleus at 20°C. Blue lines in panels A, B, and C indicate steps detected by the computer programming. (D) Histogram of step sizes of the movements toward the nucleus. Data was fitted with four Gaussian curves (n = 318). The main peak values were −8.4 and 8.5, respectively. (E) Histogram of the dwell times of the movements toward the nucleus. Data were fitted with single exponential function with a time constant of 7.5 ms (n = 288).
The histogram of the step size showed peaks at multiples of 8.5 nm, suggesting that the elementary step size is 8 nm (Fig. 6 D). The dwell time of the 8-nm steps in the inward direction was analyzed as the dynein step (Fig. 6 E). The histogram could be fitted with a single exponential decay with a time constant of 7.5 ms. Using a step size of 8.5 nm and a dwell time of 7.5 ms, the velocity of inward transport without backward steps was calculated to be 1100 nm/s. This value is consistent with the data in in vitro assays (800 nm/s), when taking into consideration the 5°C difference in temperature between this study and previous studies (9,10).
The outward movement of the QD vesicles, presumably induced by kinesin, was occasionally observed after the vesicles had been transported in the inward direction. The movement was measured with binning FIONA because the velocity of the movement was faster than that of the inward movement. The outward movement also involved stepwise movements with step sizes of 6–18 nm with some steps sometimes occurring in the inward direction (Fig. 7 A and Supplementary Fig. S8). A histogram of the step size showed a major peak at −8.3 nm (outward direction) and a minor peak at 8.9 nm (inward direction) (Fig. 7 B). The histogram of the dwell times for the outward 8-nm steps was fitted to a single exponential measure with a time constant of 5.0 ms (Fig. 7 C), performed for the inward steps (Fig. 6 E). The velocity of outward transport without backward steps was calculated to be 8.3/0.005 = 1660 nm/s.
FIGURE 7.

Stepwise movements from the nucleus toward the cell membrane (outward movement). (A) Typical trace of the movement away from the nucleus at 30°C. Gray line is the raw data, and the black line is average line for the three data sets. Red lines indicate steps detected by the computer programming. (B) Histogram of the step sizes of the movement away from the nucleus. Data were fitted with four Gaussian curves (n = 238). The main peak values were −8.3 and 8.9, respectively. (C) Histogram of dwell time of the movement away from the nucleus. Data were fitted with single exponential function with the time constant of 5.0 ms (n = 180).
DISCUSSION
The anti-HER2-QDs were successfully endocytosed by the KPL4 cells. After endocytosis, the QD vesicles were transported just beneath the cell membrane. The 3D single particle tracking system showed vesicles being transported via both actin filaments and microtubules. These 3D transport processes were consistent with the results from previous studies (24,25), and the switching from actomyosin to microtubule-based motors was similar to that observed for an influenza virus being infected into a cell (42).
The movement of the vesicles underneath the membrane is most likely generated by myosin VI because the observed step size of 29 nm (Fig. 5 E) is consistent with that for recombinant myosin VI of ∼30 nm in vitro (13,16). However, it is different to the step size obtained for myosin V of 36 nm in vitro (11,12,14,18) and in living cells (46). We did not observe any successive 36-nm step underneath the cell membrane in this study. The colocalization of myosin VI and anti-HER2-QD under TIRF microscopy supported the idea that myosin VI transports the endocytic vesicle, including HER2, along the actin network beneath the membrane.
In addition to these movements, we observed an unusual behavior of presumably myosin VI in vivo. This movement consisted of two distinct step sizes, 29 and 15 nm, and was observed during transport beneath the cell membrane (Fig. 5 B). These steps were not generated by double steps of shorter step size, i.e., 8 or 15 nm because shorter step sizes were not observed at the lower temperature of 20°C when the dwell time is approximately three times longer (Supplementary Fig. S9). An interesting question from this result is whether this change in step size has any biological significance. The number of stepwise changes in the simulated traces using a random arrangement of step sizes was much smaller than the number obtained in the experiment. The difference in the number of stepwise changes between the experiment (2.4) and the simulation (0.7) is 1.7 per 2-s interval or 0.85/s. This suggests that the step size switches between 29 and 15 nm during myosin VI mediated transport.
