

Abstract
Interactions of the local anesthetic tetracaine with unilamellar vesicles made of dimyristoyl or dipalmitoyl phosphatidylcholine (DMPC or DPPC), the latter without or with cholesterol, were examined by following changes in the drug's fluorescent properties. Tetracaine's location within the membrane (as indicated by the equivalent dielectric constant around the aromatic fluorophore), its membrane:buffer partition coefficients for protonated and base forms, and its apparent pKa when adsorbed to the membrane were determined by measuring, respectively, the saturating blue shifts of fluorescence emission at high lipid:tetracaine, the corresponding increases in fluorescence intensity at this lower wavelength with increasing lipid, and the dependence of fluorescence intensity of membrane-bound tetracaine (TTC) on solution pH. Results show that partition coefficients were greater for liquid-crystalline than solid-gel phase membranes, whether the phase was set by temperature or lipid composition, and were decreased by cholesterol; neutral TTC partitioned into membranes more strongly than the protonated species (TTCH+). Tetracaine's location in the membrane placed the drug's tertiary amine near the phosphate of the headgroup, its ester bond in the region of the lipids' ester bonds, and associated dipole field and the aromatic moiety near fatty acyl carbons 2–5; importantly, this location was unaffected by cholesterol and was the same for neutral and protonated tetracaine, showing that the dipole-dipole and hydrophobic interactions are the critical determinants of tetracaine's location. Tetracaine's effective pKa was reduced by 0.3–0.4 pH units from the solution pKa upon adsorption to these neutral bilayers, regardless of physical state or composition. We propose that the partitioning of tetracaine into solid-gel membranes is determined primarily by its steric accommodation between lipids, whereas in the liquid-crystalline membrane, in which the distance between lipid molecules is larger and steric hindrance is less important, hydrophobic and ionic interactions between tetracaine and lipid molecules predominate.
INTRODUCTION
Many drugs interact with membranes to pass into or through cells or to penetrate to hydrophobic sites on membrane-associated proteins. Local anesthetics, class I antiarrhythmic agents, and other amphipathic amines exert their primary actions on ionic Na+ channels either from the cytosol, after permeating the plasma membrane, or from within the membrane phase itself (1–6). Equally important may be the proposed role of the membrane in controlling the rate-limiting step for dissociation of the drug from its target protein, e.g., via the so-called “hydrophobic pathway” of escape from voltage-gated Na+ channels (3,7).
The wish to understand the molecular mechanisms by which local anesthetics block different types of ion channels (8,9) and the mechanisms by which irreversible nerve injury is caused by local anesthetics (10) has driven the study of local anesthetic-membrane interactions for over three decades. The dynamics of drug:membrane interactions have been investigated previously by others using a variety of physical methods, including x-ray diffraction (11,12), NMR (13–27), electron paramagnetic resonance (28–33), and Fourier transform infrared spectroscopy (24,34–38). These methods, although providing good spatial resolution, are very slow and cannot resolve any kinetic processes in this interaction. Furthermore, these earlier studies had not integrated the roles of membrane dynamics and drug ionization, and often the membranes were made of a heterogeneously acylated lipid, e.g., egg phostphatidyl choline, and included no cholesterol. In these studies we have used the intrinsic fluorescence of the local anesthetic tetracaine (39,40) to study its interactions with bilayer membranes made of pure lipids, sometimes including cholesterol, and to validate its use for later kinetic studies.
The interaction of tetracaine with lipid bilayers is complicated by two factors. One is that tetracaine, an amphipathic molecule, has detergent properties dependent on its concentration and on the solution pH. For example, charged tetracaine may form micelles (29,41), for which it has a different behavior than as monomers in solution, and also can dissolve membranes through its detergent action. As a consequence of the second property, at high concentrations tetracaine may induce significant changes in membrane structure and phase-transition parameters, even forming tetracaine-lipid mixed micelles (23,29,41–44). Additionally, tetracaine at high concentration can form dimers, at least in the membrane (26). This is potentially problematic since techniques used previously to study the interaction of local anesthetics with model or nerve membranes, e.g., NMR, quite frequently used high concentrations of the investigated substance and therefore would have studied phenomena that do not occur during normal local anesthesia.
Previously we examined the equilibrium interactions of a series of structurally related local anesthetics with micelles made from different detergents to weigh the importance of hydrophobicity, the dipole moment of the local anesthetics' “linker” moiety (amide versus ester or ether), and drug ionization on the affinity and the “depth” from the surface for drug binding to micelles (45), following earlier studies by Garcia-Soto and Fernandez (46). We found that both hydrophobicity and ionization of these drugs, as well as the type of and charge on the detergent, affected the equilibrium affinity and depth, with more hydrophobic local anesthetics, in the uncharged form, binding tighter and deeper within micelles, and drug protonation positioning molecules closer to the micellar surface. Drug molecules containing stronger dipoles (i.e., the amide bond in procainamide > the ester bound in procaine) had both weaker binding affinity and a shallower interfacial depth when bound. The pKa of local anesthetics bound to neutral detergents was lower than in solution.
In this work we have extended the micellar study by examining the interaction of tetracaine with model membranes made of dipalmitoylphosphatidyl choline (DPPC), without or with cholesterol, or dimyristoylphosphatidyl choline (DMPC) to answer the following questions: 1), How does the molecular composition or physical state of the membrane affect tetracaine's adsorption to and distribution within the membrane? 2), How does the state of drug ionization affect its affinity and binding position? 3), How is tetracaine's ionization altered by membrane adsorption, i.e., is the effective pKa of membrane-bound tetracaine different from that of free tetracaine in aqueous solution?
The intrinsic fluorescence of drug molecules containing conjugated electron orbitals is a useful probe of both the drug's state of ionization and the polarity of the medium surrounding the fluorophore (39,40). The fluorescence intensity (I) is a function of the quantum efficiency of photon emission, which is reduced both by increasing solvent polarity and by drug ionization. Protonation of local anesthetics causes electron shifts, which results in decreased I, primarily through the absorbance process (47) with minimal spectral shifts. By measuring pH-dependent changes in I, we are able to establish the pKa of TTC in either the aqueous or membrane-bound state. In addition, the wavelength of maximum emission (λmax) is governed by the fluorescence transition dipole (π*→π), which is reduced by interaction with polar solvent molecules. We exploited the dependence of λmax on local polarity by interpreting the blue shifts of λmax in the presence of drug-adsorbing membranes as a reflection of the average polarity of the membrane regions surrounding the aromatic moieties of the drug, calibrated by its blue shift in homogeneous organic or mixed solvents of known dielectric constant. The results give a picture of tetracaine:membrane dynamics, mostly consistent with previous reports but with some important differences, and extend the findings to show the relative importance of the physical state and composition of the membrane for drug interactions. Furthermore, the findings validate this fluorescence method for investigating interactions of many other drugs and membranes, with no special requirements for labeled drugs or exogenous probes, and establish its use for kinetic studies of these interactions.
MATERIALS AND METHODS
Reagents
Tetracaine (HCl) was obtained from Sigma-Aldrich Chemicals (St. Louis, MO). All phospholipids (PL) were obtained from Avanti Polar Lipids (Alabaster, AL), were >99% pure dissolved in CHCl3, and were stored at −20°C and used as received. Cholesterol was obtained from Avanti Polar Lipids as a powder. Radio-labeled 14C-dipalmitoyl phosphatidylcholine in toluene:methanol (1:1) was purchased from Perkin-Elmer Life and Analytical Sciences (Boston, MA).
Solutions
All experiments were conducted in a standard aqueous medium (SAM), which contained 150 mM NaCl in doubly deionized (18 Mohm-cm), highly purified, pyrogen-free water (Water Systems 5601, Millipore, Woburn, MA), buffered with a combination of 3 Good buffers (48), 2 mM CAPS (3-(cyclohexylamino)-propanesulfonic acid, pKa = 10.4), 2 mM BICINE (N,N-bis(2-hydroxyethyl) glycine, pKa = 8.35), and 2 mM MES (2-(N-morpholino)-ethanesulfonic acid, pKa = 6.15) monohydrate (all obtained from Calbiochem-Behring, La Jolla, CA) to provide a constant buffering capacity over the broad pH range 5.5–11.
Membrane preparation
Large unilamellar vesicles (LUVETs), ∼100-nm diameter, were created by the extrusion technique of Hope et al. (49), using 14C-radiolabeled DPPC as a tracer to quantify yields and determine the concentration of LUVETs. Fifty milligrams of DPPC (an additional 10 mg cholesterol in CHCl3 was added when making DPPC-cholesterol LUVETs) and tracer were placed in a round-bottom flask and vortexed together, and the chloroform in which they were delivered was evaporated under N2 gas. Twenty milliliters of SAM were then added to the lipid residue and the lipid-SAM mixture was heated in a water bath to 50°C–55°C, a temperature above the solid gel-liquid crystal transition temperature (41°C for DPPC) and repeatedly vortexed until all lipid was in solution. A sample of the resulting mixture was counted on a Beckman Model LS 6500 liquid scintillation counter (10 μL/5 ml Aquasol 2, NEN Dupont, Boston, MA, or EcoLiteliquid scintillation cocktail, ICN Biomedicals, Costa Mesa, CA) to determine the specific radioactivity (cpm/μmoles lipid), the conversion factor to be used for calculation of lipid concentration.
