

Abstract
The scorpion toxin CnErg1 binds to human ether-a-go-go related gene (hERG) K+ channels with a 1:1 stoichiometry and high affinity. However, in contrast to other scorpion toxin-ion channel interactions, the inhibition of macroscopic hERG currents by high concentrations of CnErg1 is incomplete. In this study, we have probed the molecular basis for this incomplete inhibition. High concentrations of CnErg1 had only modest effects on hERG gating that could not account for the incomplete block. Furthermore, the residual current in the presence of 1 μM CnErg1 had normal single channel conductance. Analysis of the kinetics of CnErg1 interaction with hERG indicated that CnErg1 binding is not diffusion-limited. A bimolecular binding scheme that incorporates an initial encounter complex and permits normal ion conduction was able to completely reproduce both the kinetics and steady-state level of CnErg1-hERG binding. This scheme provides a simple kinetic explanation for incomplete block; that is, relatively fast backward compared to forward rate constants for the interconversion of the toxin-channel encounter complex and the blocked toxin-channel complex. We have also examined the temperature-dependence of CnErg1 binding to hERG. The dissociation constant, Kd, for CnErg1 increases from 7.3 nM at 22°C to 64 nM at 37°C (i.e., the affinity decreases as temperature increases) and the proportion of binding events that lead to channel blockade decreases from 70% to 40% over the same temperature range. These temperature-dependent effects on CnErg1 binding correlate with a temperature-dependent decrease in the stability of the putative CnErg1 binding site, the amphipathic α-helix in the outer pore domain of hERG, assayed using circular dichroism spectropolarimetry. Collectively, our data provides a plausible kinetic explanation for incomplete blockade of hERG by CnErg1 that is consistent with the proposed highly dynamic conformation of the outer pore domain of hERG.
INTRODUCTION
Human ether-a-go-go related gene (hERG) potassium channels are critical for the maintenance of normal electrical activity in the heart (1). hERG channels are also the molecular target for the vast majority of drugs that cause drug-induced arrhythmias and cardiac death (2). There is therefore intense interest in understanding the molecular and structural basis of gating in hERG K+ channels.
Despite significant sequence homology to other members of the voltage-gated K+ channel family, hERG channels have very distinct kinetics, characterized by slow activation (time constants range from hundreds of milliseconds to many seconds) but a very rapid rate of inactivation (time constants in the range 1–10 ms) (3). Previous work from our lab (4) as well as others (5,6) has shown that the unusually rapid inactivation of hERG relative to the rate of activation is crucial for its roles in normal cardiac repolarization and suppression of propagation of premature beats.
Voltage-gated potassium channels are composed of four subunits, each with six transmembrane domains (S1–S6). The S5 and S6 domains along with the intervening pore-loop (P domain) from each of the four subunits form the ion conductance pathway. The outer pore region of the ether-a-go-go subfamily of voltage-gated K+ channels is unique among the voltage-gated ion channel family in that it has a much longer linker located between the S5 and P domains, i.e., the S5P domain, ∼40 residues long compared to 10–12 residues in other channels (7–9). The linker contains an amphipathic α-helix (9) that is critical for normal inactivation (8,10). Although we have solved the structure of the isolated S5P domain, using two-dimensional NMR spectroscopy (9), this did not provide sufficient spatial constraints to enable us to determine a unique three-dimensional structure for the outer pore region. Furthermore, Tseng and colleagues have shown, using cysteine scanning mutagenesis and monitoring rates of intersubunit disulfide bond formation, that the S5P domain of hERG is likely to have a highly dynamic structure (11).
In the absence of crystal structures, one of the methods that has been very useful for gaining insights into channel structure is toxin footprinting (12,13). Scorpion venoms have been a rich source of peptides that inhibit ion channels with high affinity and selectivity. There are a number of scorpion toxins that inhibit hERG K+ channels with high specificity and low nanomolar affinity (14). CnErg1 is one of the best characterized of these toxins. It binds to hERG K+ channels with a 1:1 stoichiometry and a Kd of ∼10 nM (9,15–17). Studies on chimeric channels composed of hERG and the closely related human ether-a-go-go channels showed that the S5P domain was the most important domain for toxin binding (15). But identifying the precise CnErg1 binding site has proved difficult. Cysteine scanning mutagenesis of the S5P domain identified a number of residues, and in particular in the amphipathic α-helix of the S5P domain, that affected CnErg1 binding (16). However, all the mutants that caused a >10-fold reduction in affinity for CnErg1 also perturbed channel function (8,11,16). Thus, it is not yet known whether the amphipathic α-helix contributes directly to the CnErg1 binding pocket or whether this helix stabilizes a nearby binding site.
Peptide toxins usually occlude the pore of the channel, either directly by occupying the selectivity filter (18) or by binding to an electrostatic ring surrounding the pore (19). As a consequence, the toxins can cause complete high affinity block and the blockade appears to be diffusion-limited (20,21). In marked contrast to this, CnErg1 does not produce complete blockade of hERG currents, even at concentrations orders-of-magnitude higher than is required for inhibition of 50% of channels (16,22). This has led to the suggestion that CnErg1 may be a gating modifier rather than pore blocker and/or bind near to the pore but not fully occlude the permeation-pathway (23,24).
In this study, we have set out to identify the mechanism underlying CnErg1 block of macroscopic hERG current. Neither gating modification nor reduced single channel conductance can account for the submaximal block of hERG current by high concentrations of CnErg1. However, a detailed analysis of the kinetics of CnErg1 binding to hERG revealed that association rates are not diffusion-limited. A kinetic scheme incorporating an on-path intermediate (i.e., a toxin-channel encounter complex that does not involve channel block) was able to reproduce all our data. This scheme provides a simple kinetic explanation for incomplete block; i.e., relatively fast backward compared to forward rate constants for the interconversion of the toxin-channel encounter complex and the blocked toxin-channel complex.
MATERIALS AND METHODS
Electrophysiology
Experiments were performed using a CHO-cell line stably transfected with WT hERG K+ channels as previously described (25). Cells were studied either at 22°C or 37°C. The cell chamber was heated using a  Bipolar temperature controller (Cell MicroControls, Wellesley Hills, MA), as previously described (26). Cells were voltage-clamped in whole cell mode using an Axopatch 200B headstage amplifier (Axon Instruments, Union City, CA). In general, currents were filtered at 2 kHz and digitized at 5 kHz using a Digidata 1322 A/D converter (Axon Instruments) operated using pClamp software. However, due to the rapidity of hERG inactivation, for protocols designed to measure rates of inactivation and recovery from inactivation and the voltage-dependence of inactivation, currents were recorded at 20 kHz and filtered at 5 kHz. The internal solution contained (in mM): 120 K gluconate, 20 KCl, 1.5 MgATP, 5 EGTA, and 10 HEPES (pH 7.3 with KOH). The standard bath solution contained 130 NaCl, 4.8 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 1 CaCl2, 10 glucose, and 10 HEPES (titrated to pH 7.4 with NaOH at room temperature). The calculated junction potential of −15 mV was adjusted for in all recordings. Series resistance was compensated by at least 80% in all recordings.
Bipolar temperature controller (Cell MicroControls, Wellesley Hills, MA), as previously described (26). Cells were voltage-clamped in whole cell mode using an Axopatch 200B headstage amplifier (Axon Instruments, Union City, CA). In general, currents were filtered at 2 kHz and digitized at 5 kHz using a Digidata 1322 A/D converter (Axon Instruments) operated using pClamp software. However, due to the rapidity of hERG inactivation, for protocols designed to measure rates of inactivation and recovery from inactivation and the voltage-dependence of inactivation, currents were recorded at 20 kHz and filtered at 5 kHz. The internal solution contained (in mM): 120 K gluconate, 20 KCl, 1.5 MgATP, 5 EGTA, and 10 HEPES (pH 7.3 with KOH). The standard bath solution contained 130 NaCl, 4.8 KCl, 1.2 MgCl2, 1.2 NaH2PO4, 1 CaCl2, 10 glucose, and 10 HEPES (titrated to pH 7.4 with NaOH at room temperature). The calculated junction potential of −15 mV was adjusted for in all recordings. Series resistance was compensated by at least 80% in all recordings.
