

Abstract
Apolipoproteins play a central role in lipoprotein metabolism, and are directly implicated in cardiovascular diseases, but their structural characterization has been complicated by their structural flexibility and heterogeneity. Here we describe the structural characterization of the N-terminal region of apolipoprotein B (apoB), the major protein component of very low-density lipoprotein and low-density lipoprotein, in the presence of phospholipids. Specifically, we focus on the N-terminal 6.4–17% of apoB (B6.4-17) complexed with the phospholipid dimyristoylphosphatidylcholine in vitro. In addition to circular dichroism spectroscopy and limited proteolysis, our strategy incorporates nanogold-labeling of the protein in the reconstituted lipoprotein complex followed by visualization and molecular weight determination with scanning transmission electron microscopy imaging. Based on the scanning transmission electron microscopy imaging analysis of ∼1300 individual particles where the B6.4-17 is labeled with nanogold through a six-His tag, most complexes contain either two or three B6.4-17 molecules. Circular dichroism spectroscopy and limited proteolysis of these reconstituted particles indicate that there are no large conformational changes in B6.4-17 upon lipoprotein complex formation. This is in contrast to the large structural changes that occur during apolipoprotein A-I-lipid interactions. The method described here allows a direct measurement of the stoichiometry and molecular weight of individual particles, rather than the average of the entire sample. Thus, it represents a useful strategy to characterize the structure of lipoproteins, which are not structurally uniform, but can still be defined by an ensemble of related patterns.
INTRODUCTION
The transportation of lipids in the body is mainly achieved through lipoproteins, a group of protein and lipid emulsions that vary from 10 to 100 nm in diameter (1). Central in lipoprotein metabolism are the apolipoproteins. Apolipoproteins are essential for lipoprotein formation, transportation, metabolic reaction, and receptor recognition (1). Structurally, these proteins are composed of similar secondary structural motifs—amphipathic α-helices and β-sheets, which are essential for lipid interaction (2,3). However, the tertiary structure of apolipoproteins and their assembly on lipoprotein particles is poorly understood. Computational modeling remains the primary tool and empirical data are lacking. The challenge lies in the structural flexibility of apolipoproteins and the intrinsic heterogeneity associated with lipoprotein samples, which are often difficult to overcome by biochemical purification.
Apolipoprotein B (apoB) is the major protein component in low-density lipoprotein (LDL), also known as the “bad cholesterol,” which is directly implicated in atherosclerosis. ApoB regulates the synthesis of chylomicrons from the small intestine and very low density lipoproteins, the precursor of LDL from the liver (4). As one of the largest proteins in humans, full-length apoB (B100) is composed of 4536 amino acids and is highly glycosylated (5). The current model of B100 suggests a βα1-β1-α2-β2-α3 pentapartite superdomain organization, in which β corresponds to domains rich in amphipathic β-sheets, and α corresponds to domains rich in amphipathic α-helices (3). These amphipathic sequences are the basic building blocks to achieve lipid interaction in lipoprotein particles. When the protein is separated from its lipid components, apoB is no longer soluble as a monomer (6).
The structure of apoB in LDL has been studied at several labs and models at >20 Å resolution have been proposed using cryo-electron microscopy and crystallography (7–10). Based on these low resolution models of LDL, apoB appears to form belts that wrap around on the surface of an LDL particle. Electron microscopic studies also indicate that the N-terminal region of apoB protrudes away from the LDL particle, suggesting a globular fold (7,9). Indeed, the N-terminal 20.5% of apoB is homologous to lipovitellin, a globular protein found in the egg yokes of lamprey, whose atomic structure has been determined by crystallography (11–13). Based on the structure of lipovitellin, several labs, including ours, have proposed comparative models of the N-terminal region of apoB (11,14,15) (Fig. 1, a and b). We further tested the homology model of B17 using limited proteolysis, chemical cross-linking, and circular dichroism. These studies showed that most empirically determined domain boundaries, domain interactions, and secondary structures are consistent with the lipovitellin-based B17 model (15).
FIGURE 1.