The SAM-lipid mixture was alternatively frozen and thawed at least five times to ensure complete hydration and equilibration (especially in the presence of cholesterol). The mixture was frozen in a dry-ice/acetone bath and thawed at 50°C–55°C in a water bath. The lipid mixture was then extruded at least 10 times through a Northern Lipids (Vancouver, B.C.) extruder fitted with two 0.1-μm polycarbonate filters (BioRad, Westbury, NY) and driven by a pressure of 400 psi, supplied by N2 gas, to produce LUVETs. The extruder was heated to 50°C for the extrusion steps. A sample of this LUVET stock was counted by liquid scintillation to determine the concentration of phospholipid in the final membrane suspension. The LUVETs were allowed to cool and were stable overnight when stored at 4°C. To obtain a higher concentration of LUVET, two or more batches of LUVETs were centrifuged and resuspended in a smaller volume of SAM. The DMPC LUVETs were made similarly with 14C-radiolabeled DPPC as a tracer, except that the water bath temperature was set at 35°C during the freezing/thawing process.
Optical measurements
To avoid light scattering caused by vesicles, front-face illumination was used with a triangular cuvette (45°, 45°, and 90°) (50). Fluorescence emission spectra were obtained with excitation at 302 nm using an Aminco spectrofluorometer (model SPF-500 C) with a xenon lamp and direct temperature control of the cuvette (Aminco, Urbana, IL).
Tetracaine's equilibrium partitioning
The affinity of tetracaine for the model membranes was quantitated by determining spectrofluorometrically the partition coefficient (Kp) of the drug into the membrane under different conditions, as described here. Fluorescence both increased in intensity and shifted to shorter wavelengths in the presence of membranes (LUVETs) (Fig. 1). The wavelength of maximum tetracaine fluorescence (λmax) is shifted from 370 nm, in aqueous solutions, to ∼350 nm when adsorbed to high concentrations of DPPC membranes (Fig. 1 A).
FIGURE 1.

(A) Typical fluorescence emission spectra (solid lines) of 1.0 μM tetracaine in the absence and presence of increasing DPPC-Chol LUVETs, pH 5.5, 23°C. From bottom to top, the total lipid concentration of DPPC-Chol LUVETs was 0, 3.88, 7.76, 10.4, 12.9, and 15.5 mM, respectively. The fluorescence emission maxima are shown for polynomial fitting of the curves. The intensity at lower wavelength, e.g., at 350 nm, increases with membrane concentration. The broken line shows the spectrum for aqueous TTCH+ scaled to the right vertical axis. (B) Solid lines, fluorescence spectra of 3.88 mM DPPC-Chol LUVET alone (bottom-most trace) and, from bottom to top, with [TTCH+] = 1, 3, 5, 7, 9, and 11 μM, respectively, at pH 5.5, 23°C; dotted lines, fluorescence difference spectra after subtraction of emission spectrum of LUVET alone in the same order as the unsubtracted spectra shown by the solid lines.
Suspensions of LUVETs containing different concentrations of lipid were formed initially in SAM at pH 5.5 or 11. Each suspension was scanned spectrofluorometrically, and then to each aliquot 1 mM tetracaine stock solution in SAM was added to bring the final total drug concentration of up to 10 or 20 μM (specific tetracaine concentrations are stated in the text). After incubation at a specific temperature, each sample was then assayed spectrofluorometrically. Difference spectra were obtained by subtracting corresponding spectra of LUVETs alone from those of tetracaine-LUVETs. Both the fluorescence intensity at 350 nm and the wavelength of maximum emission (λmax) were taken from the difference spectra smoothed by third polynomial functions, each with Origin software (version 7.5, Origin, Northampton, MA). All the experiments were repeated at least three times.
We initially determined that the relationship between fluorescence intensity at 350 nm and [tetracaine] was linear at these low concentrations (up to 20 μM, data not shown). We then analyzed the fluorescence intensity data to calculate the relative concentrations of bound and free tetracaine molecules. This was done by first normalizing each set of fluorescence spectra for different membrane/drug ratios by the spectra representing totally membrane-bound drug (high lipid/tetracaine) or drug in aqueous solution alone. From every experimental spectrum, which represented the weighted sum of the fluorescence from membrane-bound molecules plus that from free molecules, we calculated the ratio of bound to free molecules, using the following derivation:
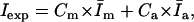 |
(1) |
where 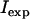 = measured fluorescence intensity,
= measured fluorescence intensity, 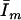 is the limiting fluorescence intensity/mole, assuming all tetracaine is in the membrane, and
is the limiting fluorescence intensity/mole, assuming all tetracaine is in the membrane, and 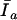 is the fluorescence intensity/mole tetracaine in SAM, i.e., when no membranes are present.
is the fluorescence intensity/mole tetracaine in SAM, i.e., when no membranes are present.
If  = moles of tetracaine in solution (“free”) and
= moles of tetracaine in solution (“free”) and 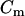 = moles of tetracaine in membrane, then the membrane:solution partition coefficient, Kp, defined in terms of mole fractions, is
= moles of tetracaine in membrane, then the membrane:solution partition coefficient, Kp, defined in terms of mole fractions, is  for the low solute/solvent ratios used here (Cm ≪ [PL] or [H2O]). Rearranging terms gives
for the low solute/solvent ratios used here (Cm ≪ [PL] or [H2O]). Rearranging terms gives
 |
(2) |
where PL/H2O = mole fraction of membrane lipid in SAM. From mass conservation,  where T = total moles of tetracaine. Substituting in Eq. 1 gives
where T = total moles of tetracaine. Substituting in Eq. 1 gives
 |
(3) |
Similarly,
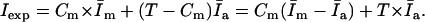 |
(4) |
Rearranging Eqs. 3 and 4 gives
 |
(5) |
 |
(6) |
Substituting Eqs. 5 and 6 into Eq. 2 gives
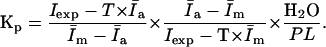 |
(7) |
Rearranging Eq. 7 gives
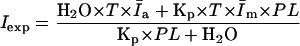 |
(8) |
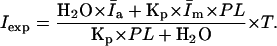 |
(9) |
The slope of Iexp versus T
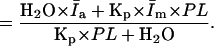 |
(10) |
Since 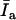 can be experimentally measured, we can obtain different slopes at different vesicle concentrations, then solve the system of equations to obtain Kp, and
can be experimentally measured, we can obtain different slopes at different vesicle concentrations, then solve the system of equations to obtain Kp, and 
 and
and  were dependent on instrumental settings, like gain factors, intensity of light sources, and on experimental conditions, like pH, membrane composition, and membrane fluidity. However, for one fluorometer with fixed instrument settings, with which fluorescence spectra were collected at multiple lipid concentrations,
were dependent on instrumental settings, like gain factors, intensity of light sources, and on experimental conditions, like pH, membrane composition, and membrane fluidity. However, for one fluorometer with fixed instrument settings, with which fluorescence spectra were collected at multiple lipid concentrations,  and
and 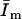 vary only with experimental conditions, like pH, membrane composition, and membrane fluidity, allowing calculation of Kp from Eq. 10, above. One emission spectrum can be collected within 1 min, and we determined that the samples used here are stable for more than 30 min. So no calibration was needed from one operating of the instrument to another and within one operating of the fluorometer. To obtain best signal/background ratio,
vary only with experimental conditions, like pH, membrane composition, and membrane fluidity, allowing calculation of Kp from Eq. 10, above. One emission spectrum can be collected within 1 min, and we determined that the samples used here are stable for more than 30 min. So no calibration was needed from one operating of the instrument to another and within one operating of the fluorometer. To obtain best signal/background ratio, 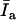 and
and 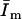 were obtained at different instrumental settings, so their values are not comparable for the different scenarios.
were obtained at different instrumental settings, so their values are not comparable for the different scenarios.
Equivalent dielectric constants
Using solutions of mixtures of dioxane (ɛ = 2.2) and buffer (ɛ = 78), as well as pure organic solvents, to produce solutions of known dielectric constant (ɛx) (see Fig. 6), a set of calibration curves was created establishing the relationship between λmax of tetracaine and dielectric constants (45). The limiting, minimum values of λmax in the fluorescence spectra from the highest lipid/tetracaine ratio, representing maximum “membrane-bound” drug molecules, were then used to calculate the equivalent bulk dielectric constant around the fluorophore, the aromatic moiety of the membrane-bound drug, providing an estimate of the “nesting position” of the local anesthetic when adsorbed to the bilayer.
FIGURE 6.

Calibration curves for 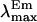 versus dielectric constant.
versus dielectric constant.
Optical determination of apparent pKa
Tetracaine's apparent pKa under the various conditions was determined using fluorescence intensity as an indicator of the proportions of charged and neutral species. Beginning at high pH values, we sequentially added aliquots of 0.25 M HCl to a tetracaine + LUVET suspension, containing enough lipid so that the fluorescence contributed by membrane-bound tetracaine-dominated total fluorescence. Fluorescence spectra were then taken at each experimentally measured pH (digital pH/millivolt meter 611, Orion Research, Boston, MA) and the corresponding changes in peak fluorescence intensity recorded. Changes in this intensity as a function of pH were fit by a Langmuir isotherm, and the apparent pKas were determined from this fit. Because total fluorescence was almost exclusively from membrane-bound tetracaine, regardless of pH, the fluorescence intensity dependence on pH reflects the ionization state of membrane-bound drug. In contrast, in the cases where lipid was increased at a fixed pH, the increase in fluorescence intensity resulted from the increase in membrane-bound tetracaine of only one ionization species, since the buffered pH was 2–3 pH units above or below the apparent pKa.