Voltage-clamp protocols
For toxin binding studies, cells were depolarized from a holding potential of −80 mV to +40 mV for 500 ms, then repolarized to 0 mV for 100 ms and then to −120 mV for 1 s. This pulse protocol was repeated every 5 s. Current amplitudes were measured from the peak inward current at −120 mV.
The voltage dependence of steady-state inactivation and steady-state activation as well as the rates of inactivation, recovery from inactivation, and deactivation were measured as previously described (26). The rate of activation was measured using an envelope-of-tails protocol (27), cells were depolarized to +40 mV for variable durations in the range 1.6–1000 ms before stepping to −160 mV where tail currents were recorded. Specific details for each voltage protocol are presented in the relevant figures and legends.
Peptides
The CnErg1 toxin and a peptide corresponding to S581–S599 of the hERG S5P linker were synthesized manually on a 0.50 mmol scale using HBTU activation of Boc-amino acids with in situ neutralization chemistry as previously described (22). Toxins were dissolved directly in bath solution at concentrations ranging from 5 nM to 3 μM and applied using a Picospritzer Perfusion Device (Intracel, Cambridge, UK).
Circular dichroism spectropolarimetry
Circular dichroism spectropolarimetry (CD) spectra were recorded on a JASCO 720 spectropolarimeter (Tokyo, Japan) equipped with a Neslab RTE-111 temperature controller (Portsmouth, NH). FarUV spectra were collected using a 1 mm cuvette over the wavelength range 190–250 nm and with a resolution of 0.5 nm, a bandwidth of 1 nm, and a response time of 1 s. Final spectra were the average of three scans collected at a speed of 20 nm/min and were baseline-corrected. Thermal denaturation experiments were conducted with heating at a rate of 1°C/min, a step size of 0.5°C, a bandwidth of 1 nm, a response time of 8 s, and detection at 222 nm. Data are expressed as mean residue molar ellipticity ([θ]MRW), calculated as follows: [θ]MRW = θ (mdeg)/10 C l n, where θ is the ellipticity (in millidegrees), C is the molar concentration, l is the pathlength (in cm), and n is the number of residues.
Data analysis
Dose response curves for the toxin concentration dependence of current inhibition were fitted with a modified Hill equation,
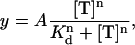 |
(1) |
where A is the maximum inhibition, Kd is the affinity of toxin for the hERG K+ channel, n is the Hill slope, and T is toxin.
Conductance voltage curves were fitted with the Boltzmann equation,
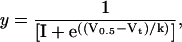 |
(2) |
where V0.5 is the half-activation voltage, Vt is the test potential, and k is the slope factor.
All data are presented as mean ± SE.
Nonstationary noise analysis
For nonstationary noise analysis, cells were depolarized from a holding potential of −80 mV to +40 mV for 500 ms then repolarized to −120 mV for 1 s. This protocol was repeated every 3 s. All data were acquired at 20 KHz and filtered at 5 KHz. Mean and variance of the mean at each isochrone was calculated using Excel 2003 (Microsoft, Redmond, WA). For this, >40 subsequent records were used to construct the mean under each experimental condition. Leak was subtracted off-line. The variance of the records with respect to the mean current was computed by pairs to compensate for time-dependent shifts in the mean. (28,29). The relation between mean and variance is described by the equation
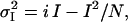 |
(3) |
where at a given holding potential, 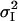 is the variance; i is the single channel current amplitude; I is the macroscopic mean current; and N is the number of channels. This equation describes a parabola with its roots at I = 0 and I = i N. Mean current and variance data were binned with respect to current amplitude and mean variance within each bin plotted against the corresponding mean current. Standard error for each data point was calculated taking into account only data within that bin. Data was then fitted with Eq. 3, weighted according to the inverse of the sum of the squares of the standard errors of the variance and mean allowing estimation of i.
is the variance; i is the single channel current amplitude; I is the macroscopic mean current; and N is the number of channels. This equation describes a parabola with its roots at I = 0 and I = i N. Mean current and variance data were binned with respect to current amplitude and mean variance within each bin plotted against the corresponding mean current. Standard error for each data point was calculated taking into account only data within that bin. Data was then fitted with Eq. 3, weighted according to the inverse of the sum of the squares of the standard errors of the variance and mean allowing estimation of i.
Modeling
The kinetic model describing the bimolecular reaction scheme was set up in MS Excel 2003 (Microsoft). The Premium Solver Platform for Microsoft Excel (Frontline Systems, Seattle, WA) was used to find global minima of the sums of squares of the difference between the experimentally measured values and those predicted by the model. The functions to be minimized were highly nonlinear in terms of the known variables (on- and off-rates); therefore, the quadratic extrapolation method was used to estimate the unknown variables for each one-dimensional search. Central differencing was used to refine the solutions obtained. Automatic scaling in the Premium Solver platform was unable to function optimally when the parameters differed by many orders of magnitude, and so in this case manual scaling of the variables, by the use of log values, was also required.
RESULTS
CnErg1 causes incomplete block of macroscopic hERG current
Fig. 1 A illustrates typical hERG current traces recorded before and 2 min after application of 10 and 100 nM CnErg1. The best fit of the Hill equation (see Materials and Methods) to the full dose-response curve (solid line in Fig. 1 B) gave an IC50 value of 7.3 nM and a slope of 1.02. The data in Fig. 1 B also highlights that at concentrations orders-of-magnitude higher than the IC50, CnErg1 does not cause complete block of hERG current. The IC50 value of 7.3 nM, slope of Hill curve of ∼1, and 93.5% maximum block are very similar to the values reported previously for mammalian cells (22) and Xenopus oocytes (15,16).
FIGURE 1.

(A) Typical examples of current traces recorded from CHO-cells stably expressing hERG channels before and 2 min after exposure to (i) 10 nM CnErg1 and (ii) 100 nM CnErg1. Cells were depolarized from a holding potential of −80 mV to +40 mV for 500 ms, then stepped to 0 mV for 100 ms and then to −120 mV for 1 s (only the last two voltage steps are shown). (B) Toxin concentration dependence of hERG current inhibition. Data points show the mean ± SE for n = 5–14 experiments. The solid line shows best fit of the Hill equation (see Materials and Methods) to the data with IC50 = 7.3 nM, n = 1.02, and % maximum block = 93.5%.
Modification of hERG gating does not account for incomplete block
To explain incomplete block of macroscopic hERG current we first considered whether toxin-induced changes in hERG gating could account for the residual current observed at high concentrations of CnErg1. For example, a large positive shift in the V0.5 of activation would result in only a fraction of the channel population being activated by the depolarizing step in our voltage protocol, manifesting as reduced macroscopic current. Fig. 2 A illustrates typical traces recorded during a voltage-clamp protocol designed to measure steady-state activation (26). Addition of 1 μM CnErg1 (Fig. 2 A, ii) reduced current amplitude compared to control (Fig. 2 A, i) and caused a shift in the V0.5 of activation from −15.1 ± 1.6 mV to −1.6 ± 2.9 mV (n = 5, p < 0.05, student's t-test). Despite this shift, all channels were still fully activated at +40 mV.
FIGURE 2.

Effect of 1 μM CnErg1 on voltage dependence of activation of hERG channels. (A) Typical example of current traces recorded in the absence (i) and presence (ii) of 1 μM CnErg1 during 4 s depolarizing pulses to voltages in the range +40 mV to −60 mV followed by a 500 ms step to −60 mV. (B) Plot of normalized peak tail currents (see arrows in A) plotted against the voltage of the preceding test pulse in the absence (▴) and presence (□) of 1 μM CnErg1. The line of best fit is the Boltzmann function (see Materials and Methods) giving V0.5 for activation of 15.1 ± 1.6 mV to −1.6 ± 2.9 mV and slope factors of 8.9 ± 0.8 mV and 9.6 ± 1 mV for control and 1 μM CnErg1, respectively (mean ± SE, n = 5).