Homology model of B17. (a) Comparison of domain structures of lipovitellin (LV) and apoB. Only the N-terminal 20% of apoB is colored by proposed domains. β-barrel domain, green; α-helical domain, cyan; C-sheet, red; A-sheet, dark blue; missing regions in the structure of LV, white. A domain diagram of B17 has been colored by phospholipid remodeling efficiencies. Lighter color corresponds to more efficient phospholipid remodeling activities as determined previously (24). (b) Ribbon representation of the B17 model colored by domain as in panel a. (c) Ribbon representation of B17 colored by the phospholipid remodeling sequences as in panel a.
Although the N-terminal region of apoB may not directly interact with lipids in LDL, it clearly plays an indispensable role in the assembly of an apoB-containing lipoprotein. Through the expression of C-terminally truncated apoB constructs, it has been demonstrated that the N-terminal 19.5 or 22% of apoB was sufficient to support the secretion of spherical lipoprotein particles (16,17). Moreover, the N-terminal β-barrel and α-helical domains of apoB are the only identified locations responsible for the interaction with microsomal triglyceride transfer protein (MTP), a critical chaperone for apoB assembly in the endoplasmic reticulum (11,18). Interestingly, as a protein of 894 amino acids, MTP shares homology with lipovitellin and B17 (11). Although the exact role of MTP in vivo is not yet clear, this chaperone is critical for efficient lipoprotein assembly. In vitro, MTP transfers neutral lipids from donor vesicles to acceptor vesicles, which suggests its potential lipid shuttling function during the lipidation of apoB (19). Besides its shuttling capability for neutral lipids, MTP has been also suggested to play structural roles to assist the stabilization of lipoprotein particles during the synthesis of the nascent apoB polypeptide (20). In addition, studies by Rava et al. (21) suggested that the phospholipid transferring activity of MTP is sufficient to support lipoprotein assembly and secretion.
The N-terminal region of apoB contains intrinsic lipid remodeling activities, although to what extent these activities are involved in lipoprotein assembly has not been established. It has been demonstrated that B17 binds phosphatidylcholine vesicles in vitro and has the capability to convert dimyristoylphosphatidylcholine (DMPC) multilamellar vesicles into discoidal particles (22,23). Using DMPC as a model lipid, we have shown that, in B17, the DMPC binding sequences largely reside in the N-terminal half of the α-helical (B6.4-10) and C-sheet (B13-17) domains, but not in the N-terminal β-barrel (B5.9) or the C-terminal half of the α-helical domain (B9-13) (Fig. 1, a and c) (24). Although the reconstituted particles obtained in vitro do not exist in vivo, the structure of these reconstituted particles may provide important insights into the biophysical properties and potential conformation of apoB domains during the initiation of lipoprotein assembly. In this article, we continue our study of the phospholipid-binding regions in B17 and characterize the structure of B6.4-17 in reconstituted lipoprotein particles using a novel strategy that incorporates spectroscopic methods, limited proteolysis, and scanning transmission electron microscopy (STEM). These methods allow us to propose an in vitro working model of the conformation of B6.4-17 in a reconstituted DMPC particle.
MATERIALS AND METHODS
Protein cloning, expression, and purification
Protein constructs, including B6.4-10 (residues 292–469), B6.4-13 (residues 292–593), B6.4-17 (residues 292–782), and B9-13 (residues 430–593), were cloned into the pET24a vector (EMD Chemicals, San Diego, CA) and expressed in BL21 DE3 Escherichia coli cells using standard protocols. Each construct has a 6-His tag on its C-terminus. Cells were grown at 37°C to an optical density of 0.6–0.8 at 600 nm in Luria broth supplemented with kanamycin (34 μg/ml). Expression was then induced with 1 mM isopropyl-β-D-thiogalactopyranoside and the cells were grown for an additional 3 h. Cell pellets were lysed with 1 mg/ml lysozyme at room temperature for 30 min and then sonicated with a probe sonicator (Branson, Danbury, CT). The protein-containing inclusion bodies were dissolved in 8 M urea after washing with 1% Triton X-100 and 1 M Urea. Soluble proteins were loaded on a Ni-NTA Sepharose column (Qiagen, Valencia, CA) and eluted with 250 mM imidazole and 6 M guanidine hydrochloride (GuHCl). Protein refolding was achieved by slowly adding concentrated protein stocks in 6 M GuHCl into a refolding buffer containing 50 mM tris(hydroxmethy)aminomethane (Tris), 800 mM arginine, 10 mM reduced glutathione, 2 mM oxidized glutathione, and 0.02% sodium azide at pH 8.0. Protein at 1–2 μM in the refolding buffer was incubated at 4°C overnight and then dialyzed extensively against 10 mM Tris and 150 mM sodium chloride, pH 7.5 (TS buffer). The final protein volume and concentration were adjusted using an Amicon Ultra concentrating apparatus (Millipore, Billerica, MA) and the protein concentration was determined by UV absorbance at 280 nm (25).