This change in step size during transport along myosin VI is the first report of this unusual behavior in living cells. The ∼15 nm steps of myosin VI observed in this study have not been previously observed in in vitro assays. The movements of myosin VI in almost all previous in vitro assays were recorded in conditions that were not physiological. Artificial conditions optimized the performance of myosin VI in the assays. For example, to dimerize the monomers of myosin VI, a leucine zipper can be inserted into the neck domain of myosin VI or an antibody against myosin VI can be added in the assay solution (13,16,47). Wild-type myosin VI is a monomer in vitro, and its step size decreased dependent on the load (48). Such studies discussed how the step size of myosin VI monomers decreased to ∼20 nm at 1.5–2.0 pN load, while that of the myosin VI dimer with the artificial neck domain remained constant at ∼30 nm independent of the load. Although myosin VI is thought to exist as a monomer in cells, there is a current debate as to whether monomers of myosin VI dimerize in living cells (47–49). Therefore, two possible models of how the step size changes in a dimer or monomer of myosin VI must be discussed.
In the first model, monomers of myosin VI dimerize and function as a dimer molecule in a cell. The dimers may take two stable state steps using either the short and long steps. For example, recombinant dimers used to zip the long neck domain of myosin VI exhibit a short step size of 12 ± 2 nm (50). This result suggests that the transfer between zipping and unzipping of the neck domain regulates the step size of myosin VI. In this study, the small step size of myosin VI in cells (15 nm) was consistent with that of the zipped dimer (12 nm). The steps of ∼29 and ∼15 nm could be induced by unzipping and zipping of the dimer of myosin VI, respectively. This model is consistent with the result that the dwell time of the small and large steps was similar, suggesting that the same dimer molecule produces both the small and large steps. Furthermore, the variance of the step sizes also changed in correlation with the average step size (Fig. 5, B, D, and E). The variation of the step sizes at 0 to ∼500 ms was distinctly different from the next ∼500 ms, as shown in Fig. 5 B. The flexibility of the forward head of myosin VI may decide the step size.
In the second model, myosin VI transports the vesicle as a monomer in cell. A monomer of myosin VI could move processively by means of cargo binding, but not without cargoes (48). The step size of the monomer myosin VI at high load was ∼20 nm, close to the 16 nm obtained in this study, and the distribution was also narrow when the step size was decreased. Furthermore, the load did not affect the dwell time whereas step size was decreased. These results are consisted with the results in Fig. 6. Monomer myosin VI transports a vesicle in large steps without any load. When myosin VI gets the vesicle hung up on the actin network near the membrane, the step size is decreased with the increase in load.
We are not able at present to decide which model is the correct depiction of what takes place in cells. However, the change of step sizes in this study is remarkable for cell biology in general because it is to our knowledge the first such finding and represents unusual behavior in living cells.
The inward movement, which is presumably generated by cytoplasmic dynein, occurred in successive 8-nm steps (Fig. 6, B–D), which is consistent with previous reports (20,26). Kural et al. discussed the possibility that many dynein molecules work together because the maximum velocity of the vesicular movements was >10 μm/s. However, in our study, vesicles were observed moving with a mean velocity of ∼1.1 μm/s and a maximum velocity of ∼2 μm/s at 30°C. The 8-nm steps observed in the inward transport are also consistent with the result that single molecules of dynein purified from porcine brain moved using 8-nm steps (10). The outward movement, which is generated by kinesin, was also composed of successive 8-nm steps (Fig. 7, A and B). The velocity of kinesin of 1.66 μm/s was slightly faster than that, 1.1 μm/s, observed for dynein. These values are consistent with those obtained in in vitro assay of kinesin-1 (930 nm/s) and dynein (800 nm/s) in the previous report, considering the temperature for the assays in this study was 5°C higher than assays in the previous studies (7,10).