Bulk separation method to determine Kp
Bulk separation of membrane-bound tetracaine from free tetracaine was used to obtain the partition coefficient directly, for comparison with that obtained by the fluorescence method described above. When making DPPC LUVET for this procedure, 30 μl of 3H2O was added to the DPPC suspension before extruding LUVETs to determine the aqueous volume entrapped in the pelleted LUVETs after centrifugation. Tetracaine was added to 1.5 ml of DPPC LUVET (2.94 mM) at pH 11 to a final nominal concentration of 4 μM and incubated at 23°C for 2 min, then centrifuged at 23°C with Beckman Avanti J-25, rotor JA20.1 at 51,500 × g for 30 min. The supernatant was decanted and mixed with an equal volume of 40 mM sodium lauryl sulfate (SDS) in SAM, and the pellet was also dissolved in 2 ml of 20 mM SDS in SAM. At this concentration SDS forms micelles which both greatly enhance the fluorescence of the tetracaine (45) and also free it by dissolving the membranes. The fluorescence spectra of the processed supernatant and pellet were scanned, a parallel control experiment conducted without added tetracaine, and the respective control spectra subtracted from those with tetracaine; tetracaine concentrations in supernatant and pellet were calculated using a standard curve of fluorescence intensity at 350 nm of tetracaine, 0–10 μM, in 20 mM SDS in SAM. The aqueous volume entrapped in the pellet was calculated from the pelleted 3H2O, using liquid scintillation counting, and the aqueous entrapped tetracaine was subtracted from the total tetracaine in the pellet. Kp could be calculated by the definition of Eq. 2 (see above experimental design).
Statistical analysis
The significance of differences between parameters measured in different membranes or between physical conditions was determined by applying Student's unpaired t-test, with p < 0.05 as the criterion for a significant difference.
RESULTS
General fluorescence changes with membrane adsorption
The adsorption of tetracaine to all of the different membranes used here caused generally similar changes in the drug's fluorescence. The general pattern was an increase in the intensity and a “blue” shift to shorter emission wavelengths, with increasing concentration of membrane (Fig. 1 A). An increase in tetracaine concentration, from 1 to 20 μM, a range that had no detectable self-quenching, led to a proportional increase in I (Fig. 1 B), which was used to determine partition coefficients (see below). These fluorescence emission changes reached limiting λmax values at high relative concentrations of lipid (Fig. 2), whereas their intensity, Iexp, saturated at even higher lipid concentrations (Fig. 1 A) for which total fluorescence was almost exclusively from membrane-bound drug.
FIGURE 2.

Maximum emission wavelength (λmax) versus total lipid concentration. (▪) DPPC-Chol LUVET at constant total concentration of 1.0 μM tetracaine, pH 5.5, 23°C (data from polynomial fit of the curves in Fig. 1 A); (•) DPPC LUVET at constant concentration of 4.0 μM tetracaine, pH 11, 45°C. The curves through the data points are empirical functions used to show the asymptotes of 351 ± 0.3 nm (•) and 351 ± 0.6 nm (▪).
The disparity between the membrane concentration dependence of λmax and of Iexp is a predictable outcome when the quantum efficiency of membrane-bound drug, fluorescing at the lower wavelength, is far greater than that of aqueous, free drug. We simulated this effect in Fig. 3 by adding the emission spectra from membrane-bound and -free tetracaine, modeled by Gaussian functions fit to the experimental results at high and at zero lipid, respectively, and weighted by their calculated concentrations (as mole fractions) using a partition coefficient that we calculated (Kp = 1.76 × 104) from experimental data. Just as in the experimental results (cf. Figs. 1 A and 2 A), simulated fluorescence intensity continued to increase with increasing lipid after λmax had reached its limiting value, finally saturating at ∼10-fold higher concentration.
FIGURE 3.

Simulation of λmax and fluorescence intensity dependence on lipid concentration. Two Gaussian curves centered at 370 nm and 350 nm (created using Origin software version 7.5) model the fluorescence spectra of aqueous tetracaine and of membrane-bound tetracaine, the latter with higher fluorescence quantum yield. The net fluorescence spectra were created by using an experimentally determined Kp and a lipid concentration, and the simulated λmax and fluorescence intensity at 350 nm, graphed versus total membrane lipid. The simulation was carried out for DPPC, pH 11, 45°C, Kp = 1.76 × 104, and [Tetracaine] = 4 μM.
These changes in fluorescence parameters were used to quantitate tetracaine:membrane interactions, as described in Materials and Methods, above. The distribution of tetracaine into the membranes of different composition and/or phase was expressed as the partition coefficient for neutral or protonated drug species. The physicochemical properties of membrane-bound drug were investigated under conditions of high lipid:tetracaine. pH-dependent changes of fluorescence intensity were used to determine apparent pKas in different membranes, and the limiting minimal emission wavelengths were interpreted as average solvent polarities (dielectric constant) around the membrane-bound fluorophore, the aromatic ring of the local anesthetic molecule.
Membrane partitioning of tetracaine
By monitoring tetracaine's relative fluorescence intensity at 350 nm, which increases markedly for membrane-bound molecules (see Fig. 1 A), it was possible to calculate the membrane-bound and free drug. Graphs of I350 against total [tetracaine] at a high lipid/drug ratio (cf. Fig. 1 B) yield a linear relation with a slope that depends on the specific lipid concentration (Fig. 4; see Materials and Methods). Solving the system of Eq. 10 at two different lipid concentrations yields the Kp. When measured at pH values several units below and above the apparent pKa, this partition coefficient closely approaches the partition coefficients of the respective protonated and neutral drug species. (At intermediate pH, where both species of tetracaine coexist, the Kp equals the distribution coefficient from membrane binding of both forms.) This method was applied for different lipids and at different temperatures, allowing comparisons of Kp between liquid-crystal and solid-gel membranes.
FIGURE 4.

Plots of I350 versus total tetracaine concentration. The data were obtained from DPPC-Chol LUVET at pH 5.5, 23°C, and fit by linear regression analysis. ▪ represents data obtained with a membrane (LUVET) concentration of 3.9 mM DPPC-Chol, with a slope of 2.54 × 10−2 μM−1 and a correlation coefficient of 0.989; = represents data obtained with 7.8 mM DPPC-Chol LUVET, with a slope of 4.24 × 10−2 μM−1 and a correlation coefficient of 0.999.
Table 1 lists all the partition coefficients determined in this way. For each membrane composition and temperature, Kp values were determined for TTCH+ (pH 5.5) and TTC (pH 11), at pH values ∼3 pH units below and 2.5 pH units above the apparent pKa for aqueous solutions, 8.48 (see below). Membrane phase was established by adjusting the temperature or by using a different phospholipid and was modified by addition of cholesterol. For example, pure DPPC forms membranes with a phase transition temperature, Tp, of 41°C (51,52) so that the solid-gel phase exists at 23°C and the liquid-crystalline phase at 45°C. Membranes of pure DMPC have a Tp of 23°C (51,52), and the partitioning of tetracaine into these membranes in the solid-gel phase was measured at 18°C and in the liquid-crystal phase at 30°C.
TABLE 1.
Partition coefficients of tetracaine into model membranes (mean ± SE; number of independent experiments)
| LIPID | pH | 18°C | 23°C | 30°C | 45°C |
|---|---|---|---|---|---|
| DMPC | 11 | 1.29 ± 0.09 × 104(10) | 1.89 ± 0.15 × 104 (23) | ||
| 5.5 | 6.56 ± 0.64 × 103(14) | 1.18 ± 0.2 × 104 (30) | |||
| DPPC | 11 | 1.24 ± 0.09 × 104 (45) | 1.76 ± 0.11 × 104 (28) | ||
| 5.5 | 4.76 ± 0.8 × 103 (20) | 1.31 ± 0.13 × 104 (30) | |||
| DPPC:Chol | 11 | 7.49 ± 0.6 × 103 (22) | 1.11 ± 0.1 × 104 (28) | ||
| 5.5 | 3.84 ± 0.5 × 103 (13) | 1.10 ± 0.09 × 104 (30) |
Comparison with Kp determined by bulk separation
The Kp value obtained from bulk separation of DPPC LUVETs, at pH 11, 23°C, was 1.67 ± 0.21 × 104 (n = 2), the same as the value of 1.24 ± 0.09 × 104 (n = 45) obtained by the fluorescence method (p < 0.05, Student's t-test).
Effects of membrane phospholipid composition and phase
With the exception of the cholesterol-containing LUVETs, which lack a well-defined phase transition, the phosphatidylcholine (PC) bilayer membranes undergo discrete phase transitions, and subtransitions, from a tightly packed, highly ordered solid-gel phase to a less ordered liquid-crystalline “fluid” phase (52,53). We compared the partitioning of tetracaine into the two model membranes, above and below their transition temperatures, to discern the role of the membrane's physical state on tetracaine uptake.
The larger values of Kp in liquid-crystalline membranes than in solid-gel membranes indicate the preferred adsorption of tetracaine into the more fluid phase (Table 2). Partitioning for both protonated and uncharged species into the solid-gel phase of DMPC membranes (at 18°C) was the same as that into solid-gel DPPC membranes (at 23°C) (Student's t-test). Partition coefficients of charged and neutral local anesthetic into DMPC membranes in the liquid-crystal state (30°C) was also the same as their respective values into liquid-crystal DPPC membranes (45°C), showing that membrane phase rather than the lipid's fatty acyl composition primarily determines tetracaine's partitioning.