DMPC vesicle preparation and reconstituted particle formation
To prepare multilamellar 1,2-dimyristoyl-sn-glycero-3-phosphocholine (DMPC) suspensions, 10 mg DMPC (Avanti Polar Lipids, Alabaster, AL) in chloroform was transferred to a round bottom flask and connected to a rotary evaporator (Buchi, New Castle, DE) to form a uniform DMPC thin layer, which was then placed under vacuum for 1 h to remove residual chloroform. The lipid was hydrated with 5 ml TS buffer and gently agitated with small glass beads at room temperature to form multilamellar DMPC suspensions. To form reconstituted lipoprotein particles, B6.4-17 at 1 mg/ml was mixed with the DMPC suspension at the desired ratio in TS buffer and incubated at 24°C for at least 16 h.
Size exclusion chromatography analysis
B6.4-17 or B6.4-17/DMPC particles were prepared at 0.5–1 mg/ml protein concentration at the specified L/P ratios and then injected onto a Superdex GL-200 column (GE HealthCare, Piscataway, NJ). The column was eluted with TS buffer at 0.5 ml/min and 4°C and the elution was monitored by absorbance at 280 nm.
Circular dichroism analysis
Circular dichroism (CD) data were collected on an AVIV 215 instrument (AVIV, Lakewood, NJ). Protein or B6.4-17/DMPC samples at 1–2 μM (protein concentration) were dialyzed against 5 mM potassium phosphate at pH 7.5 before the experiment. The exact protein concentration was determined by absorbance at 280 nm immediately before the wavelength scan. Each reported wavelength scan was an average of four scans taken in a 1 mm cuvette with a 5-s averaging time at every nm. CD scans for the buffer and cuvette were measured right before the data collection for background correction. For chemical denaturation experiments, protein samples at 0.1–0.2 μM were prepared in 5 mM potassium phosphate, pH 7.5. Protein unfolding was achieved using an AVIV titration accessory by the addition of the same concentration of protein in 7 M GuHCl, pH 7.5, in 0.1 M steps into 2.0 ml native protein solution in a 1 cm cuvette. The sample volume was maintained constant throughout the titration, and protein unfolding was monitored at 222 nm at 25°C. After each injection of denaturant, the sample was stirred for 3 min, equilibrated for 20 s and the data were collected with an averaging time of 20 s. Since most of the unfolding curves do not exhibit typical two-state transitions, the percentage unfolding at each GuHCl concentration (%Unfolded [x]) was calculated using a modified equation:
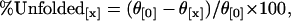 |
in which θ[0] is the CD signal in the absence of GuHCl. This modification allows the comparison of curves without arbitrary determination of the folded and unfolded baselines. For comparison, a conversion using the first 10 data points as the folded baseline and the CD signal at 6 M GuHCl concentration as the unfolded reference was used to calculate the inset plot in Fig. 3 c.
FIGURE 3.