Fig. 3 illustrates typical examples of currents recorded during an envelope-of-tails protocol to measure the rate of activation at +40 mV for control (Fig. 2 A, i) and in the presence of 1 μM CnErg1 (Fig. 2 A, ii). The rate of activation at +40 mV in control (τact 176.2 ± 19.1 ms) was not significantly different to the rate of activation in the presence of 1 μM CnErg1 (τact 210.3 ± 22.4 ms, mean ± SE, n = 5, p < 0.05). In the voltage protocol used to assay toxin binding, channels were activated by a 500 ms step to +40 mV (see Fig. 1). Based on the data in Figs. 2 and 3, we would expect this protocol to elicit a very similar (and near maximal) level of channel activation in both control conditions and in the presence of 1 μM CnErg1. Thus, submaximal activation of channels in the presence of 1 μM CnErg1 cannot explain reduced macroscopic current.
FIGURE 3.

Effect of 1 μM CnErg1 on rate of activation of hERG channels. (A) Typical examples of currents recorded in the absence (i) and presence (ii) of 1 μM CnErg1 during envelope-of-tails voltage-clamp protocols to measure rates of activation at +40 mV (see Materials and Methods). Insets are plots of the peak tail current amplitude versus the duration of the test pulse from the traces shown. Solid lines are fits of single exponentials to the data giving time constants of 214.5 ms and 227.2 ms for control and CnErg1, respectively. (B) Normalized current plotted against duration of the test pulse for control (▴) and 1 μM CnErg1 (□). Mean τact were not significantly different (P < 0.05).
Typical examples of currents recorded during a voltage-protocol to measure steady-state inactivation for both control cells and cells in the presence of 1 μM CnErg1 are shown in Fig. 4 A. The V0.5 for inactivation was shifted from −82.7 ± 5.7 mV (mean ± SE, n = 6) for control to −105.9 ± 2.9 mV (mean ± SE, n = 4) in the presence of 1 μM CnErg1 (p < 0.05, student's t-test). CnErg1, 1 μM, also caused a slight slowing in the rates of inactivation (in the voltage range +60 to +20 mV; Fig. 5, A–C) and recovery from inactivation (in the voltage range 0 to −20 mV; Fig. 5 F). However, 1 μM CnErg1 did not affect the rate of deactivation over the voltage range tested (Fig. 5 E). Given that CnErg1 binds, at least in part, to the S5P linker domain (15,16), a site that is involved in hERG inactivation (8,10), it is not surprising that there are at least some changes in inactivation gating. Nevertheless, these changes in hERG inactivation gating are insufficient to the incomplete block of whole cell currents by 1 μM CnErg1.
FIGURE 4.

Effect of 1 μM CnErg1 on voltage dependence of inactivation of hERG channels. (A) Typical example of current traces recorded in the absence (i) and presence (ii) of 1 μM CnErg1 during a 1 s activating pulse to +40 mV followed by a 500 ms step in the range +40 mV to −170 mV. Tails current recorded below −80 mV show the characteristic hooked appearance reflecting recovery from inactivation followed by deactivation. The inset shows a typical tail current recorded at −140 mV with the dashed shaded line indicating how peak current was corrected for deactivation. A single exponential was fitted to the timecourse of deactivation at each holding potential and extrapolated back to the origin of the voltage step (*). (B) Plots of corrected peak tail currents in the absence (▴) and presence (□) of 1 μM CnErg1 for the traces shown in panel A. (C) Data from panel B replotted as conductance versus voltage. (D) Summary of the effect of 1 μM CnErg1 on the voltage dependence of hERG inactivation. The lines of best fit are the Boltzmann function (see Materials and Methods) giving V0.5 for inactivation of −82.7 ± 5.7 mV (mean ± SE, n = 6) and −105.9 ± 2.9 mV (mean ± SE, n = 4) for control and 1 μM CnErg1, respectively.
FIGURE 5.

Effect of 1 μM CnErg1 on rates of inactivation, recovery from inactivation, and deactivation. (A) Typical examples of currents recorded during a protocol to measure rate of inactivation in the absence (i) and presence (ii) of 1 μM CnErg1. (B) A representative sweep recorded at +30 mV from the trace in (Ai) illustrating how time constants for inactivation were measured. The dashed shaded line is a single exponential fitted to the timecourse of inactivation giving a time constant of 3.5 ms. (C) Summary of changes in τinact between 0 and +50 mV in the absence (▴) and presence (□) of 1 μM CnErg1. Below 0 mV exponentials could not be accurately fitted to the data in the presence of CnErg1 due to the small magnitude of the currents. (D) Measurement of time constants for recovery from inactivation (τrecov) and deactivation (τdeact). Characteristically hooked tail currents recorded in response to the voltage protocol shown (see traces in Fig. 1 A) were fitted with a double exponential (dashed shaded line). The example shown is recorded at −170 mV. (E) Summary of τdeact over the voltage range −100 mV to −170 mV in the absence (▴) and presence (□) of 1 μM CnErg1. (F) Summary of τrecov over the voltage range −150 mV to −180 mV for control (▴) and 1 μM CnErg1 (□). Above −150 mV exponentials could not be accurately fitted to τrecov due to the small magnitude of the currents in the presence of CnErg1. P < 0.05.
Reduced single channel conductance does not account for reduced macroscopic current in the presence of CnErg1
A second hypothesis that has been proposed, to explain incomplete block of hERG channels by CnErg1, is that the toxin binds near the pore but in an “off center” location and thereby causes a reduced channel conductance. Since it is not possible to accurately resolve the small unitary current amplitude of hERG channels using conventional single channel recording in the presence of low external [K+], to investigate whether CnErg1 binding affected the single channel conductance we used nonstationary noise analysis (28,29). Fig. 6 A shows theoretical curves for the variance versus mean current, which would be expected from (i) a reduction in single channel current amplitude from I to I/5 pA; and (ii) a reduction in open probability of the channel population (nPo) from 1 to 0.2 (black lines show control curves and shaded lines show curves for the reduced macroscopic current). In the first instance, the gradient of the parabola at the first root is reduced by a factor of 5, defining a reduced single channel conductance. The second root, at i.n pA (where i is single channel current amplitude and n is the number of channels) is also reduced by a factor of 5 corresponding to the decrease in I. Importantly, the entire parabola is still evident since the same number of activated channels are being examined, albeit with reduced conductance. In the second panel, where nPo is reduced, we only see a fraction of the parabola since only a fraction of the channel population is active. However, the gradient at the first root is the same (since single channel current amplitude is unaltered). Fig. 6 C shows a typical example of a plot of ensemble variance as a function of mean current recorded in response to the voltage protocol shown in Fig. 6 B in the absence (solid square) and presence (shaded square) of 1 μM CnErg1. The CnErg1 data clearly falls on the same line as the control data, resembling the theoretical curve in Fig. 6 A, ii, indicating a reduction in nPo in response to addition of CnErg1 rather than a reduction in the single channel current amplitude. The mean calculated single channel conductance at −120 mV was 3.2 ± 0.2 pS in control cells (mean ± SE, n = 5) and 3.8 ± 0.3 pS in the presence of 1 μM CnErg1 (mean ± SE, n = 5; p = not significant compared to control). The corollary of this is that conductance of the CnErg1 blocked fraction of the channel population (∼93%) is zero, i.e., CnErg1 causes complete block of individual hERG channels but ∼7% of the population remains unblocked at saturating concentrations of CnErg1.
FIGURE 6.