The unitary step size of cytoplasmic dynein is still being debated (10,20,26,51). In this study successive 8-nm steps were observed during the inward transport. Dynein produced mainly 8-nm steps that were not excessively large (24 or 32 nm) steps using laser trapping nanometry and FIONA in vitro (10). Other groups have reported large steps as well as 8-nm steps (51). This discrepancy may arise from the difference of the coating of the beads or proteins. Mallik and his colleagues combined dynein molecule and bead nonspecifically without any coating despite the possibility of inactivating the dynein (51). In our previous study, the beads were coated by protein A to maintain the activity of dynein (10). It is possible that two molecules of dynein, whose one head was inactive, took large steps because 16, 24-nm steps were observed with multiple one-headed kinesin molecules (52). Nan et al. also reported that dynein takes successive 16-nm steps in cells, while Kural et al. and we observed mainly 8-nm steps (20,26). We did not observe the successive 16-nm steps during the inward and outward transports. Dynein might change the step size corresponding to surrounding circumstances, i.e., the kinds of cargoes or by the number of dynein molecules interacted.
The chemical processes involved in the stepwise movement in vivo (in cell) with in vitro techniques need to be discussed. The single exponential decay of the dwell time during the inward and outward steps (Figs. 6 E and 7 C) indicates that the 8-nm steps are coupled to the chemical reaction with a single rate-limiting step, such as the ATPase cycle. Kinesin is known to be a tight coupling motor in that single ATP cycles are coupled with single 8-nm steps (7,53). The inward motor, dynein, also seems to be a tight-coupled motor. Thus, similar dwell times were recorded for both dynein and kinesin in vivo and in vitro, and the viscosity of the cytoplasm in cells did not significantly affect the velocity of the movement. This indicates the single molecular measurements in vitro can be used to understand the biochemical processes of the protein in vivo.
The size of the steps generated by purified myosin VI, kinesin, and dynein in in vitro studies is reasonably consistent with those in the present living cell (7,10,13,15–17). The measurements using binning FIONA with nanometer and submicrosecond accuracy reveals new insights into the function of motor proteins in living cells. These new methods provide a link between the results obtained on purified proteins and in living cells.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We thank Sachiko Higuchi (Tohoku University Biomedical Engineering Research Organization) for technical assistance on the immunofluorescence experiments. We gratefully acknowledge J. M. West for critical reading of this manuscript.
This work was supported by Grants-in-Aid for Scientific Research in Priority Areas from the Japan MEXT (H.H.), and Special Coordination Funds for Promoting Science and Technology of JST (H.H. and T.M.W.).
References
- 1.Hirokawa, N. 1998. Kinesin and dynein superfamily proteins and the mechanism of organelle transport. Science. 279:519–526. [DOI] [PubMed] [Google Scholar]
- 2.Tuxworth, R. I., and M. A. Titus. 2000. Unconventional myosins: anchors in the membrane traffic relay. Traffic. 1:11–18. [DOI] [PubMed] [Google Scholar]
- 3.Funatsu, T., Y. Harada, M. Tokunaga, K. Saito, and T. Yanagida. 1995. Imaging of single fluorescent molecules and individual ATP turnovers by single myosin molecules in aqueous solution. Nature. 374:555–559. [DOI] [PubMed] [Google Scholar]