Circular dichroism studies of B6.4-17 and its subdomains. (a) CD wavelength scans of B6.4-13 (⋄), B6.4-10 (◃), and B9-13 (▹). Samples contained ∼5 μM protein in 5 mM potassium phosphate at pH 7.5, in a 1 mm cuvette at 25°C. Four scans with an averaging time of 5 s at every nm were collected and the average data are reported. (b) GuHCl titration of B6.4-13 (⋄), B6.4-10 (◃), and B9-13 (▹). (c) GuHCl titration of B6.4-17 (▵) and B6.4-17/DMPC complex (▴) at 1.2:1 L/P ratio. Native proteins in a 1 cm cuvette were titrated by the protein at the same concentration in 7 M GuHCl at 0.1 M per step at 25°C. After each injection of the denaturant, the sample was stirred for 3 min and the CD signal at 222 nm was averaged for 20 s. The raw CD data were converted to percentage unfolded by calculating the ratio between each data point and the CD signal at 0 M GuHCl as described in Materials and Methods. In the inset figure, the percentage unfolded curve was calculated by applying a folded baseline using the first 10 data points and using the CD signal at 6 M GuHCl as the unfolded reference.
Limited proteolysis, HPLC, and mass spec analysis
Trypsin from bovine pancreas (Sigma, TPCK-treated; St. Louis, MO) was freshly prepared at 1 mg/ml in 1 mM HCl and added to 250 μl of 1 mg/ml B6.4-17 or B6.4-17/DMPC complexes in TS buffer to produce the final trypsin to protein ratio of 1:1000 (w/w). The mixtures were incubated at room temperature for 15 min. The proteolytic digestion was terminated by the addition of 2-mercaptoethanol to a final concentration of 10 mM and acidification with glacial acetic acid to a final concentration of 5%. Digestion products were either analyzed by SDS-tricine (26) gels or high performance liquid chromatography (HPLC) on an analytical C4 reverse phase column (Grace Vydac, Columbia, MD). The column was eluted at 1 ml/min with a 1%/min gradient from water to 90% acetonitrile with 0.1% trifluoroacetic acid throughout the gradient and monitored at 220 nm. Samples for N-terminal sequencing were purified either from SDS-tricine gels or by HPLC. Peak fractions from HPLC were analyzed with matrix-assisted laser desorption ionization time-of-flight mass spectrometry to determine the molecular weight of each fragment at the Molecular Biology Core Facility of the Dana Farber Cancer Institute.
Ni-NTA-Nanogold labeling
Ni-NTA-Nanogold (Nanoprobes, Yaphank, NY) at 10 μM was incubated with 3.3 μM (protein concentration) reconstituted B6.4-17/DMPC particles for 15 min at room temperature. The mixture was then loaded on a Superdex GL-200 column to remove excess protein and Ni-NTA-Nanogold from the reconstituted particles. The column was eluted with TS buffer at 0.5 ml/min at 4°C.
Negative stain electron microscopy
For negative stain electron microscopy, reconstituted B6.4-17/DMPC particles (4 μl) at 0.1–0.5 mg/ml (protein concentration) were loaded on a carbon-coated and glow-discharged copper grid (SPI Supplies, West Chester, PA) for 1 min, washed with 10 drops of distilled water and stained with 1% sodium phosphotungstate, pH 7.5 or Nanovan (Nanoprobes, Yaphank, NY) for 30 s (27). All samples were imaged on a Philips CM12 transmission electron microscope (FEI Company, Hillsboro, OR) operated at 120 kV with a LaB6 filament and recorded on SO-163 EM (Kodak, New Haven, CT) films at 45,000× magnifications under minimal electron dose conditions. The film was processed with undiluted Kodak D-19 developer for 12 min and Kodak rapid fixer for 5 min. Electron micrographs were digitized on a Creo IQ Smart2 Scanner (Kodak, Rochester, NY) at 1270 dpi.
Sample preparation and STEM imaging
Grids for the STEM were prepared by the wet film technique as described previously (28,29). Briefly, 2.3 mm titanium grids, coated with a thick holey film, were placed on a floating thin (2–3 nm) carbon film prepared by ultra-high vacuum evaporation onto freshly cleaved rock salt. Both the sample and the tobacco mosaic virus, an internal standard, were allowed to adsorb to the carbon film for 1 min. The grids were washed extensively, ending with a volatile buffer, blotted to a very thin layer of liquid and plunged into liquid nitrogen slush. They were later transferred to an ion-pumped freeze-drier, freeze-dried overnight, and transferred under vacuum to the STEM.