Nonstationary noise analysis. (A) Theoretical curves showing the effect of a reduction in single channel current amplitude from i to i/5 (i) and a reduction in nPo from 1 to 0.2 (ii) on mean current versus variance plots (see results for explanation). (B) Representative examples of leak-corrected mean current and ensemble variance as a function of time in the absence (solid traces) and presence (shaded traces) of 1 μM CnErg1. (C) Mean variance versus current plot for the traces in panel B for control (solid squares) and 1 mM CnErg1 (shaded squares). The solid line is a best fit of Eq. 3 (see Materials and Methods). The inset is a magnification of the boxed area highlighting that the data obtained in the presence of CnErg1 falls on the same line as the control data. (D) Bar graph showing mean single channel conductances calculated at −120 mV. There was no significant difference in conductance between control and in the presence of 1 μM CnErg1 (3.2 ± 0.2 pS and 3.8 ± 0.3 pS, respectively (mean ± SE, n = 5, P < 0.01)).
Kinetics of CnErg1 binding to hERG
Since neither modification of gating (Figs. 2–5) nor conductance (Fig. 6) could account for incomplete block of macroscopic current in the presence of 1 μM CnErg1, we next investigated whether the incomplete block could be caused by a kinetic mechanism. To measure the kinetics of toxin binding and dissociation we used an Intracel Picospritzer Rapid Perfusion System. Theoretically this device permits solution changes within 10 ms; however, our own tests with altering external K+ concentrations indicated that the time constant for solution change was 24 ± 2.5 ms (mean ± SE, n = 9), thus limiting us to the measurement of on-rates with time constants >∼100 ms.
Concentration dependence of toxin binding and unbinding at 22°C
In the example illustrated in Fig. 7 A, 100 nM CnErg1 resulted in a rapid onset of block that could be well described by a single exponential process with a time constant, τon, of 4.5 s. Similarly, toxin unbinding after washout of the toxin could be well described by a single exponential process with time constant, τoff, of 82.5 s.
FIGURE 7.

(A) Typical example of the timecourse of current inhibition during exposure to 100 nM CnErg1. Current values were measured from the peak during the −120 mV step (see voltage protocol in Fig. 1). Dashed and dashed-dotted lines are single exponential functions fitted to the data for the onset and offset of current block. (B) Summary of toxin concentration dependence of the association (k+1, [toxin], •) and dissociation (k−1, □) rate constants for CnErg1 block of hERG currents. Data points show mean ± SE for n = 5–14 experiments and the dashed line shows a straight line of best fit to the association rate constant data at low toxin concentrations. (C) Magnification of the boxed region from panel B showing straight line of best fit to rate constant data at low toxin concentrations.
The observation that the binding of CnErg1 to hERG can be fitted by a single exponential function is consistent with diffusion-limited binding (30); however, it does not prove it. To investigate whether CnErg1 binding is really diffusion-limited we determined the on-rates of CnErg1 binding over a wide range of concentrations. If binding is truly diffusion-limited, then the on-rates should have a linear dependence on toxin concentration over the entire range of concentrations (30). On-rates were calculated assuming that binding occurs via simple bimolecular mechanism,
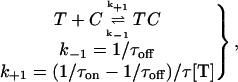 |
(Scheme 1) |
where T is CnErg1 toxin, C is channel, TC is the toxin bound channel, k+1 is the association rate constant (M−1 s−1), k−1 is the dissociation rate constant, and τon and τoff are the measured time constants for the onset and recovery from block (see Fig. 7 A).
The plot of [T] k+1 against [T] (shown in Fig. 7, B and C) is clearly nonlinear, indicating that binding of CnErg1 to hERG is not diffusion-limited. As expected, the value of k−1 is independent of toxin concentration.
Ligand binding to a macromolecule is more accurately described by the scheme
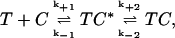 |
(Scheme 2) |
where T, C, and TC are as defined above (see Scheme 1), and TC* is the toxin channel encounter complex that represents the entire set of possible initial contact orientations formed between the toxin and the channel (21). The values k+1 and k−1 are the rate constants for diffusion up to and away from the encounter complex; and k+2 and k−2 are the rate constants for formation and dissociation of the toxin blocked state. If k+2 is considerably larger than k−1 then Scheme 2 effectively becomes Scheme 1 above, and this is the criterion for diffusion-limited binding.
The kinetic scheme shown in Scheme 2 was fitted to the timecourses for the onset and removal of block of hERG channels during wash-on and wash-off of CnErg1. To determine a unique set of values for k+1, k−1, k+2, and k−2, we simultaneously fitted the timecourses for onset and offset of block of hERG channels at concentrations ranging from 5 nM to 300 nM. The data shown in Fig. 8 represent the mean ± SE for the normalized timecourses of channel block and recovery (derived from timecourses such as that shown in Fig. 7 A) and the fitted lines represent the best fits for the simultaneous fitting of the model described in Scheme 2 to all five sets of data.
FIGURE 8.

Mean changes in hERG current magnitude during wash-on and wash-off of CnErg1 at concentrations ranging from 5 nM to 300 nM. Data points are mean ± SE for normalized current values from n = 5–14 experiments. The lines of best fit are derived from kinetic Scheme 2 using values for k+1, k−1, k+2, and k−2, shown in Table 1.
The values for k+1, k−1, k+2, and k−2 for the best fit of the model at 22°C are shown in Table 1 (see below). These values of k+1, k−1, k+2, and k−2 were used to derive “modeled” data for the toxin concentration dependence of channel inhibition (Fig. 9 A, solid line) and the toxin concentration dependence of the on-rates and off-rates (Fig. 9 B, solid lines). Individual data points in Fig. 9 are the original experimental data (from Figs. 1 B and 7 B) shown for comparison to the modeled curves.
TABLE 1.
Model derived rate constants for binding of CnErg1 to hERG channels at 22°C and 37°C
| k+1 (M−1 s−1) | k−1 (s−1) | k+2 (s−1) | k−2 (s−1) | |
|---|---|---|---|---|
| 22°C | 3.2 × 106 | 0.28 | 0.66 | 0.053 |
| 37°C | 1.4 × 106 | 1.1 | 0.67 | 0.064 |
FIGURE 9.

Model predicted values for (A) steady-state current block and (B) association and dissociation rates for CnErg1 block of hERG currents. Solid lines show simulated values and points are the experimental data reproduced from Figs. 1 B and 7 B.
The model accurately reproduces the incomplete block of hERG currents and suggests a mechanism by which this could occur. At high [toxin], TC* will be effectively permanently occupied since k+1 is many orders-of-magnitude greater than k−1. Under conditions where all channels are bound to toxin there will then be a simple equilibrium between channels in the encounter complex (TC*) and the channels blocked by bound toxin such that
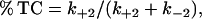 |
(4) |
i.e., from our model we would predict a maximum block of 92.6%. This value is very close to the experimentally determined value of 93.5%.
We can also use our kinetic model to predict the proportion of toxin-channel interactions that do not proceed to the blocked state, i.e.,
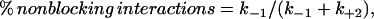 |
(5) |
which is ∼30% of interactions at 22°C.
Effect of temperature on kinetics of CnErg1 binding to hERG
Fig. 10 A shows a plot of the timecourse of changes in hERG current amplitude measured at 37°C during a 2-min exposure to 100 nM CnErg1 and subsequent washout. In this example, 100 nM CnErg1 caused a 58.6% inhibition of hERG current, which is considerably less than the 83.7% inhibition observed at 22°C (see Fig. 7 A). The best fit of the Hill equation (see Materials and Methods) to the full dose-response curve at 37°C (solid line in Fig. 10 B) gave an IC50 value of 64 nM and a slope of 1.02. Thus, the affinity of hERG channels for CnErg1 is approximately ninefold lower at 37°C compared to 22°C. Despite the lower affinity, the mechanism of block at 37°C appears to be fundamentally similar to that at 22°C. There is still incomplete block at μM concentrations of toxin (90.8% at 37°C compared to 93.5% at 22°C). Furthermore, the plot of [toxin], k+1 versus [toxin] is clearly nonlinear at 37°C (Fig. 10 C) as it was at 22°C (see Fig. 7, B and C). We therefore used the same model (Scheme 2) to fit the timecourses for the onset and offset of block of hERG channels during wash-on and wash-off of CnErg1 at 37°C (see Fig. 11). The unique set of values for k+1, k−1, k+2, and k−2, obtained by fitting the 37°C data are summarized in Table 1.