- 4.Vale, R. D., T. Funatsu, D. W. Pierce, L. Romberg, Y. Harada, and T. Yanagida. 1996. Direct observation of single kinesin molecules moving along microtubules. Nature. 380:451–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tokunaga, M., K. Kitamura, K. Saito, A. H. Iwane, and T. Yanagida. 1997. Single molecule imaging of fluorophores and enzymatic reactions achieved by objective-type total internal reflection fluorescence microscopy. Biochem. Biophys. Res. Commun. 235:47–53. [DOI] [PubMed] [Google Scholar]
- 6.Ishijima, A., H. Kojima, T. Funatsu, M. Tokunaga, H. Higuchi, H. Tanaka, and T. Yanagida. 1998. Simultaneous observation of individual ATPase and mechanical events by a single myosin molecule during interaction with actin. Cell. 92:161–171. [DOI] [PubMed] [Google Scholar]
- 7.Nishiyama, M., H. Higuchi, and T. Yanagida. 2002. Chemomechanical coupling of the forward and backward steps of single kinesin molecules. Nat. Cell Biol. 4:790–797. [DOI] [PubMed] [Google Scholar]
- 8.Yildiz, A., J. N. Forkey, S. A. McKinney, T. Ha, Y. E. Goldman, and P. R. Selvin. 2003. Myosin V walks hand-over-hand: single fluorophore imaging with 1.5-nm localization. Science. 300:2061–2065. [DOI] [PubMed] [Google Scholar]
- 9.Hirakawa, E., H. Higuchi, and Y. Y. Toyoshima. 2000. Processive movement of single 22S dynein molecules occurs only at low ATP concentrations. Proc. Natl. Acad. Sci. USA. 97:2533–2537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toba, S., T. M. Watanabe, L. Yamaguchi-Okimoto, Y. Y. Toyoshima, and H. Higuchi. 2006. Overlapping hand-over-hand mechanism of single molecular motility of cytoplasmic dynein. Proc. Natl. Acad. Sci. USA. 103:5751–5755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mehta, A. D., R. S. Rock, M. Rief, J. A. Spudich, M. S Mooseker, and R. E. Cheney. 1999. Myosin-V is a processive actin-based motor. Nature. 400:590–593. [DOI] [PubMed] [Google Scholar]
- 12.Rief, M., R. S. Rock, A. D. Mehta, M. S. Mooseker, R. E. Cheney, and J. A. Spudich. 2000. Myosin-V stepping kinetics: a molecular model for processivity. Proc. Natl. Acad. Sci. USA. 97:9482–9486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rock, R. S., S. E. Rice, A. L. Wells, T. J. Purcell, J. A. Spudich, and H. L. Sweeney. 2001. Myosin VI is a processive motor with a large step size. Proc. Natl. Acad. Sci. USA. 98:13655–13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Uemura, S., H. Higuchi, A. O. Olivares, E. M. De La Cruz, and S. Ishiwata. 2004. Mechanochemical coupling of two substeps in a single myosin V motor. Nat. Struct. Mol. Biol. 11:877–883. [DOI] [PubMed] [Google Scholar]
- 15.Kaseda, K., H. Higuchi, and K. Hirose. 2003. Alternate fast and slow stepping of a heterodimeric kinesin molecule. Nat. Cell Biol. 5:1079–1082. [DOI] [PubMed] [Google Scholar]
- 16.Okten, Z., L. S. Churchman, R. S. Rock, and J. A. Spudich. 2004. Myosin VI walks hand-over-hand along actin. Nat. Struct. Mol. Biol. 11:884–887. [DOI] [PubMed] [Google Scholar]
- 17.Yildiz, A., M. Tomishige, R. D. Vale, and P. R. Selvin. 2004. Kinesin walks hand-over-hand. Science. 303:676–678. [DOI] [PubMed] [Google Scholar]
- 18.Warshaw, D. M., G. G. Kennedy, S. S. Work, E. B. Krementsova, S. Beck, and K. M. Trybus. 2005. Differential labeling of myosin V heads with quantum dots allows direct visualization of hand-over-hand processivity. Biophys. J. 88:L30–L32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gelles, J., B. J. Schnapp, and M. P. Sheetz. 1988. Tracking kinesin-driven movements with nanometer-scale precision. Nature. 331:450–453. [DOI] [PubMed] [Google Scholar]
- 20.Kural, C., H. Kim, S. Syed, G. Goshima, V. I. Gelfand, and P. R. Selvin. 2005. Kinesin and dynein move a peroxisome in vivo: a tug-of-war or coordinated movement? Science. 308:1469–1472. [DOI] [PubMed] [Google Scholar]
- 21.Kubitscheck, U., O. Kuckmann, T. Kues, and R. Peters. 2000. Imaging and tracking of single GFP molecules in solution. Biophys. J. 78:2170–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hibino, K., T. M. Watanabe, J. Kozuka, A. H. Iwane, T. Okada, T. Kataoka, T. Yanagida, and Y. Sako. 2003. Single- and multiple-molecule dynamics of the signaling from H-Ras to cRaf-1 visualized on the plasma membrane of living cells. Chem. Phys. Chem. 4:748–753. [DOI] [PubMed] [Google Scholar]