The STEM was operated in a dark-field mode. The scattered electrons were collected in two annular detectors from each pixel and the number of scattered electrons (in each pixel) was directly proportional to the mass thickness in that pixel. By summing the scattered electrons over a particle and subtracting the background from the thin carbon film, the mass of the particle can be determined. B6.4-17/DMPC particles were imaged by the STEM at the Brookhaven National Laboratory, which is described in more detail at http://www.biology.bnl.gov/stem/stem.html.
Analysis of STEM images
A total of 1382 well-dispersed particles were manually picked from 72 independent STEM images using PCMass28 (30) and were categorized into 1 of the 16 geometrical groups as defined in Fig. 6. The mass of each particle was determined using two background correction methods available in PCMass28: the entire image background and the local background. Two types of particles were excluded in the statistical analysis: 1), particles that did not fall into any of the 16 geometrical groups; and 2), particles with molecular weight differences >20% using the two background correction methods. A total of 1315 particles met these two criteria and were used in the statistical analysis. Statistical analysis was performed in Origin 7.5 (OriginLab, Northampton, MA).
FIGURE 6.

Categories of nanogold-labeled particles. (a) Histogram of the number of nanogold on each particle. A total of 1315 particles were analyzed, and are categorized based on the number of nanogolds attached on each particle. (b) Accumulated plot of the nanogold count. Each data point represents the sum of the number of particles in this group plus the accumulated number of particles in the previous groups. (c) Histogram of the 16 geometrical groups. Nanogold-labeled particles were categorized into 16 geometrical groups as indicated at the bottom of the histogram. The number of particles counted in each group was plotted with shaded bars.
Protein modeling and graphics presentation
The homology model of B17 was generated with MODELLER as described previously (15). Molecular images were generated in MOLMOL (31). Pov-Ray 3.6 (www.povray.org) was used to generate the cylinder-ribbon representation of the proposed B6.4-17 tertiary folding in a reconstituted DMPC particle. The lipid core and each B6.4-17 domains were kept approximately to scale.
RESULTS
Preparation of B6.4-17 and B6.4-17/DMPC reconstituted particles
B6.4-17 contains all phospholipid remodeling sequences within B17, including the proposed α-helical and C-sheet domains (Fig. 1, a and b) (24). This construct encompasses the central regions in the corresponding lipovitellin structure, but lacks the N-terminal β-barrel domain (B5.9) that is difficult to refold in high yield and does not interact with lipids (15). In lipovitellin, the α-helical domain serves as a clamp that maintains the relative position of the C-sheet and the A-sheet (12,13). B6.4-17 forms inclusion bodies when expressed in E. coli, but it can be routinely refolded to a monomeric state using the method previously described (15). B6.4-17 clears turbid DMPC multilamellar vesicles and forms reconstituted lipoprotein particles in vitro (24). These DMPC-reconstituted particles can be separated from lipid-free proteins using size exclusion chromatography (Fig. 2 a). Similar to the behavior of apoA-I, the size of the reconstituted particles correlates with the initial lipid-to-protein ratio (L/P) (32). Higher L/P ratios gives rise to larger particles and the reconstituted particle peak elutes earlier (Fig. 2 a). When the peak fractions from the size exclusion column are analyzed with negative stain electron microscopy, the images of these particles are reminiscent of the reconstituted high density lipoprotein (HDL) particles using apolipoprotein A-I (apoA-I) (Fig. 2, b and c) (32,33). We also found that when the protein is in excess (L/P < 1.25, wt/wt), the B6.4-17/DMPC complex reaches a minimal size (24). This observation suggests that a minimal, fixed amount of DMPC is required to form a stable complex in excess protein. Because the size and composition of these minimal-sized particles are the most uniform and are not influenced by slight differences in protein and lipid concentrations, they are the focus of this study.
FIGURE 2.

B6.4-17 forms reconstituted particles with DMPC. (a) Size exclusion chromatography of reconstituted B6.4-17/DMPC particles. Thin shaded line, lipid-free B6.4-17; thick shaded line, B6.4-17/DMPC complex at 1:1 L/P weight ratio; thick solid line, B6.4-17/DMPC complex at 4:1 L/P weight ratio; dotted shaded line, B6.4-17/DMPC complex at 0.8:1 L/P weight ratio with Ni-NTA-Nanogold labels (AU); B6.4-17/DMPC complex at 4:1 L/P weight ratio with Ni-NTA-Nanogold labels. (b,c) Negative stain electron microscopic images of reconstituted B6.4-17/DMPC particles at 1:1 L/P weight ratio (b) and 4:1 L/P weight ratio (c). The magnification bar corresponds to 50 nm in both images.