FIGURE 10.

Concentration-dependence of CnErg1 block of hERG currents at 37°C. (A) Typical example of the timecourse of current block and recovery during 120 s exposure to 100 nM CnErg1. Dashed lines are single exponential functions fitted to the onset and recovery from block. (B) Summary of the concentration dependence of steady-state block at 37°C (▴) and at 22°C (○, reproduced from Fig. 1 B). (C) Summary of toxin concentration dependence of the association (k+1.[toxin], •) and dissociation (k−1, □) rate constants for CnErg1 block of hERG currents at 37°C. Data points show mean ± SE for n = 4–7 experiments and the dashed line shows a straight line of best fit to the association rate constant data at low toxin concentrations.
FIGURE 11.

Mean changes in hERG current magnitude recorded at 37°C during wash-on and wash-off of CnErg1 at concentrations ranging from 30 nM to 3 μM. Data points are mean ± SE for normalized current values from n = 4–7 experiments. The lines of best fit are derived from kinetic Scheme 2 using the 37°C values for k+1, k−1, k+2, and k−2, shown in Table 1.
The data shown in Table 1 illustrates that there are only modest increases in the values of k+2 and k−2 between 22°C and 37°C. However, more significant changes are evident for k+1 and k−1, which showed a 2.6-fold decrease and 3.9-fold increase, respectively, between 22°C and 37°C. The decreased affinity for CnErg1 at 37°C compared to 22°C is therefore due to a combination of a reduction in the initial association rate, k+1, and an increase in the proportion of binding events that do not result in a blocked channel, i.e., the value of k−1/(k−1 + k+2) has increased from 30% to 60%.
Effect of temperature on secondary structure of CnErg1 and the CnErg1 binding site
The above kinetic data clearly indicate that at any given timepoint, in the presence of saturating concentrations of CnErg1, 6–9% of hERG channels exist as toxin-channel encounter complexes that are not blocked. The two major candidates for the nonproductive encounter complexes are CnErg1 binding in a nonproductive orientation or the toxin binding to a conformation of the channel protein distinct from the final channel blocked state. Given that the steady-state occupancy of the nonproductive encounter complexes is higher at higher temperatures, we investigated, using circular dichroism spectropolarimetry, whether the conformation of either the CnErg1 molecule, or the hERG S5P linker domain, which forms part of the CnErg1 binding site (8,10), varied with temperature.
The farUV CD spectrum of CnErg1 reflects the mixed α-helix and β-sheet structure of the folded toxin. The spectra obtained from CnErg1 at 22°C and 37°C are essentially indistinguishable, indicating that the secondary structure of the toxin is unaffected by temperature over this temperature range (Fig. 12 A, i). A synthetic peptide corresponding to residues S581–S599 of the hERG S5P linker shows an α-helical signal in the presence of SDS micelles (Fig. 12 B, i), but is mainly unstructured in aqueous solution (Fig. 12 C, i). Increasing the temperature of the sample from 22°C to 37°C results in small but significant changes in the α-helical content of the S5P peptide in both SDS and aqueous solutions. These changes can be most clearly seen from the difference spectra in Fig. 12, B, ii, and C, ii. It is important to note that the difference curves calculated for aqueous and SDS solutions were in the opposite direction, i.e., 22–37°C for the SDS micelle samples (indicating that the helical content has decreased with heating) but 37–22°C for the aqueous samples (indicating that the helical content has increased with heating under these conditions). The different effects of temperature on the helical content of CnErg1 and the hERG S5P peptide are summarized in Fig. 12 D, which shows plots for thermal melt curves where the ellipticity was monitored at 222 nm (negative peak for α-helix content) while the sample was heated. The α-helix content of CnErg1 is essentially unaltered over the temperature range 20–50°C while the α-helix content of S5P decreases steadily in SDS micelles ([θ]MRW −9050 and −8340 deg cm−2 dmol−1 at 22°C and 37°C, respectively) but increases steadily in aqueous solution ([θ]MRW −1420 and −1740 deg cm−2 dmol−1 at 22°C and 37°C, respectively). This suggests that the temperature dependence of changes in the occupancy of the toxin-channel encounter complex is more likely due to changes in the structure of the CnErg1 binding site than to changes in the toxin itself.
FIGURE 12.

CD spectra for (A) CnErg1, (B) hERG S5P helix peptide in SDS micelles, and (C) hERG S5P peptide in aqueous solution, at 22°C and 37°C. Insets to panels A (i) and B (i) show the structures of the respective peptides, determined by two-dimensional NMR spectroscopy. Panel (ii) shows the difference spectrum for each peptide and condition. (D) Thermal melt curves for CnErg1, hERG S5P helix peptide in SDS micelles, and hERG S5P helix peptide in water. The shaded area indicates the temperature range 22°C to 37°C corresponding to the temperatures at which the kinetics of CnErg1 binding were assayed in this study. Over this range the α-helix content of the CnErg1 peptide (measured from the negative molar ellipticity at 222 nm) is essentially unchanged (as it is over the entire range tested). Between 22°C and 37°C the α-helix content of the hERG S5P helix peptide steadily increases in the aqueous solution ([θ]MRW increases from −1420 to −1740 deg cm−2 dmol−1) but decreases in SDS micelles ([θ]MRW decreases from −9050 to −8340 deg cm−2 dmol−1).
DISCUSSION
Toxins are a very valuable tool for probing ion channel structure and function (23,31). The discovery of a series of highly selective and high affinity toxins, including CnErg1 (17), that inhibit hERG channels by binding to the outer pore domain region (15,16) has opened up the possibility of using these toxins to obtain the experimental constraints required to permit modeling of the complete pore domain of the hERG channel structure (32). There are, however, a number of questions about how CnErg1 binds to hERG that need to be answered before we can use this toxin to gain insights into the structure of the unique outer pore domain of hERG. Most important among these is to determine the mechanism by which CnErg1 blocks hERG current and to explain the observation that high concentrations of CnErg1 cause incomplete block of the channel.
Is CnErg1 a pore blocker or a gating modifier?
There are two major classes of toxins that have been used to probe K+ channel structure and function—the pore-blocking toxins and the gating-modifying toxins. Typical pore-blocking toxins, including, e.g., charybdotoxin, kaliotoxin, and iberiotoxin, bind in the outer vestibule of the channel and block ion conduction by physically occluding the pore (23,31). Gating-modifier toxins typically bind to the voltage-sensor domain (33–35) and shift the voltage range for channel activation. One of the features of voltage-sensor gating modifier toxins is that the apparent channel block can be overcome by increasing the voltage driving activation (34) and so if sufficiently positive voltages are used, residual currents can always be observed.
The typical features of pore-blocking toxins include binding to the outer pore domain (36–38), binding that is sensitive to changes in permeant ion concentrations (39), and to tetraethyl ammonium an external pore blocker (40). The association of pore-blocking toxins also typically involves electrostatic interactions (19) and this results in toxin-channel association rates closely approximating diffusion-limited control (20). CnErg1 binds to the outer vestibule of hERG (15,16) similar to the site for other pore blocking toxins and it is sensitive to tetraethyl ammonium (16). However, CnErg1 binding to hERG is not sensitive to changes in [K+] and is relatively insensitive to changes in ionic strength of the extracellular solutions (16). Furthermore, CnErg1 does not cause complete block of channels (16,22). These atypical features of CnErg1 binding to hERG are very similar to those for BeKm-1 binding to hERG (41). It has therefore been suggested that CnErg1 and BeKm-1 bind to overlapping sites in the turret region rather than inserting into the selectivity filter (16,23,24) and two hypotheses have been proposed for their mechanisms of action: either they act as “unconventional” gating modifiers (41) or cause only partial occlusion thereby permitting ion permeation albeit with a lower conductance (24,42).