TABLE 2.
The effect of membrane physical state: Kp (liquid-crystalline)/Kp (solid-gel) for tetracaine in model membranes
The reduction of Kp values caused by DDPC membrane condensation to the solid-gel phase is about twice as large for TTCH+ (52% decrease) as for uncharged TTC (33% decrease). The same order occurs for this phase transition in DMPC membranes (decrease in Kp of 45% vs. 32%, respectively). Both examples show the relatively larger contribution of ionic interactions in the changes of affinity that accompany membrane melting.
Cholesterol's effects on partitioning
The inclusion of cholesterol in DPPC membranes lowers tetracaine's partition coefficients (Table 3). The addition of cholesterol into DPPC in its solid-gel state, at 23°C, reduces partitioning of neutral TTC by a smaller amount (38%) than the reduction of partitioning into the liquid-crystal state (58%). For the charged species of tetracaine the reductions in partitioning caused by cholesterol have the opposite “state dependence”, 21% for the solid-gel state, slightly larger than the 15% for the liquid-crystal. These data also demonstrate that the reduction in partitioning caused by cholesterol is larger for uncharged TTC (by 38% at 23°C, 58% at 45°C), than for TTCH+ (by 21% at 23°C, 15% at 45°C). Therefore, cholesterol's addition affects the two species of tetracaine, uncharged and protonated, in the opposite order of the liquid-crystal to solid-gel transition (compare Tables 2 and 3). Although liquid-crystal membranes become “less fluid” from both the solid-gel transition (52) and the inclusion of cholesterol (54,55), the different molecular “ordering” that underlie these two types of fluidity changes have different effects on tetracaine's membrane interactions (see Discussion).
TABLE 3.
The effect of cholesterol: Kp (without cholesterol)/Kp (with cholesterol) for tetracaine in model membranes
Effect of TTC ionization state on adsorption affinity
The effects of tetracaine's ionization on partitioning are collected in Table 4. In almost all cases the ratio of Kp for neutral to charged tetracaine exceeds unity, showing the preferred partitioning of the neutral form. This ratio is highest (2.6) for solid-gel DPPC membranes and lowest, essentially 1.0, for DPPC-cholesterol at the high temperature, 45°C. The high ratio for solid-gel DPPC primarily results from the low absolute partitioning of TTCH+ into these membranes (4.8 × 103), whereas the low ratio for DPPC-cholesterol results from the low absolute partitioning of TTC into DPPC-cholesterol at high temperature (1.1 × 104). Although both cholesterol-free and cholesterol-containing membranes generally show a higher affinity for neutral over charged TTC, at high and low temperatures, there is a consistently higher ratio (approximately twofold) in the solid-gel compared to the liquid-crystal phase of DPPC membranes, but a smaller difference that does not reach significance between these ratios for DMPC membranes (Table 4).
TABLE 4.
Effect of drug ionization on membrane partitioning: 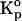 (neutral TTC)/
(neutral TTC)/ (protonated TTCH+) in model membranes
(protonated TTCH+) in model membranes
Ionization of membrane-bound tetracaine
The apparent pKa of tetracaine adsorbed to bilayers of various compositions was determined using fluorescence methods described above and in Desai et al. (45). Titrations were conducted from pH 6.0–11.0; extreme acidity was avoided to eliminate titration of the secondary amino group on the aromatic ring, and alkalinity above pH 11 was avoided to minimize hydrolysis of tetracaine's ester linkage (56). The fluorescence versus pH data are well fit by an equation for a single proton binding to one site with association constant Ka (Fig. 5). Apparent pKas, determined from fits of this equation to these data, are collected in Table 5. At the same temperature (23°C or 45°C), the pKa of tetracaine in DPPC membranes, be they solid-gel or liquid-crystal, is ∼0.3–0.4 pH units below that in aqueous solution, in agreement with the changes reported for tetracaine in multilamellar membranes made from egg PC (57). The presence of cholesterol causes no, or insignificant (0.1 pH unit), decreases in the membrane-bound tetracaine's pKa at these respective temperatures.
FIGURE 5.

Typical pH-dependent fluorescence intensity (I) of membrane-bound tetracaine (302 nm excit./350 nm em.) and of aqueous tetracaine (302 nm excit./370 nm em.). The equation, I = I2 + (I1 − I2)/(1 + exp((pH − pKa)/pH)) was used to fit the data to obtain pKa values, (▪) 8.17 ± 0.02 (N = 10) for tetracaine bound to DPPC membranes at 23°C; (•) 8.48 ± 0.05 (N = 15) for tetracaine in SAM at 23°C. The data for aqueous TTC were normalized to unity.
TABLE 5.
pKa of tetracaine in solution and membranes (mean ± SE; number of measurements)
| Membranes
|
|||||||
|---|---|---|---|---|---|---|---|
| Solid-gel
|
Liquid-crystal
|
||||||
| T (°C) | Solution (SAM) | DPPC | DPPC-Chol* | DMPC | DPPC | DPPC-Chol* | DMPC |
| 18 | 8.70 ± 0.02 (16) | 8.30 ± 0.02 (16) | |||||
| 23 | 8.48 ± 0.05 (15) | 8.17 ± 0.02 (10) | 8.15 ± 0.06 (18) | ||||
| 30 | 8.50 ± 0.03 (18) | 8.12 ± 0.02 (12) | |||||
| 45 | 8.20 ± 0.03 (11) | ||||||
| 7.91 ± 0.03 (21) | 7.80 ± 0.01 (19) | ||||||
No phase transition in cholesterol-containing membranes.
Estimating tetracaine's location in bilayers
The depth of TTC's penetration into the bilayer at equilibrium binding was estimated from the limiting value of λmax at high lipid concentrations (cf. Fig. 2), using calibration curves for tetracaine's fluorescence in bulk solvents of known dielectric constant (see Materials and Methods and Fig. 6). From these λmax values we determined an “equivalent bulk dielectric constant” around the local anesthetic's aromatic ring.
These adsorption studies were conducted in membranes of pure DPPC and of DPPC-cholesterol at pH 5.5 and 11.0. In all cases, and regardless of the phase of the membrane, at high lipid concentrations tetracaine fluoresced maximally at ∼351 nm (Figs. 1 A and 2). This corresponds to a dielectric constant, ɛ, of >6 and <10 (Fig. 6), an apolar milieu but with values greater than a saturated alkane, implying that the aromatic fluorophore of bound TTC and TTCH+ is localized well away from the aqueous interface (ɛ = 80), closer to but not within the fatty acyl region of the membrane. The constancy of this value, as much as its meaning for membrane location, shows that protonation of the local anesthetic does not change its position with respect to that of the lipid's in the bilayer.
DISCUSSION
Spectrofluorometry of low concentrations of tetracaine in a suspension of membrane vesicles can be used to characterize tetracaine-membrane interactions without separating membrane-bound from free drug. Where the results of this work can be compared to previous reports using other methods, the findings are generally similar (see below). Partitioning measured for one condition by bulk separation in this study was the same as that determined by fluorescence, further validating the fluorescence method, which thus provides a satisfactory means to determine partitioning into the bilayer using micromolar concentrations of drug that minimally perturb membrane structure.
Experiments then proceeded to inquire about the influence of membrane phase and membrane composition of drug partitioning and about the role of cholesterol when added to DPPC bilayers in the solid-gel or the liquid-crystal state. The importance of drug charge for partitioning among the different membrane types was assessed and, reciprocally, the effect of membrane binding on tetracaine's ionization equilibrium. A physicochemical measure of “location” in the different membranes used here was also determined. This is the first time that these different behaviors have been addressed in one systematic study, and their integration allows a more complete analysis of the interactions between local anesthetic and membrane lipids. In this Discussion we will relate these data to published results, both for membranes and from our earlier work on detergent micelles and then analyze the findings in terms of the known structural differences among the different membrane states studied here.
Tetracaine's partitioning into the membrane
Tetracaine partitioned more strongly into liquid-crystal than solid-gel membranes, and the neutral species of the drug partitioned more strongly than the charged one. (Although local anesthetics have been reported to lower the transition temperature of pure DPPC membranes (42,58), the concentrations of tetracaine used here would cause a <1°C reduction.) Thermodynamically consistent with the latter behavior, membrane-adsorbed tetracaine was less protonated than in solution at the same bulk pH, resulting in a lower effective apparent pKa in the membrane. Partition coefficients were lowered when cholesterol was included in the membrane of both solid-gel and liquid-crystalline phases (22), but this effect of cholesterol was greater on the neutral species, whereas the phase change from liquid-crystal to solid-gel membranes caused by temperature reduction reduced the partitioning of the charged species more than the neutral one. All tetracaine adsorbed to membranes, regardless of phase or composition, had the same equivalent bulk dielectric constant at its aromatic fluorophore, showing that the general locus for binding was unchanged, even though the energy of the bound state was altered.
Partition coefficients ranged around 104 = χm/χs, the ratio of mole fraction in the membrane:mole fraction in solution, in general agreement with earlier reports (e.g., de Paula and Schreier (59)). At the 1–10 μM tetracaine concentration used in these experiments, the respective drug concentrations in the membrane will be 1 tetracaine molecule for approximately every 500–5000 lipid molecules. At this stoichiometry the effects of the drug on the bulk bilayer properties should be minimal, unless there are extended effects, e.g., ramifying through the condensed solid-gel phase. We found, however, that the changes of fluorescent intensity with increasing [tetracaine] were consistently linear, indicating a constant partitioning behavior over the drug range used here, unlikely to occur if bulk properties were altered by the drug.