Characterization of protein conformation by circular dichroism spectroscopy
Both homologous modeling and biophysical studies suggest that B6.4-13 is a largely α-helical domain (15). The α-helical domain in lipovitellin contains an extensively hydrophobic interface between the two layers of α-helices, which maintains the integrity of the helix bundle (Fig. 1 b). Yet, we noticed that the N-terminal half (B6.4-10) and C-terminal half (B9-13) of the α-helical domain behave differently when they are exposed to phospholipid bilayers, suggesting potential differences in their biophysical properties (24). To compare these two potential subdomains within B6.4-13, the protein conformation of B6.4-10 and B9-13 were characterized using circular dichroism (CD). Both truncated constructs give typical α-helical spectra, with minima at 222 nm and 209 nm. The mean residue α-helical content of either truncated construct is 25–30% less than that of B6.4-13 by an approximate calculation based on the ellipticity at 222 nm (Fig. 3 a). Despite the similar overall secondary structure between B6.4-10 and B9-13, guanidine hydrochloride (GuHCl) denaturation monitored by CD reveals a significant difference in their chemical stability. The unfolding of B6.4-10 (left facing triangles) with increasing GuHCl concentration is almost linear, suggesting a molten globule conformation that lacks a fixed hydrophobic core (Fig. 3 b). In contrast, B9-13 (right facing triangles) has a clear transition point at 2.1 M GuHCl, consistent with a cooperative protein unfolding model. The unfolding curve of B6.4-13 (diamonds) appears to reflect the combined unfolding of both subdomains.
The potential conformational changes of B6.4-17 upon the formation of reconstituted lipoproteins are examined by CD as well. The far-UV CD wavelength scan of B6.4-17 exhibits the typical characteristics of α-helical proteins. The incorporation of B6.4-17 into a DMPC complex does not change the overall shape of the far-UV CD spectrum, suggesting that no large changes in the protein secondary structure have occurred (24). The protein tertiary structure was probed by GuHCl denaturation and monitored by CD at 222 nm, which primarily tracks changes in α-helical structures. B6.4-17 in DMPC complexes appears to exhibit unfolding curves comparable to the lipid-free protein (Fig. 3 c), but the extent of unfolding is ∼10% less than that of the lipid-free protein. This observation suggests that the binding to DMPC may stabilize slightly more α-helical segments, but does not cause dramatic changes in the unfolding cooperativity. Indeed, when both curves are fitted by their folded based line, the shapes of the transitions are identical (inset in Fig. 3 c). The similar shape of these two curves suggests that DMPC binding does not change the unfolding cooperativity of B6.4-17, which is largely achieved by the tertiary structure and the formation of hydrophobic cores.
Conformational changes probed by limited proteolysis
Protease susceptibility is an indication of unstructured protein sequences, as folded structure is usually protected from proteolytic degradation. Therefore, a comparison of limited proteolysis patterns in lipid-free and lipid-bound forms serves as a sensitive tool in identifying unstructured regions or lipid-interacting sites. Both lipid-free B6.4-17 and the B6.4-17/DMPC complex were incubated with trypsin (1000:1 wt/wt, respectively) for 15 min, and their proteolysis patterns were compared (Fig. 4). Similar fragments (1a and 2a, 2b) at ∼35 kDa were observed for both samples. In the proteolysis of the B6.4-17/DMPC complex, an additional fragment at ∼20 kDa (2c) and a less significant fragment at a smaller molecular weight (2d) were observed. In contrast, no significant fragment of a similar size was observed in lipid-free B6.4-17.
FIGURE 4.