The experimental data presented in this study disproves both of these hypotheses with respect to CnErg1 binding to hERG. Firstly, we have presented a comprehensive description of the effect of CnErg1 on hERG gating and found that while there are changes in the voltage dependence of several gating parameters in the presence high concentrations of CnErg1 (see Figs. 2–5), they are insufficient to cause the degree of macroscopic current inhibition observed with 1 μM CnErg1. Therefore the inhibition of macroscopic hERG current by CnErg1 cannot be explained by modification of channel gating behavior. Secondly, the single channel conductance for the residual current seen in the presence of 1 μM CnErg1 was identical to that observed for control hERG currents (see Fig. 6 d). Therefore a reduced conductance of hERG channels bound to CnErg1 cannot account for the residual macroscopic current.
Incomplete block of hERG by CnErg1 can be explained by a kinetic mechanism
If incomplete block is not due to gating modification and not due to reduced single channel conductance, then how can it be explained? The binding of toxins to channels involves both diffusional and nondiffusional steps: the toxin first must diffuse up to its receptor site on the channel before it can bind. The subsequent binding step then involves rearrangement of amino-acid side chains (on the channel or the toxin), displacement of hydration water, and/or formation of hydrogen bonds necessary to produce the bound state. Similarly, dissociation must involve the disruption of favorable short-range interactions and then diffusion of toxin away from the receptor site. This is summarized in Scheme 2 (see above), where k+1 and k−1 are the rate constants for diffusion up to and away from the encounter complex, and k+2 and k−2 are the rate constants for formation and dissociation of the toxin blocked state. If k+2 ≫ k−1, then the first step becomes the rate-limiting step, i.e., binding is said to be diffusion-limited and Scheme 2 can be approximated by Scheme 1 (see above). A key observation we have made in this study is that binding of CnErg1 to hERG is not diffusion-limited (Figs. 7 and 10). Therefore, to fully understand CnErg1 binding to hERG we need to understand not just the blocked toxin-channel complex but also the toxin-channel encounter complex.
To derive unique global solutions for the values of k+1, k−1, k+2, and k−2, it was necessary to simultaneously fit the model to the timecourses for block and unblock at multiple toxin concentrations (see Fig. 8, Table 1). Given that the hERG channel can exist in multiple different conformational states, including a series of closed states, at least one open state and at least one inactivated state (3), the kinetic model shown in Scheme 2 is undoubtedly a simplification. Nevertheless, this scheme was able to reproduce the data accurately for the timecourses of toxin binding and dissociation at all toxin concentrations examined (see Fig. 9). Furthermore, the model was able to reproduce the incomplete blockade of channels at high concentrations of toxin. It should be noted that the binding of CnErg1 (see Supplementary Material) and the related toxin BeKm1 (45) to hERG varies according to the voltage protocol used to elicit current during toxin binding. Thus the values obtained for the rate constants in the kinetic model shown in Scheme 2 are specific for the voltage protocol used in this study. However, whatever voltage protocol is used, the basic mechanism of block is the same, i.e., block of macroscopic current is incomplete at high concentrations of toxin, as seen in this study and in the literature (16,22,41,45). The mechanistic insight gained from this study is, therefore, generally applicable to CnErg1 binding to hERG, and not limited to the specific voltage protocol used.
It is also important to highlight that incorporating a toxin-channel encounter complex in our model does not in itself necessarily produce incomplete block. Rather, incomplete block is explained by the relatively fast dissociation rate from the blocked channel conformation relative to the rate of conversion of the toxin-channel encounter complex to the blocked channel conformation, i.e., the value of k−2 is of similar order of magnitude to k+2 in our model (see Table 1). Thus at saturating concentrations of toxin (>1 μM at 22°C) where all channels will be toxin-bound, there is a simple equilibrium between the toxin-channel encounter complex and blocked channels with the percentage of blocked channels given by Eq. 4 (see above). From our modeling data we would therefore predict maximum block at 22°C and 37°C to be 92.6% and 91.2%, respectively. These values are very close to the experimentally determined values of 93.5 and 90.8%, respectively. Thus, despite its simplicity, this kinetic scheme provides an accurate model of CnErg1 binding to hERG channels.
Temperature-dependence of CnErg1 block of hERG
The Kd for CnErg1 block of hERG channels increased approximately ninefold between 22°C and 37°C. When we fitted the Scheme 2 model to timecourses of channel block and unblock at 37°C (Fig. 11) and compared the values for the rate constants at 22°C and 37°C, the major differences were seen in the values for k+1 and k−1 (see Table 1). Consequently, at 37°C, ∼60% of encounters are unproductive compared to ∼30% at 22°C.
This suggests that the temperature-dependent changes in the CnErg1 binding to hERG could be explained by decreased stability of either the toxin and/or the toxin binding site on the channel at higher temperatures. Our CD data shows the hERG S5P linker domain (a region previously shown to be an important component of the CnErg1 binding site (15,16)) is thermally labile, whereas the structure of CnErg1 is stable over a wide range of temperatures (see Fig. 12). The correlation between temperature-dependent changes in S5P secondary structure and binding affinity is consistent with decreased stability of the toxin binding site on the channel being the explanation for the decreased CnErg1 affinity at higher temperatures. Our data is also consistent with the suggestion that the S5P linker domain has a highly dynamic structure (11).
Possible model of CnErg1 interaction with hERG
The cartoon in Fig. 13 depicts a schematic model that can explain our data and the incomplete block of macroscopic hERG currents. In our model, CnErg1 binds to the S5P linker, as is suggested by previous site-directed mutagenesis studies (15,16) and by the correlation between binding affinity and temperature-dependent structural changes in the S5P domain described in this study. However, it should be noted that the precise binding site for CnErg1 on hERG remains to be definitively determined. The three panels in Fig. 13 depict the three species in Scheme 2, i.e., toxin + free toxin (T + C), the toxin-channel encounter complex (TC*) and the toxin-blocked channel (TC). The toxin binds to the amphipathic α-helix in the hERG S5P linker (15,16) (Fig. 13 B). However, unlike previous models (24), we propose that the amphipathic α-helix must be sufficiently peripheral that the bound toxin molecule does not occlude the ion conduction pathway. We propose that there is a subsequent conformational rearrangement that brings the toxin close to the central axis of the pore where it can block ion conduction (Fig. 13 C). The initial interaction is very temperature-sensitive, and our CD data suggests that this is due to thermal lability of the amphipathic α-helix in the hERG S5P linker (depicted in black in Fig. 13). The interactions that stabilize the blocked conformation (Fig. 13 C) must be relatively weak and hence have a rapid dissociation rate, relative to the association rate; this explains the incomplete block of macroscopic current and also explains the low temperature sensitivity of this second step.
FIGURE 13.

Cartoon depicting proposed model of CnErg1 binding to hERG. Panel A depicts the free toxin (striped) and channel (two subunits shown) drawn approximately to scale based on the NMR structure for CnErg1 (22) and crystal structure of KcsA (46). The hERG S5P amphipathic α-helix, that forms part of the CnErg1 binding site, is shown in black. Panel B depicts the toxin channel encounter complex (TC* in Scheme 2). Panel C depicts the toxin-blocked channel (TC in Scheme 2). The rate constants at 22°C (shaded) and at 37°C are those shown in Table 1. The two important features of the scheme are that 1), the initial encounter of toxin with the channel occurs at a site that does not overlap the central axis and so permits ion conduction, depicted by the long arrow; and 2), the toxin-channel encounter complex undergoes a conformational change that results in occlusion of the pore. The nature of that conformational change, however, is speculative. In this model, the temperature-dependence of CnErg1 binding to hERG is explained by the hERG S5P amphipathic α-helix (putative CnErg1 binding site) being thermally labile and so increasing temperature primarily affects the values for k+1 and k−1.