Although TTCH+ partitioned less than TTC, partitioning of the charged species was still substantial. Electrophysiological studies have shown the importance of the uncharged species of smaller local anesthetics, e.g., lidocaine, for permeating from the extracellular space to the cytoplasmic compartment, the apparent locus from which blockade of ionic Na+ channels occurs (1–3,60). Both direct studies of isotopically labeled tetracaine in membranes (17,24,61), bulk separations of multilamellar membranes from free drug in solution (57), and indirect measurements, such as monitoring the local anesthetic-induced quenching of an intramembrane probe (62) or studying the electrophoretic mobility of multilamellar vesicles in the presence of drug (63), have shown that charged local anesthetics bind to membranes. These and other earlier studies (15) clearly contradict claims that only the neutral form of amine local anesthetics interacts with a particular phase of membranes (58). In fact, under the most physiological of conditions used here, DPPC with cholesterol at a mole fraction (0.28), like that in many plasma membranes (64), and near physiological temperature, protonated and neutral tetracaine partitioned almost identically. The charged species does permeate through membranes considerably more slowly than the neutral one, however, showing that surface adsorption is not the rate-limiting step for drug permeation (65,66).
High concentrations of tetracaine, 5–10 mg tetracaine per milliliter buffer (17–33 mM), equilibrated with planar dispersions of egg phosphatidyl ethanolamine (16), or 10 mg tetracaine per milliliter equilibrated with DMPC (22), have been used previously to determine Kp by bulk separation methods. These high drug concentrations may cause membrane disruption, as 10 mg/mL tetracaine (equal to 33 mM) has been shown to disrupt model membrane integrity because of its detergent property (10). Such an effect has been implicated in the irreversible nerve injury caused by local anesthetics (67,68), whereas the concentrations of TTC (<20 μM) used in this study are close to those of the IC50 for sodium channel blockade (69) or action potential inhibition (70). The much higher local anesthetic concentrations used to effect drug-induced changes in membrane parameters such as phase transition temperature and bulk viscosity/fluidity, or even tetracaine-lipid mixed micelle formation, resulting in drug/lipid ratios of 1:10 or higher (23,29,41,44). Although the gross changes in membranes under these conditions have been proposed as mechanisms of anesthetic action, it is likely that such phenomena are minimal at “physiological” drug concentrations, the concentrations that exist at the plasma membranes of the target cells which are usually 50–100-fold lower than drug concentrations that are injected clinically to overcome the very poor efficiency for delivery of these drugs to their active compartment (71,72).
The partitioning behavior reported here agrees qualitatively with previous reports. The favored partitioning of neutral TTC over the protonated form has been measured by spectroscopic methods for a variety of unilamellar phospholipid membranes (73) and multilamellar preparations (16) where lipid bilayer dispersions, not enclosed vesicles, were used (13,62,74). Valid quantitative comparisons are not possible, however, because many of these earlier studies used mixtures of different phosphatidyl cholines with undefined acyl compositions, e.g., from egg yolk, and most studies were conducted with membranes in the solid-gel phase. Furthermore, the values reported by others as “partition coefficients” were often measured at pH values where both charged and neutral species contribute to the membrane uptake and therefore are truly “distribution coefficients”.
In an earlier study of tetracaine's binding to SDS detergent micelles, we found that, as with membranes, neutral TTC adsorbed 3–4 times more strongly than TTCH+, but the bound drug had an equivalent dielectric constant, calibrated by the method used here, of ∼10–15 for TTCH+, 33–38 for TTC, showing the micellar-bound drug's localization in a more polar domain than in membranes and one that changed with drug ionization. The micelles are an imperfect model for membranes, having only the most general features of a hydrophobic core and polar surface that will adsorb and orient amphipathic molecules such as local anesthetics. The poor comparison of absolute values of parameters between micellar- and membrane-bound drug illustrates this disparity.
Location of tetracaine in lipid bilayers
The location of membrane-bound local anesthetics has been explored previously. Using deuterated PC membranes, Boulanger et al. (14) and Smith et al. (24) found NMR spectral changes which suggested that the charged tertiary amine group of TTCH+ resided in the area of the polar head of the phospholipid, causing an ordering of the headgroup region, which was confirmed by Watts and Poile (75) from quadrupole splitting of the methylenes of the choline headgroup. This conformational change at the interface was accompanied by a disordering of the fatty acyl chain region, creating an overall increase in phospholipid cross sectional area and a necessary decrease in membrane thickness. The neutral form of TTC, however, appeared to penetrate more deeply into the membrane, as evidenced by minimal changes in headgroup signal but similar disordering in the fatty acyl core. Boulanger et al. proposed a model in which the two ionization states of TTC occupy distinct locations in the bilayer.
In Watts and Poile's (75) investigation, however, the 2H-NMR studies using specifically deuterated phospholipids showed similar disturbances of α and β methylene groups of choline, by both protonated and neutral tetracaine, suggesting that either ionization state had the same depth of membrane penetration and that the local anesthetic's tertiary amine lies close to both methylene groups. This implies a superficially bound membrane location for the drug in either state. Kelusky and Smith (17,18) studied the same interaction using specifically deuterated tetracaine. They found no pH dependence of the quadrupole splittings of the NMR spectral peaks representing membrane-bound TTC and they too concluded that both ionization states of the drug acquire a similar depth of penetration. However, Kelusky et al. (20) undertook studies of tetracaine's position in negatively charged PS vesicles using the same techniques and found a significant pH dependence of quadrupole splittings. This result suggests different membrane locations for the two ionization states of TTC, the neutral form nesting more closely to the glycerol backbone, with its aromatic moiety experiencing a much more highly ordered environment. Experiments employing ESR-labeled local anesthetic analogs suggest a location in dioleoyl PC where both charged and neutral species' amine nitrogens are near the membrane-aqueous interface (76).
Our findings show that the aromatic group of tetracaine experienced the same equivalent dielectric around the aromatic fluorophore, in any state of membranes made from PC, for both TTC and TTCH+. Calibrations with bulk solvents showed this “equivalent” dielectric constant to be 6–10. Although a membrane's physical properties are anistropic compared to those of small, mobile solvents, the effective polarity can be related to charge density within membranes, as calculated by White and Wimley (77,78) to better understand the interactions between lipids and membrane proteins; the same thermodynamic arguments can be applied to small, reversibly bound drug molecules. The dielectric constant of H2O is ∼80, and even the structured H2O of hydration at the membrane's surface contains molecules that can interact with solute molecules (79,80). Mobile ions at the aqueous interface account for a relatively high charge density there, which falls with distance into the membrane, nearing a zero value at the acyl chain core. This charge density gradient, which accounts for the polar property within the membrane, establishes the “equivalent dielectric constant” reported by bound tetracaine; the steepest gradient of charge density occurs at the region of the glycerol backbone and the ester carbonyls of phospholipids. Molecular models of tetracaine that localize the aromatic fluorophore near the lower end of this region, around carbons 2–5 of the acyl tails of PC, place the tertiary amine N within 1–2 Å of the negative phosphate in the headgroup and the dipole from tetracaine's ester bond in the middle of the membrane dipole field, caused by the aligned esters of the fatty acid-glycerol ester bonds and of the oriented interfacial H2O molecules (81,82). More important than an absolute location, however, the pH independence of this equivalent ɛ means that both charged and neutral local anesthetic molecules are located at the same place in any membrane made predominantly of PC, whether cholesterol is present or not.
This conclusion is inconsistent with a charge-dependent position for membrane-bound tetracaine (see above) and requires that the hydrophobic and dipole-dipole field interactions define the location of the drug among the phospholipids such that addition of an ionic bond between TTCH+ and PC changes the adsorption energy without altering the position.
The energetics of tetracaine's membrane interactions
Equilibrium partitioning is determined by the free energy differences between the two environments, water and the membrane. Local anesthetic molecules have three physicochemical properties that provide potential free energy differences between water and membranes: 1) hydrophobicity, due to acyl moieties on the tertiary amine nitrogen (3° N) and on the p-amino group as well as the aromatic ring; 2) polar and charged properties due to the unshared electron pair on the 3° N of neutral TTC or the charged amine group in protonated TTCH+, and the carbonyl oxygen of the ester bond; and 3) a dipolar property due to the charge separation at the ester bond. Interactions of hydrophobic solutes with H2O organize the dynamic intermolecular structures of that fluid, inducing ordered ice-like clathrates around the hydrophobic moieties, and so entropically favoring the partitioning into the membrane by “hydrophobic forces” (77,83,84). In contrast, there is the potential for energy-gaining interactions between charged or polar tetracaine and both H2O and phospholipid molecules. Protonated TTCH+ in the membrane might form ionic bonds with the anionic phosphate moiety of the lipid headgroup (while being more weakly repelled by the cationic choline moiety; see Wimley et al. (85) Ben-Tal (86)) as well as being hydrated by the polar H2O molecules, although hydrogen bonds to tetracaine's carbonyl oxygen and unprotonated amine nitrogen are possible only in H2O, not lipid (87). Finally, dipole-dipole interactions are possible between tetracaine in solution and the mobile, dipolar H2O molecules and also between tetracaine in the membrane and the strong membrane dipole field caused by the anisotropic alignment of all the ester bonds of the membrane phospholipids (88) as well as H2O molecules semiordered at the membrane-solution interface (81,82). Such dipole fields have been estimated to have potentials of several hundred millivolts which, even with drug-contained dipoles of fractions of a debye, would make a substantial contribution to the overall partition energy (89).