Limited proteolysis of B6.4-17 and B6.4-17/DMPC particles. Lipid-free B6.4-17 and B6.4-17/DMPC complexes (1.2:1 wt/wt L/P ratio) were prepared at 1 mg/ml protein concentration in 20 mM Tris-HCl and 150 mM sodium chloride, pH 7.5. Freshly prepared Trypsin in 1 mM HCl was added to a protein to trypsin ratio of 1000:1 (wt/wt) and incubated for 15 min at room temperature. The digestion was stopped by the addition of 10 mM β-mercaptoethanol and acetic acid to 5%. Lane 1, protein standards; lane 2, B6.4-17 before digestion; lane 3, B6.4-17 after digestion; lane 4, B6.4-17/DMPC after digestion. The major proteolytic products are labeled to the left of the gel and are identified in Table 1.
To identify these fragments, proteolytic products were purified on a C4 reverse phase column and subjected to N-terminal sequencing and mass spectrometry analysis. A common cleavage after Lys614 is observed for both lipid-free and DMPC-bound B6.4-17 (Table 1). This cleavage, located in the middle of the first proposed β-strand in the C-sheet, was also observed in the trypsin digestion of B17 as previously reported (15). Fragments 1a and 2a correspond to the α-helical domain and fragment 2b corresponds to the α-helical domain missing 15 (or 16) residues at the N-terminus and 10 (or 9) residues at the C-terminus. Interestingly, the incorporation of B6.4-17 into DMPC particles protects the protein from trypsin cleavage in the C-sheet, as fragment 2c and 2d are almost absent in the digestion of lipid-free B6.4-17. Fragment 2c is indeed the proposed C-sheet domain after the cleavage at Lys614, while fragment 2d is missing the N-terminal 14 amino acids from fragment 2c (Table 1). The protection of the C-sheet in the digestion of the DMPC-bound B6.4-17 indicates that this domain is stabilized against proteolysis by DMPC probably through direct involvement in lipid binding.
TABLE 1.
Identification of proteolytic digest products
| Fragment* | N-terminal sequence | Experimental MW | Predicted fragment | Calculated MW† | Calc. vs. Exp. MW error |
|---|---|---|---|---|---|
| 1a | PKQAE | 36851.5 | 292–614 | 36783.5 | −0.18% |
| 2a | PKQAE | 36820.3 | 292–614 | 36783.5 | −0.10% |
| 35558.8 | 292–605 | 35583.1 | 0.07% | ||
| 2b | KLTIS | 33767.0 | 307–604 | 33777.0 | 0.03% |
| LTISE | 308–605 | 0.03% | |||
| 2c | SVSLP | 19240.9 | 615–782 | 19219.0† | −0.11% |
| 2d | IEGNL | 17985.2 | 628–782 | 17965.6† | −0.11% |
The identity of each fragment is defined in Fig. 4.
The calculated molecular weight includes six histidines at the C-terminus of the protein.
Ni-NTA-Nanogold labeling of the reconstituted B6.4-17/DMPC particles
Previously, we have described the morphology of the reconstituted B6.4-17/DMPC particles imaged by negative stain electron microscopy (EM) (24). The contrast in a negative stain image comes primarily from the stain, thus it fails to differentiate the protein and lipid moieties. Based on the observation of the plate- and rod-shaped morphologies of B6.4-17/DMPC complexes, a discoidal DMPC complex model has been proposed (Fig. 2, b and c) (24). However, an alternative vesicle interpretation is difficult to exclude since the staining process inevitably introduces artifacts and could potentially collapse fragile vesicles. Furthermore, although only a 15% standard deviation is observed in the average diameter of the minimal-sized particles, the negative stain method cannot be used to determine variations in protein/lipid stoichiometry or the geometry of proteins on individual particles.
To overcome these problems, we used a Ni-NTA-Nanogold probe to label the protein component of the reconstituted lipoprotein particles. Ni-NTA-Nanogold contains a 1.8 nm gold particle with multiple nickel-nitrilotriacetic acid (Ni-NTA) groups on the surface (34). This probe specifically labels the C-terminus of B6.4-17 through its 6-His tag and allows the direct localization of each individual protein molecule. To generate gold-labeled particles, a threefold molar excess of nanogold was incubated with the reconstituted DMPC complexes and the unbound nanogold was removed by purification on a size exclusion column. Three peaks were observed, corresponding to the excess nanogold, nanogold-labeled complex and nanogold-labeled protein (Fig. 2 a). The nanogold labeled proteins elute earlier than unlabeled proteins on a size exclusion column as expected due to the increase in mass from the large gold particle.