- 23.Wu, X., H. Liu, J. Liu, K. N. Haley, J. A. Treadway, J. P. Larson, N. Ge, F. Peale, and M. P. Bruchez. 2002. Immunofluorescent labeling of cancer marker Her2 and other cellular targets with semiconductor quantum dots. Nat. Biotechnol. 21:41–46. [DOI] [PubMed] [Google Scholar]
- 24.Lidke, D. S., P. Nagy, R. Heintzmann, D. J. Arndt-Jovin, J. N. Post, H. E. Grecco, E. A. Jares-Erijman, and T. M. Jovin. 2004. Quantum dot ligands provide new insights into erbB/HER receptor-mediated signal transduction. Nat. Biotechnol. 22:198–203. [DOI] [PubMed] [Google Scholar]
- 25.Lidke, D. S., K. A. Lidke, B. Rieger, T. M. Jovin, and D. J. Arndt-Jovin. 2005. Reaching out for signals: filopodia sense EGF and respond by directed retrograde transport of activated receptors. J. Cell Biol. 170:619–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nan, X., P. A. Sims, P. Chen, and X. S. Xie. 2005. Observation of individual microtubule motor steps in living cells with endocytosed quantum dots. J. Phys. Chem. B Condens. Matter Mater. Surf. Interfaces Biophys. 109:24220–24224. [DOI] [PubMed] [Google Scholar]
- 27.Carpenter, G. 2000. The EGF receptor: a nexus for trafficking and signaling. Bioessays. 22:697–707. [DOI] [PubMed] [Google Scholar]
- 28.Aschenbrenner, L., S. N. Naccache, and T. Hasson. 2004. Uncoated endocytic vesicles require the unconventional myosin, Myo6, for rapid transport through actin barriers. Mol. Biol. Cell. 15:2253–2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roberts, R., I. Lister, S. Schmitz, M. Walker, C. Veigel, J. Trinick, F. Buss, and J. Kendrick-Jones. 2004. Myosin VI: cellular functions and motor properties. Philos. Trans. R. Soc. Lond. B Biol. Sci. 359:1931–1944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Goodson, H. V., C. Valetti, and T. E. Kreis. 1997. Motors and membrane traffic. Curr. Opin. Cell Biol. 9:18–28. [DOI] [PubMed] [Google Scholar]
- 31.Alroy, I., and Y. Yarden. 1997. The ErbB signaling network in embryogenesis and oncogenesis: signal diversification through combinatorial ligand-receptor interactions. FEBS Lett. 410:83–86. [DOI] [PubMed] [Google Scholar]
- 32.Lenferink, A. E., R. Pinkas-Kramarski, M. L. van de Poll, M. J. van Vugt, L. N. Klapper, E. Tzahar, H. Waterman, M. Sela, E. J. van Zoelen, and Y. Yarden. 1998. Differential endocytic routing of homo- and hetero-dimeric ErbB tyrosine kinases confers signaling superiority to receptor heterodimers. EMBO J. 17:3385–3397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Worthylake, R., L. K. Opresko, and H. S. Wiley. 1999. ErbB-2 amplification inhibits down-regulation and induces constitutive activation of both ErbB-2 and epidermal growth factor receptors. J. Biol. Chem. 274:8865–8874. [DOI] [PubMed] [Google Scholar]
- 34.Lapierre, L. A., R. Kumar, C. M. Hales, J. Navarre, S. G. Bhartur, J. O. Burnette, D. W. Provance Jr., J. A. Mercer, M. Bahler, and J. R. Goldenring. 2001. Myosin vb is associated with plasma membrane recycling systems. Mol. Biol. Cell. 12:1843–1857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.van Ijzendoorn, S. C. 2006. Recycling endosomes. J. Cell Sci. 119:1679–1681. [DOI] [PubMed] [Google Scholar]
- 36.Yarden, Y. 2001. Biology of HER2 and its importance in breast cancer. Oncology. 61:1–13. [DOI] [PubMed] [Google Scholar]
- 37.Marmor, M. D., K. B. Skaria, and Y. Yarden. 2001. Signal transduction and oncogenesis by ErbB/HER receptors. Int. J. Radiat. Oncol. Biol. Phys. 58:903–913. [DOI] [PubMed] [Google Scholar]