Both hydrophobic and ionic interactions appear to contribute energy to tetracaine's partitioning. Hydrophobic interactions dominate the overall energetics, as it is well known that more hydrophobic local anesthetics have a higher membrane partition coefficient (56). Hydrophobic forces probably also account for the preferred partitioning of both TTC and TTCH+ into liquid-crystal over solid-gel membranes; the greater fluidity of the liquid-crystal phase will allow more intimate associations between the N-buytl-substituted aromatic moiety of the drug and the fatty acyl groups of phospholipids (and the larger spacing between phospholipid headgroups in the liquid crystal will better accommodate the tertiary amine of the local anesthetic). That adsorption of tetracaine into the lipid bilayer leads to membrane volume expansion is supported by the fact that pressure induces exclusion of tetracaine from both model membranes and nerve membranes (34). Thus, a more expanded membrane is better able to accommodate the bound drug.
In addition, hydration of the membrane interface is greater in the liquid-crystal phase (79), allowing charged TTCH+ to be stabilized by these semimobile H2O molecules. In liquid crystalline PC the zwitterionic headgroups are aligned more perpendicular to the membrane surface and bound TTCH+ will reside closer to the negatively charged phosphate. Such ionic interactions add to the binding energy of TTCH+, even as hydrophobic factors will reduce it.
In this study, membrane “fluidity” was reduced in two ways: by a temperature-induced phase transition and by the inclusion of cholesterol. Both procedures reduced tetracaine's partitioning, but they had opposite preferences for the two species of the drug; liquid-crystal to solid-gel phase transitions lowered the partitioning of charged TTCH+ more than that of neutral TTC, whereas the addition of cholesterol lowered the partitioning of the neutral species more than that of the charged one. These results can be understood by the changes in membrane dynamics known to be caused by these two procedures. Solid-gel membranes have both more closely spaced headgroups and more closely packed acyl chains with less rotational mobility in the hydrocarbon core of the membrane than do the liquid-crystal membranes (52). Membranes containing cholesterol, at the mole fraction used here, 0.28, have less space at the steroid's location in the region of the phospholipids' glycerol moiety, ester linkage, and nearer regions of the acyl chains but have more rotational fluidity near the ends of the acyl chains in the hydrocarbon core (90).
We interpret the selective reduction of TTCH+'s partitioning by solid-gel formation as a reflection of the more intimate interactions of the polar headgroup or of interfacial H2O with bound TTCH+; condensation of this outer region of the membrane corresponds to more closely packed zwitterionic headgroups, lying at a more acute angle to the membrane surface, and the expulsion of some interfacial H2O; for TTCH+ to partition into such a membrane requires the disruption of these PL interactions. The loss of binding energy at this polar region is larger than the reduction of the hydrophobic bonding of the drug's aromatic moiety, which is the major factor in the binding of uncharged TTC, thus accounting for the larger decrease in partitioning of the charged species upon solid-gel formation. In contrast, cholesterol causes an expansion between inner regions of the acyl tails of phospholipids but simultaneously resides in the membrane near the loci for tetracaine's hydrophobic and dipole-dipole field interactions. Perturbation by cholesterol of polar-charged interactions between PC and tetracaine will be less than its steric interference at this inner location, resulting in a greater reduction of binding of neutral TTC than of charged TTCH+. Cholesterol lowers tetracaine's binding energy but it does not change the membrane location of where the drug is most stable.
According to this analysis, the binding of tetracaine with membranes below the transition temperature is dominated by favorable electrostatic interactions and above it by unfavorable hydrophobic considerations in water. One would predict, accordingly, that partitioning into solid-gel membranes is predominantly an enthalpy-driven process, whereas partitioning into liquid-crystal membranes is predominantly entropy driven.
Acknowledgments
We thank Florence Mujenda, B.A. for technical assistance in making membranes.
This work was supported by National Institutes of Health grant R01GM064792.
Theresa Hadlock's present address is Massachusetts Eye and Ear Infirmary, Massachusetts General Hospital, Boston, MA 02115.
References
- 1.Narahashi, T., T. Frazier, and M. Yamada. 1970. The site of action and active form of local anesthetics. I. Theory and pH experiments with tertiary compounds. J. Pharmacol. Exp. Ther. 171:32–44. [PubMed] [Google Scholar]
- 2.Strichartz, G. R. 1973. The inhibition of sodium channels in myelinated nerve by quaternary derivatives of lidocaine. J. Gen. Physiol. 62:37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hille, B. 1997. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor interaction. J. Gen. Physiol. 69:497–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hille, B. 1977. The pH-dependent rate of action of local anesthetics on the node of Ranvier. J. Gen. Physiol. 69:475–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Trumbore, M., D. W. Chester, J. Moring, D. Rhodes, and L. G. Herbette. 1988. Structure and location of amiodarone in a membrane bilayer as determined by molecular mechanics and quantitative x-ray diffraction. Biophys. J. 54:535–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang, G. K., C. Russell, and S. Y. Wang. 2004. State-dependent block of voltage-gated Na+ channels by amitriptyline via the local anesthetic receptor and its implication for neuropathic pain. Pain. 110:166–174. [DOI] [PubMed] [Google Scholar]
- 7.Courtney, K., and G. Strichartz. 1984. Structural elements which determine local anesthetic activity. In Handbook of Experimental Pharmacology. Local Anesthetics, Vol. 81. G. R. Strichartz editor. Springer-Verlag, Berlin. 53–94.
- 8.Guo, X. T., N. A. Castle, D. M. Chernoff, and G. R. Strichartz. 1991. Comparative inhibition of voltage-gated cation channels by local anesthetics. Ann. N. Y. Acad. Sci. 625:181–199. [DOI] [PubMed] [Google Scholar]
- 9.Yanagidate, F., and G. Strichartz. 2006. Local Anesthetics. In Analgesia. (Handbook of Experimental Pharmacology Vol. 177). C. Stein, editor. Springer-Verlag, Berlin. 95–127. [DOI] [PubMed]
- 10.Kitagawa, N., M. Oda, and T. Totoki. 2004. Possible mechanism of irreversible nerve injury caused by local anesthetics: detergent properties of local anesthetics and membrane disruption. Anesthesiology. 100:962–967. [DOI] [PubMed] [Google Scholar]
- 11.Herbette, L. G., P. E. Mason, K. R. Sweeney, M. W. Trumbore, and R. P. Mason. 1994. Favorable amphiphilicity of nimodipine facilitates its interactions with brain membranes. Neuropharmacology. 33:241–249. [DOI] [PubMed] [Google Scholar]
- 12.Barcelo, F., J. Prades, S. S. Funari, J. Frau, R. Alemany, and P. V. Escriba. 2004. The hypotensive drug 2-hydroxyoleic acid modifies the structural properties of model membranes. Mol. Membr. Biol. 21:261–268. [DOI] [PubMed] [Google Scholar]
- 13.Boulanger, Y., S. Schreier, L. Leitch, and I. Smith. 1980. Multiple binding sites for local anesthetics in membranes: characterization of the sites and their equilibria by deuterium NMR of specifically deuterated procaine and tetracaine. Can. J. Biochem. 58:986–995. [DOI] [PubMed] [Google Scholar]
- 14.Boulanger, Y., S. Schreier, and I. Smith. 1981. Molecular details of the anesthetic-lipid interaction as seen by deuterium and phophorous-31 NMR. Biochemistry. 20:6824–6830. [DOI] [PubMed] [Google Scholar]
- 15.Westman, J., Y. Boulanger, A. Ehrenberg, and I. Smith. 1982. Charge and pH dependent drug binding to model membranes. A 2H-NMR and light adsorption study. Biochim. Biophys. Acta. 685:315–328. [DOI] [PubMed] [Google Scholar]
- 16.Kelusky, E. C., and I. C. Smith. 1983. Characterization of the binding of the local anesthetics procaine and tetracaine to model membranes of phosphatidylethanolamine: a deuterium nuclear magnetic resonance study. Biochemistry. 22:6011–6017. [DOI] [PubMed] [Google Scholar]
- 17.Kelusky, E. C., and I. C. Smith. 1984. Anesthetic-membrane interaction: a 2H NMR study of the binding of specifically deuterated tetracaine and procaine to phosphatidylcholine. Can. J. Biochem. Cell Biol. 62:178–184. [DOI] [PubMed] [Google Scholar]
- 18.Kelusky, E. C., and I. C. Smith. 1984. The influence of local anesthetics on molecular organization in phosphatidylethanolamine membranes. Mol. Pharmacol. 26:314–321. [PubMed] [Google Scholar]
- 19.Siminovitch, D. J., M. F. Brown, and K. R. Jeffrey. 1984. 14N NMR of lipid bilayers: effects of ions and anesthetics. Biochemistry. 23:2412–2420. [DOI] [PubMed] [Google Scholar]
- 20.Kelusky, E. C., Y. Boulanger, S. Schreier, and I. C. Smith. 1986. A 2H-NMR study of the interactions of the local anesthetic tetracaine with membranes containing phosphatidyl serine. Biochim. Biophys. Acta. 856:85–90. [DOI] [PubMed] [Google Scholar]
- 21.Kuroda, Y., and Y. Fujiwara. 1987. Locations and dynamical perturbations for lipids of cationic forms of procaine, tetracaine, and dibucaine in small unilamellar phosphatidylcholine vesicles as studied by nuclear Overhauser effects in 1H nuclear magnetic resonance spectroscopy. Biochim. Biophys. Acta. 903:395–410. [DOI] [PubMed] [Google Scholar]
- 22.Auger, M., H. C. Jarrell, and I. C. Smith. 1988. Interactions of the local anesthetic tetracaine with membranes containing phosphatidylcholine and cholesterol: a 2H NMR study. Biochemistry. 27:4660–4667. [DOI] [PubMed] [Google Scholar]