Do other hERG toxins bind by a similar mechanism?
CnErg1 is only one of dozens of toxins that can inhibit hERG channels (14). All of the toxins for which hERG binding has been characterized in detail, CnErg1 (this study; (16)), BeKm-1 (42), APETX1 (43), and BmTx3 (44) cause incomplete blockade of hERG currents. In the absence of detailed kinetic experiments, such as those performed in this study, it is not possible to determine whether all of these toxins interact with hERG in the same way as CnErg1. However, Milnes and colleagues (45) have shown that the affinity of BeKm-1 for hERG decreases five-to-tenfold at 37°C compared to 22°C, which is very similar to that reported here for CnErg1. It is therefore possible that the binding of BeKm-1 to hERG occurs via the same mechanism as that for CnErg1. Zhang and colleagues (41) examined the effects of high concentrations of BeKm1 on hERG gating and reported substantial alterations in gating behavior. Though superficially this seems at odds with the changes we have reported with CnErg1, we believe that the effects are fundamentally similar. While we report a smaller positive shift in the V0.5 of activation (13.5 mV, Fig. 2) compared to that reported by Zhang et al. for BeKm-1 (54.9 mV, (41)), we believe that both data sets are unavoidably skewed. Both CnErg1 and BeKm-1 show voltage-dependent unbinding from hERG at depolarized potentials (41,45) and this is more prominent for BeKm-1 than CnErg1 (45). Therefore at the more depolarized sweeps of the steady-state activation protocols used in both studies, toxin unbinding progressively increases resulting in increasing current amplitude. These effects manifest as an apparent rightward shift in the V0.5 of activation of the steady-state activation curve and a decrease in slope of the Boltzmann function. Due to the faster dissociation rate for BeKm-1 unbinding compared to CnErg1 (45) the apparent shift in the voltage-dependence of activation would be expected to be greater in the presence of BeKm-1 than CnErg1, exactly as observed (compare (41) with the data presented here).
SUMMARY
In this study we have systematically examined the possible mechanisms for incomplete block of hERG by CnErg1, namely: 1), modification of hERG gating; 2), incomplete block of single channel conductance; and 3), a kinetic mechanism. Our data indicates that the last of these is correct. Specifically, we have shown that incomplete block is due to the forward and backward rate constants for the final rearrangements, which must be made for the toxin-channel encounter complex to form the blocked state (k+2 and k−2 in Scheme 2) being of similar magnitude.
The kinetic scheme for CnErg1 binding to hERG channels presented in this study provides a framework to interpret the binding of mutant toxins and the analysis of toxin footprinting data. For example, we anticipate that different mutants will have differential effects on the first and second components of the reaction scheme shown in Scheme 2, and so should enable us to gain insights into conformational changes that take place during the transition between the toxin-channel encounter complex and the blocked conformations of the channel toxin complex.
SUPPLEMENTARY MATERIAL
An online supplement to this article can be found by visiting BJ Online at http://www.biophysj.org.
Acknowledgments
We gratefully acknowledge the expert technical assistance of Jane Bursill and Ken Wyse. We thank Paramjit Bansal and Paul Alewood for the supply of peptides and toxins and Allan Torres, Philip Kuchel, and Cath Clarke for valuable discussions.
This work was supported in part by an Australian Research Council project grant (to P.W.K. and J.I.V.) and in part by a National Health and Medical Research Council project grant (to J.I.V. and T.J.C.). J.I.V. is an NHMRC Senior Research Fellow and M.S. is an NHMRC R.D. Wright Career Development Fellow.
References
- 1.Sanguinetti, M. C., and M. Tristani-Firouzi. 2006. hERG potassium channels and cardiac arrhythmia. Nature. 440:463–469. [DOI] [PubMed] [Google Scholar]
- 2.Vandenberg, J. I., B. D. Walker, and T. J. Campbell. 2001. hERG K+ channels: friend and foe. Trends Pharmacol. Sci. 22:240–246. [DOI] [PubMed] [Google Scholar]
- 3.Vandenberg, J. I., A. M. Torres, T. J. Campbell, and P. W. Kuchel. 2004. The hERG K+ channel: progress in understanding the molecular basis of its unusual gating kinetics. Eur. Biophys. J. 33:89–97. [DOI] [PubMed] [Google Scholar]
- 4.Lu, Y., M. P. Mahaut-Smith, A. Varghese, C. L. Huang, P. R. Kemp, and J. I. Vandenberg. 2001. Effects of premature stimulation on hERG K+ channels. J. Physiol. 537:843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith, P. L., T. Baukrowitz, and G. Yellen. 1996. The inward rectification mechanism of the hERG cardiac potassium channel. Nature. 379:833–836. [DOI] [PubMed] [Google Scholar]
- 6.Spector, P. S., M. E. Curran, A. Zou, M. T. Keating, and M. C. Sanguinetti. 1996. Fast inactivation causes rectification of the IKr channel. J. Gen. Physiol. 107:611–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Trudeau, M. C., J. W. Warmke, B. Ganetzky, and G. A. Robertson. 1995. hERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 269:92–95. [DOI] [PubMed] [Google Scholar]
- 8.Liu, J., M. Zhang, M. Jiang, and G. N. Tseng. 2002. Structural and functional role of the extracellular s5-p linker in the hERG potassium channel. J. Gen. Physiol. 120:723–737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Torres, A. M., P. S. Bansal, M. Sunde, C. E. Clarke, J. A. Bursill, D. J. Smith, A. Bauskin, S. N. Breit, T. J. Campbell, P. F. Alewood, P. W. Kuchel, and J. I. Vandenberg. 2003. Structure of the hERG K+ channel S5P extracellular linker: role of an amphipathic α-helix in C-type inactivation. J. Biol. Chem. 278:42136–42148. [DOI] [PubMed] [Google Scholar]
- 10.Clarke, C. E., A. P. Hill, J. Zhao, M. Kondo, R. N. Subbiah, T. J. Campbell, and J. I. Vandenberg. 2006. Effect of S5P {α}-helix charge mutants on inactivation of hERG K+ channels. J. Physiol. 573:291–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jiang, M., M. Zhang, I. V. Maslennikov, J. Liu, D. M. Wu, Y. V. Korolkova, A. S. Arseniev, E. V. Grishin, and G. N. Tseng. 2005. Dynamic conformational changes of extracellular S5-P linkers in the hERG channel. J. Physiol. 569:75–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hidalgo, P., and R. MacKinnon. 1995. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 268:307–310. [DOI] [PubMed] [Google Scholar]
- 13.Gross, A., and R. MacKinnon. 1996. Agitoxin footprinting the Shaker potassium channel pore. Neuron. 16:399–406. [DOI] [PubMed] [Google Scholar]
- 14.Corona, M., G. B. Gurrola, E. Merino, R. R. Cassulini, N. A. Valdez-Cruz, B. Garcia, M. E. Ramirez-Dominguez, F. I. Coronas, F. Z. Zamudio, E. Wanke, and L. D. Possani. 2002. A large number of novel Ergtoxin-like genes and ERG K+-channels blocking peptides from scorpions of the genus Centruroides. FEBS Lett. 532:121–126. [DOI] [PubMed] [Google Scholar]