- 38.Carter, P., L. Presta, C. M. Gorman, J. B. Ridgway, D. Henner, W. L. Wong, A. M. Rowland, C. Kotts, M. E. Carver, and H. M. Shepard. 1992. Humanization of the anti-p185HER2 antibody for human cancer therapy. Proc. Natl. Acad. Sci. USA 89:4285–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Lipkowitz, S. 2003. The role of the ubiquitination-proteasome pathway in breast cancer: ubiquitin mediated degradation of growth factor receptors in the pathogenesis and treatment of cancer. Breast Cancer Res. 5:8–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kurebayashi, J., T. Otsuki, C. K. Tang, M. Kurosumi, S. Yamamoto, K. Tanaka, M. Mochizuki, H. Nakamura, and H. Sonoo. 1999. Isolation and characterization of a new human breast cancer cell line, KPL-4, expressing the Erb B family receptors and interleukin-6. Br. J. Cancer. 79:707–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Swanson, J. A., M. T. Jsohnson, K. Beningo, P. Post, M. Mooseker, and N. Araki. 1999. A contractile activity that closes phagosomes in macrophages. J. Cell Sci. 112:307–316. [DOI] [PubMed] [Google Scholar]
- 42.Lakadamyali, M., M. J. Rust, H. P. Babcock, and X. Zhuang. 2003. Visualizing infection of individual influenza viruses. Proc. Natl. Acad. Sci. USA. 100:9280–9285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yanai, M., J. P. Butler, T. Suzuki, H. Sasaki, and H. Higuchi. 2004. Regional rheological differences in locomoting neutrophils. Am. J. Physiol. Cell Physiol. 287:C603–C611. [DOI] [PubMed] [Google Scholar]
- 44.Buss, F., S. D. Arden, M. Lindsay, J. P. Luzio, and J. Kendrick-Jones. 2001. Myosin VI isoform localized to clathrin-coated vesicles with a role in clathrin-mediated endocytosis. EMBO J. 20:3676–3684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Austin, C. D., A. M. De Maziere, P. I. Pisacane, S. M. van Dijk, C. Eigenbrot, M. X. Sliwkowski, J. Klumperman, and R. H., Scheller. 2004. Endocytosis and sorting of ErbB2 and the site of action of cancer therapeutics trastuzumab and geldanamycin. Mol. Biol. Cell 15:5268–5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Levi, V., V. I. Gelfand, A. S. Serpinskaya, and E. Gratton. 2006. Melanosomes transported by myosin-V in Xenopus melanophores perform slow 35 nm steps. Biophys. J. 90:L07–L09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park, H., B. Ramamurthy, M. Travaglia, D. Safer, L. Q. Chen, C. Franzini-Armstrong, P. R. Selvin, and H. L. Sweeney. 2006. Full-length myosin VI dimerizes and moves processively along actin filaments upon monomer clustering. Mol. Cell. 21:331–336. [DOI] [PubMed] [Google Scholar]
- 48.Iwaki, M., H. Tanaka, A. H. Iwane, E. Katayama, M. Ikebe, and T. Yanagida. 2006. Cargo-binding makes a wild-type single-headed myosin-VI move processively. Biophys. J. 90:3643–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lister, I., S. Schmitz, M. Walker, J. Trinick, F. Buss, C. Veigel, and J. Kendrick-Jones. 2004. A monomeric myosin VI with a large working stroke. EMBO J. 23:1729–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rock, R. S., B. Ramamurthy, A. R. Dunn, S. Beccafico, B. R. Rami, C. Morris, B. J. Spink, C. Franzini-Armstrong, J. A. Spudich, and H. L. Sweeney. 2005. A flexible domain is essential for the large step size and processivity of myosin VI. Mol. Cell. 17:603–609. [DOI] [PubMed] [Google Scholar]
- 51.Mallik, R., B. C. Carter, S. A. Lex, S. J. King, and S. P. Gross. 2004. Cytoplasmic dynein functions as a gear in response to load. Nature. 427:649–652. [DOI] [PubMed] [Google Scholar]
- 52.Inoue, Y., Y. Y. Toyoshima, A. H. Iwane, S. Morimoto, H. Higuchi, and T. Yanagida. 1997. Movements of truncated kinesin fragments with a short or an artificial flexible neck. Proc. Natl. Acad. Sci. USA. 94:7275–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schnitzer, M. J., and S. M. Block. 1997. Kinesin hydrolyses one ATP per 8-nm step. Nature. 388:386–390. [DOI] [PubMed] [Google Scholar]