- 23.Auger, M., I. C. Smith, and H. C. Jarrell. 1989. Interactions of the local anesthetic tetracaine with glyceroglycolipid bilayers: a 2H-NMR study. Biochim. Biophys. Acta. 981:351–357. [DOI] [PubMed] [Google Scholar]
- 24.Smith, I. C., M. Auger, and H. C. Jarrel. 1991. Molecular details of anesthetic-lipid interaction. Ann. N. Y. Acad. Sci. 625:668–684. [DOI] [PubMed] [Google Scholar]
- 25.Peng, X., and J. Jonas. 1992. High-pressure 31P NMR study of dipalmitoylphosphatidylcholine bilayers. Biochemistry. 31:6383–6390. [DOI] [PubMed] [Google Scholar]
- 26.Wakita, M., Y. Kuroda, Y. Fujiwara, and T. Nakagawa. 1992. Conformations of dibucaine and tetracaine in small unilamellar phosphatidylcholine vesicles as studied by nuclear Overhauser effects in 1H nuclear magnetic resonance spectroscopy. Chem. Phys. Lipids. 62:45–54. [DOI] [PubMed] [Google Scholar]
- 27.Peng, X., A. Jonas, and J. Jonas. 1995. One and two dimensional 1H-NMR studies of pressure and tetracaine effects on sonicated phospholipid vesicles. Chem. Phys. Lipids. 75:59–69. [DOI] [PubMed] [Google Scholar]
- 28.Ondrias, K., L. I. Horvath, P. Balgavy, and S. Stolc. 1984. Effects of tertiary amine local anaesthetics on the phase behaviour of the dipalmitoylphosphatidylcholine model membrane. Electron spin resonance tetrametylpiperidinyloxyl partition study. Physiol. Bohemoslov. 33:489–494. [PubMed] [Google Scholar]
- 29.Frezzatti, W. A. Jr., W. R. Toselli, and S. Schreier. 1986. Spin label study of local anesthetic-lipid membrane interactions. Phase separation of the uncharged form and bilayer micellization by the charged form of tetracaine. Biochim. Biophys. Acta. 860:531–538. [DOI] [PubMed] [Google Scholar]
- 30.Bianconi, M. L., A. T. do Amaral, and S. Schreier. 1988. Use of membrane spin label spectra to monitor rates of reaction of partitioning compounds: hydrolysis of a local anesthetic analog. Biochem. Biophys. Res. Commun. 152:344–350. [DOI] [PubMed] [Google Scholar]
- 31.Ondrias, K., and A. Stasko. 1992. Perturbation effect of the diheptanoyl phosphatidylcholine on rat brain total lipid liposomes. An electron paramagnetic resonance spectroscopy study. Chem. Biol. Interact. 84:143–151. [DOI] [PubMed] [Google Scholar]
- 32.Bartucci, R., P. Mollica, P. Sapia, and L. Sportelli. 1998. Procaine interaction with DPPC multilayers: an ESR spin label investigation. Appl. Magn. Reson. 15:181–195. [Google Scholar]
- 33.Pinto, L. M., D. K. Yokaichiya, L. F. Fraceto, and E. de Paula. 2000. Interaction of benzocaine with model membranes. Biophys. Chem. 87:213–223. [DOI] [PubMed] [Google Scholar]
- 34.Auger, M., H. C. Jarrell, I. C. Smith, P. T. Wong, D. J. Siminovitch, and H. H. Mantsch. 1987. Pressure-induced exclusion of a local anesthetic from model and nerve membranes. Biochemistry. 26:8513–8516. [DOI] [PubMed] [Google Scholar]
- 35.Auger, M., H. C. Jarrell, I. C. Smith, D. J. Siminovitch, H. H. Mantsch, and P. T. Wong. 1988. Effects of the local anesthetic tetracaine on the structural and dynamic properties of lipids in model membranes: a high-pressure Fourier transform infrared study. Biochemistry. 27:6086–6093. [DOI] [PubMed] [Google Scholar]
- 36.Auger, M., I. C. Smith, H. H. Mantsch, and P. T. Wong. 1990. High-pressure infrared study of phosphatidylserine bilayers and their interactions with the local anesthetic tetracaine. Biochemistry. 29:2008–2015. [DOI] [PubMed] [Google Scholar]
- 37.Chong, P. L., S. Capes, and P. T. Wong. 1989. Effects of hydrostatic pressure on the location of PRODAN in lipid bilayers: a FT-IR study. Biochemistry. 28:8358–8363. [DOI] [PubMed] [Google Scholar]
- 38.Shibata, A., K. Ikawa, and H. Terada. 1995. Site of action of the local anesthetic tetracaine in a phosphatidylcholine bilayer with incorporated cardiolipin. Biophys. J. 69:470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Elferink, J. G. 1977. Fluorescence and membrane-action of tetracaine. Z. Naturforsch. [C]. 32:135–136. [DOI] [PubMed]
- 40.Santos, N. C., M. Prieto, and M. A. Castanho. 2003. Quantifying molecular partition into model systems of biomembranes: an emphasis on optical spectroscopic methods. Biochim. Biophys. Acta. 1612:123–135. [DOI] [PubMed] [Google Scholar]
- 41.Hata, T., H. Matsuki, and S. Kaneshina. 2000. Effect of local anesthetics on the bilayer membrane of dipalmitoylphosphatidylcholine: interdigitation of lipid bilayer and vesicle-micelle transition. Biophys. Chem. 87:25–36. [DOI] [PubMed] [Google Scholar]
- 42.Lee, A. G. 1978. Effects of charged drugs on phase transition temperatures of phospholipid membranes. Biochim. Biophys. Acta. 514:95–104. [DOI] [PubMed] [Google Scholar]
- 43.Kaminoh, Y., C. Tashiro, H. Kamaya, and I. Ueda. 1988. Depression of phase-transition temperature by anesthetics: nonzero solid membrane binding. Biochim. Biophys. Acta. 946:215–220. [DOI] [PubMed] [Google Scholar]
- 44.Racansky, V., E. Bederova, and L. Piskova. 1988. The influence of local anesthetics on the gel-liquid crystal phase transition in model dipalmitoylphosphatidylcholine membranes. Gen. Physiol. Biophys. 7:217–221. [PubMed] [Google Scholar]
- 45.Desai, S., T. Hadlock, C. Messam, R. Chafetz, and G. Strichartz. 1994. Ionization and adsorption of a series of local anesthetics in detergent micelles: studies of drug fluorescence. J. Pharmacol. Exp. Ther. 271:220–228. [PubMed] [Google Scholar]
- 46.Garcia-Soto, J., and M. S. Fernandez. 1983. The effect of neutral and charged micelles on the acid-base dissociation of the local anesthetic tetracaine. Biochim. Biophys. Acta. 731:275–281. [DOI] [PubMed] [Google Scholar]
- 47.Sanchez, V., G. R. Arthur, and G. R. Strichartz. 1987. Fundamental properties of local anesthetics. I. The dependence of lidocaine's ionization and octanol:buffer partitioning on solvent and temperature. Anesth. Analg. 66:159–165. [PubMed] [Google Scholar]
- 48.Good, N. E., G. D. Winget, W. Winter, T. N. Connolly, S. Izawa, and R. M. Singh. 1966. Hydrogen ion buffers for biological research. Biochemistry. 5:467–477. [DOI] [PubMed] [Google Scholar]
- 49.Hope, M., M. Bally, G. Webb, and P. Cullis. 1985. Production of large unilamellar vesicles by a rapid extrusion technique. Biochim. Biophys. Acta. 812:55–65. [DOI] [PubMed] [Google Scholar]
- 50.Eisinger, J., and J. Flores. 1979. Front-face fluorometry of liquid samples. Anal. Biochem. 94:15–21. [DOI] [PubMed] [Google Scholar]
- 51.Vauhkonen, M., M. Sassaroli, P. Somerharju, and J. Eisinger. 1990. Dipyrenylphosphatidylcholines as membrane fluidity probes. Relationship between intramolecular and intermolecular excimer formation rates. Biophys. J. 57:291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Koyonova, R., and M. Caffrey. 1998. Phases and phase transitions of the phosphatidylcholines. Biochim. Biophys. Acta. 1376:91–145. [DOI] [PubMed] [Google Scholar]
- 53.Lewis, R., N. Mak, and R. McElhaney. 1987. A differential scanning calorimetric study of the thermotropic phase behavior of model membranes composed of phosphatidylcholines containing linear saturated fatty acyl chains. Biochemistry. 26:6118–6126. [DOI] [PubMed] [Google Scholar]
- 54.Soubias, O., F. Jolibois, V. Réat, and A. Milon. 2004. Understanding sterol-membrane interactions, Part II: Complete 1H and 13C assignments by solid-state NMR spectroscopy and determination of the hydrogen-bonding partners of cholesterol in a lipid bilayer. Chem. Eur. J. 10:6005–6014. [DOI] [PubMed] [Google Scholar]
- 55.Zhang, J., H. Cao, B. Jing, P. F. Almeida, and S. L. Regen. 2006. Cholesterol-phospholipid association in fluid bilayers: a thermodynamic analysis from nearest-neighbor recognition measurements. Biophys. J. 91:1402–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Strichartz, G. R., V. Sanchez, G. R. Arthur, R. Chafetz, and D. Martin. 1990. Fundamental properties of local anesthetics. II. Measured octanol:buffer partition coefficients and pKa values of clinically used drugs. Anesth. Analg. 71:158–170. [DOI] [PubMed] [Google Scholar]
- 57.Malheiros, S. V., L. M. Pinto, L. Gottardo, D. K. Yokaichiya, L. F. Fraceto, N. C. Meirelles, and E. de Paula. 2004. A new look at the hemolytic effect of local anesthetics, considering their real membrane/water partitioning at pH 7.4. Biophys. Chem. 110:213–221. [DOI] [PubMed] [Google Scholar]
- 58.Ueda, J., C. Tashiro, and K. Arakaiwa. 1977. Depression of phase-transition temperature in a model cell membrane by local anesthetics. Anesthesiology. 46:327–332. [DOI] [PubMed] [Google Scholar]
- 59.de Paula, E., and S. Schreier. 1995. Use of a novel method for determination of partition coefficients to compare the effect of local anesthetics on membrane structure. Biochim. Biophys. Acta. 1240:25–33. [DOI] [PubMed] [Google Scholar]
- 60.Frazier, D. T., T. Narahashi, and M. Yamada. 1970. The site of action and active form of local anesthetics. II. Experiments with quaternary compounds. J. Pharmacol. Exp. Ther. 171:45–51. [PubMed] [Google Scholar]
- 61.Yeagle, P., W. Hutton, and R. Martin. 1977. Molecular dynamics of the local anesthetic tetracaine in phospholipid vesicles. Biochim. Biophys. Acta. 465:173–178. [DOI] [PubMed] [Google Scholar]
- 62.Surewicz, W., and W. Leyko. 1982. Interactions of local anesthetics with model phospholipid membranes. The effect of pH and phospholipid composition studied by quenching of an intramembrane fluorescent probe. J. Pharm. Pharmacol. 34:359–363. [DOI] [PubMed] [Google Scholar]
- 63.Ohki, S. 1984. Adsorption of local anesthetics on phospholipid membranes. Biochim. Biophys. Acta. 777:56–66. [DOI] [PubMed] [Google Scholar]
- 64.Strichartz, G. R. 1977. The composition and structure of excitable nerve membrane. In Mammalian Cell Membranes, Vol. 3. G. A. Jamieson and D. M. Robinson, editors. Butterworths, London. 172–205.