- 15.Pardo-Lopez, L., J. Garcia-Valdes, G. B. Gurrola, G. A. Robertson, and L. D. Possani. 2002. Mapping the receptor site for Ergtoxin, a specific blocker of ERG channels. FEBS Lett. 510:45–49. [DOI] [PubMed] [Google Scholar]
- 16.Pardo-Lopez, L., M. Zhang, J. Liu, M. Jiang, L. D. Possani, and G. N. Tseng. 2002. Mapping the binding site of a human ether-a-go-go-related gene-specific peptide toxin (ErgTx) to the channel's outer vestibule. J. Biol. Chem. 277:16403–16411. [DOI] [PubMed] [Google Scholar]
- 17.Gurrola, G. B., B. Rosati, M. Rocchetti, G. Pimienta, A. Zaza, A. Arcangeli, M. Olivotto, L. D. Possani, and E. Wanke. 1999. A toxin to nervous, cardiac, and endocrine ERG K+ channels isolated from Centruroides noxius scorpion venom. FASEB J. 13:953–962. [PubMed] [Google Scholar]
- 18.Ranganathan, R., J. H. Lewis, and R. MacKinnon. 1996. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 16:131–139. [DOI] [PubMed] [Google Scholar]
- 19.Park, C. S., and C. Miller. 1992. Mapping function to structure in a channel-blocking peptide: electrostatic mutants of charybdotoxin. Biochemistry. 31:7749–7755. [DOI] [PubMed] [Google Scholar]
- 20.Miller, C. 1990. Diffusion-controlled binding of a peptide neurotoxin to its K+ channel receptor. Biochemistry. 29:5320–5325. [DOI] [PubMed] [Google Scholar]
- 21.Escobar, L., M. J. Root, and R. MacKinnon. 1993. Influence of protein surface charge on the bimolecular kinetics of a potassium channel peptide inhibitor. Biochemistry. 32:6982–6987. [DOI] [PubMed] [Google Scholar]
- 22.Torres, A. M., P. Bansal, P. F. Alewood, J. A. Bursill, P. W. Kuchel, and J. I. Vandenberg. 2003. Solution structure of CnErg1 (Ergtoxin), a hERG specific scorpion toxin. FEBS Lett. 539:138–142. [DOI] [PubMed] [Google Scholar]
- 23.Rodriguez de la Vega, R. C., E. Merino, B. Becerril, and L. D. Possani. 2003. Novel interactions between K+ channels and scorpion toxins. Trends Pharmacol. Sci. 24:222–227. [DOI] [PubMed] [Google Scholar]
- 24.Xu, C. Q., S. Y. Zhu, C. W. Chi, and J. Tytgat. 2003. Turret and pore block of K+ channels: what is the difference? Trends Pharmacol. Sci. 24:446–449. [DOI] [PubMed] [Google Scholar]
- 25.Walker, B. D., C. B. Singleton, J. A. Bursill, K. R. Wyse, S. M. Valenzuela, M. R. Qiu, S. N. Breit, and T. J. Campbell. 1999. Inhibition of the human ether-a-go-go-related gene (hERG) potassium channel by cisapride: affinity for open and inactivated states. Br. J. Pharmacol. 128:444–450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vandenberg, J. I., A. Varghese, Y. Lu, J. A. Bursill, M. P. Mahaut-Smith, and C. L. Huang. 2006. Temperature dependence of human ether-a-go-go-related gene K+ currents. Am. J. Physiol. Cell Physiol. 291:C165–C175. [DOI] [PubMed] [Google Scholar]
- 27.Subbiah, R. N., C. E. Clarke, D. J. Smith, J. Zhao, T. J. Campbell, and J. I. Vandenberg. 2004. Molecular basis of slow activation of the human ether-a-go-go related gene potassium channel. J. Physiol. 558:417–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sigg, D., E. Stefani, and F. Bezanilla. 1994. Gating current noise produced by elementary transitions in Shaker potassium channels. Science. 264:578–582. [DOI] [PubMed] [Google Scholar]
- 29.Sigworth, F. J. 1980. The variance of sodium current fluctuations at the node of Ranvier. J. Physiol. 307:97–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fersht, A. 1999. Structure and Mechanism in Protein Science: A Guide to Enzyme Catalysis and Protein Folding. W.H. Freeman, New York.
- 31.Garcia, M. L., Y. Gao, O. B. McManus, and G. J. Kaczorowski. 2001. Potassium channels: from scorpion venoms to high-resolution structure. Toxicon. 39:739–748. [DOI] [PubMed] [Google Scholar]
- 32.Tseng, G. N., and H. R. Guy. 2005. Structure-function studies of the outer mouth and voltage sensor domain of hERG. Novartis Found. Symp. 266:19–45. [PubMed] [Google Scholar]
- 33.Swartz, K. J., and R. MacKinnon. 1997. Mapping the receptor site for hanatoxin, a gating modifier of voltage-dependent K+ channels. Neuron. 18:675–682. [DOI] [PubMed] [Google Scholar]
- 34.Swartz, K. J., and R. MacKinnon. 1997. Hanatoxin modifies the gating of a voltage-dependent K+ channel through multiple binding sites. Neuron. 18:665–673. [DOI] [PubMed] [Google Scholar]
- 35.Wang, J., J. J. Salata, and P. B. Bennett. 2003. Saxitoxin is a gating modifier of hERG K+ channels. J. Gen. Physiol. 121:583–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.MacKinnon, R., L. Heginbotham, and T. Abramson. 1990. Mapping the receptor site for charybdotoxin, a pore-blocking potassium channel inhibitor. Neuron. 5:767–771. [DOI] [PubMed] [Google Scholar]
- 37.Aiyar, J., J. P. Rizzi, G. A. Gutman, and K. G. Chandy. 1996. The signature sequence of voltage-gated potassium channels projects into the external vestibule. J. Biol. Chem. 271:31013–31016. [DOI] [PubMed] [Google Scholar]
- 38.MacKinnon, R., and C. Miller. 1988. Mechanism of charybdotoxin block of the high-conductance, Ca2+-activated K+ channel. J. Gen. Physiol. 91:335–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldstein, S. A., and C. Miller. 1993. Mechanism of charybdotoxin block of a voltage-gated K+ channel. Biophys. J. 65:1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Miller, C. 1988. Competition for block of a Ca2+-activated K+ channel by charybdotoxin and tetraethylammonium. Neuron. 1:1003–1006. [DOI] [PubMed] [Google Scholar]
- 41.Zhang, M., Y. V. Korolkova, J. Liu, M. Jiang, E. V. Grishin, and G. N. Tseng. 2003. BeKm-1 is a hERG-specific toxin that shares the structure with ChTx but the mechanism of action with ErgTx1. Biophys. J. 84:3022–3036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Korolkova, Y. V., G. N. Tseng, and E. V. Grishin. 2004. Unique interaction of scorpion toxins with the hERG channel. J. Mol. Recognit. 17:209–217. [DOI] [PubMed] [Google Scholar]
- 43.Diochot, S., E. Loret, T. Bruhn, L. Beress, and M. Lazdunski. 2003. APETx1, a new toxin from the sea anemone Anthopleura elegantissima, blocks voltage-gated human ether-a-go-go-related gene potassium channels. Mol. Pharmacol. 64:59–69. [DOI] [PubMed] [Google Scholar]
- 44.Huys, I., C. Q. Xu, C. Z. Wang, H. Vacher, M. F. Martin-Eauclaire, C. W. Chi, and J. Tytgat. 2004. BmTx3, a scorpion toxin with two putative functional faces separately active on A-type K+ and hERG currents. Biochem. J. 378:745–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Milnes, J. T., C. E. Dempsey, J. M. Ridley, O. Crociani, A. Arcangeli, J. C. Hancox, and H. J. Witchel. 2003. Preferential closed channel blockade of hERG potassium currents by chemically synthesized BeKm-1 scorpion toxin. FEBS Lett. 547:20–26. [DOI] [PubMed] [Google Scholar]
- 46.Doyle, D. A., J. Morais Cabral, R. A. Pfuetzner, A. Kuo, J. M. Gulbis, S. L. Cohen, B. T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]