- 65.Ohki, S., C. Gravis, and H. Pant. 1981. Permeability of axon membranes to local anesthetics. Biochim. Biophys. Acta. 643:495–507. [DOI] [PubMed] [Google Scholar]
- 66.Mayer, L. D., K. F. Wong, K. Menon, C. Chong, P. R. Harrigan, and P. R. Cullis. 1988. Influence of ion gradients on the transbilayer distribution of dibucaine in large unilamellar vesicles. Biochemistry. 27:2053–2060. [DOI] [PubMed] [Google Scholar]
- 67.Lambert, L. A., D. H. Lambert, and G. R. Strichartz. 1994. Conduction block in isolated nerve by high concentrations of local anesthetics. Anesthesiology. 80:1082–1093. [DOI] [PubMed] [Google Scholar]
- 68.Bainton, C. R., and G. R. Strichartz. 1994. Concentration dependence of lidocaine-induced irreversible conduction loss in frog nerve. Anesthesiology. 81:657–667. [DOI] [PubMed] [Google Scholar]
- 69.Courtney, K. 1980. Structure-activity relations for frequency-dependent sodium channel block in nerve by local anesthetics. J. Pharm. Exp. Ther. 213:114–119. [PubMed] [Google Scholar]
- 70.Butterworth 4th, J. F., J. R. Moran, G. M. Whitesides, and G. R. Strichartz. 1987. Limited nerve impulse blockade by “leashed” local anesthetics. J. Med. Chem. 30:1295–1302. [DOI] [PubMed] [Google Scholar]
- 71.Popitz-Bergez, F. A., S. Leeson, G. R. Strichartz, and J. G. Thalhammer. 1995. Relation between functional deficit and intraneural local anesthetic during peripheral nerve block: a study in the rat sciatic nerve. Anesthesiology. 83:583–592. [DOI] [PubMed] [Google Scholar]
- 72.Nakamura, T., F. Popitz-Bergez, J. Birknes, and G. R. Strichartz. 2003. The critical role of concentration for lidocaine block of peripheral nerve in vivo: studies of function and drug uptake in the rat. Anesthesiology. 99:1189–1197. [DOI] [PubMed] [Google Scholar]
- 73.Kaminoh, Y., H. Kamaya, and I. Ueda. 1989. Differential affinity of charged local anesthetics to solid-gel and liquid-crystalline states of dimyristoylphosphatidic acid vesicle membranes. Biochim. Biophys. Acta. 987:63–68. [DOI] [PubMed] [Google Scholar]
- 74.Constantinescu, A., M. Ionescu, D. Milhailescu, and D.-G. Margineanu. 1988. Partition coefficients of tertiary amines between aqueous solutions and unilamellar liposomes. Rev. Roum. Biochim. 25:269–274. [Google Scholar]
- 75.Watts, A., and T. Poile. 1986. Direct determination by 2H-NMR of the ionization state of phospholipids and of a local anesthetic at the membrane surface. Biochim. Biophys. Acta. 861:368–372. [DOI] [PubMed] [Google Scholar]
- 76.Limbacher, H. P., G. Blickenstaff, J. Bowen, and H. Wang. 1985. Multiequilibrium binding of a spin-labeled local anesthetic in phosphatidylcholine bilayers. Biochim. Biophys. Acta. 812:268–276. [DOI] [PubMed] [Google Scholar]
- 77.White, S. H., and W. C. Wimley. 1998. Hydrophobic interactions of peptides with membrane interfaces. Biochim. Biophys. Acta. 1376:339–352. [DOI] [PubMed] [Google Scholar]
- 78.White, S. H., and W. C. Wimley. 1999. Membrane protein folding and stability: physical principles. Annu. Rev. Biophys. Biomol. Struct. 28:319–365. [DOI] [PubMed] [Google Scholar]
- 79.Mansour, H. M., and G. Zografi. 2007. The relationship between water vapor absorption and desorption by phospholipids and bilayer phase transitions. J. Pharm. Sci. 96:377–396. [DOI] [PubMed] [Google Scholar]
- 80.Higgins, M. J., M. Polcik, T. Fukuma, J. E. Sader, Y. Nakayama, and S. Jarvis. 2006. Structured water layers adjacent to biological membranes. Biophys. J. 91:2532–2542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chiu, S.-W., M. Clark, V. Balaji, S. Subramaniam, H. L. Scott, and E. Jakobsson. 1995. Incorporation of surface tension into molecular dynamics simulation of an interface: a fluid phase lipid bilayer membrane. Biophys. J. 69:1230–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Marrink, S.-J., D. P. Tieleman, A. R. van Buren, and H. J. C. Berendsen. 1996. Membranes and water: an interesting relationship. Faraday Discuss. 103:191–201. [Google Scholar]
- 83.Tanford, C. 1972. Hydrophobic free energy, micelle formation and the association of proteins with amphiphiles. J. Mol. Biol. 67:59–74. [DOI] [PubMed] [Google Scholar]
- 84.Dathe, M., M. Schűmann, T. Wieprecht, A. Winkler, M. Beyerman, E. Krause, K. Matsuzaki, O. Murase, and M. Bienert. 1996. Peptide helicity and membrane surface charge modulate the balance of electrostatic and hydrophobic interactions with lipid bilayers and biological membranes. Biochemistry. 35:12612–12622. [DOI] [PubMed] [Google Scholar]
- 85.Wimley, W. C., K. Gawrisch, T. P. Creamer, and S. H. White. 1996. A direct measurement of salt bridge solvation energies using a peptide model system: implications for protein stability. Proc. Natl. Acad. Sci. USA. 93:2985–2990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ben-Tal, N., B. Honig, R. M. Peittzsch, G. Denisov, and S. McLaughlin. 1996. Binding of small basic peptides to membranes containing acidic lipids: theoretical models and experimental results. Biophys. J. 71:561–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Ben-Tal, N., D. Sitkoff, I. A. Topol, A.-S. Yang, S. K. Burt, and B. Honig. 1997. Free energy of amide hydrogen bond formation in vacuum, in water, and in liquid alkane solution. J. Phys. Chem. B. 101:450–457. [Google Scholar]
- 88.Rothfield, L., and A. Finkelstein. 1968. Membrane biochemistry. Annu. Rev. Biochem. 37:463–496. [DOI] [PubMed] [Google Scholar]
- 89.Alkoskela, J.-M., and P. K. J. Kinnunen. 2001. Control of a redox reaction on lipid bilayer surfaces by membrane dipole potential. Biophys. J. 80:294–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Léonard, A., and E. J. Dufourc. 1991. Interactions of cholesterol with the membrane lipid matrix. A solid state NMR approach. Biochimie. 73:1295–1302. [DOI] [PubMed] [Google Scholar]