Imaging of nanogold labeled particles using negative stain EM is technically challenging. A 1.8-nm gold particle is slightly larger than the grainy noise on the electron micrograph (Fig. 5 a). Gold enhancement may increase the visibility of the nanogold, but it also lowers the homogeneity of the nanogold particles because the solvent accessibility of each nanogold can be different, depending on how the DMPC particle is oriented on the grid (Fig. 5 b). However, optimal imaging can be achieved using scanning transmission electron microscopy (STEM).
FIGURE 5.

Imaging of the reconstituted B6.4-17/DMPC particles. (a) Negative stain image of reconstituted DMPC particles labeled with Ni-NTA-Nanogold. (b) Negative stain image of reconstituted DMPC particles labeled with Ni-NTA-Nanogold after gold enhancement. Ni-NTA-Nanogold was added to a 3:1 molar ratio to the protein/lipid mixture before the preparation of EM-grids. Protein samples were stained by Nanovan and gold enhancement was performed according to the protocol from Nanoprobes. The magnification bar corresponds to 20 nm for both images. White triangles point to gold particles. (c) STEM image of reconstituted B6.4-17/DMPC particles at 0.8:1 L/P weight ratios. The reconstituted particles had been purified on a Superdex GL 200 column (Fig. 2). Tobacco mosaic virus was used as an internal standard as shown in the image. The magnification bar corresponds to 20 nm. Six reconstituted particles (i–vi) are shown in enlarged views (36 × 36 nm) on the right.
STEM does not require heavy metal stain; image contrast is solely achieved from the difference in the sample density (35). Because of the much higher density of gold, the nanogold probe is easily visible without enhancement (Fig. 5 c). After the analysis of over 1300 particles prepared at 0.8:1 L/P (wt/wt) ratio (excess protein) and 500 particles prepared 4:1 L/P (wt/wt) ratio (excess lipid), we found that the nanogold labels appear exclusively on the circumference of the particle, when the particle is in a “plate” view. Notably, fewer particles in rod views are observed and no rouleaux (stacks of particles) are spotted using STEM, which suggests that the formation of rouleaux may be promoted by the heavy metal stain. This observation provides direct support for the discoidal model of the reconstituted particle, in which the protein wraps around a DMPC bilayer core and covers the circumferential hydrocarbon chains. Otherwise, it would be expected that many nanogold labels would be seen on the particle if the reconstituted lipoprotein adopts a vesicular structure and the protein randomly docks in the phospholipid bilayer.
Assembly of proteins in a reconstituted DMPC complex
Ni-NTA-Nanogold labeling and STEM imaging provide a much more informative image of the reconstituted DMPC complex. Although we found that the distribution of nanogold on the lipoprotein particle is not uniform, certain tendencies are observed. For instance, five representative groups of particles are illustrated in Fig. 5 c. Both particles i and ii have three nanogold labels, yet the nanogold layout in particle i adopts a threefold symmetry (C3 symmetry); while particle ii appears nonsymmetrical, probably missing one nanogold in a fourfold symmetrical (C4 symmetry), which is shown in particle vi. Particles iii–v all have two gold labels. Yet, particle v differs from particles iii and iv in the way these two nanogold labels are positioned. Since the nanogold attaches to the 6-His at the protein C-terminus, particles iii and iv may adopt a head-to-tail protein assembly, while particle v may adopt a tail-to-tail assembly.
We manually picked ∼1400 particles prepared at 0.8:1 L/P (wt/wt) ratio, analyzed ∼1300 of them, and grouped them based on the number of nanogold labels observed on each particle. The excluded particles either had high error in their molecular weight, or did not did correspond to one of the 16 geometrical groups shown in Fig. 6. Almost half of the particles have two gold labels and one third of the particles have three gold labels (Fig. 6 a). An accumulative plot suggests maximum labeling occurs at three nanogolds, since it is where the curve reaches its statistical plateau (Fig. 6 b). Based on the geometry of the nanogold labels, these particles were further classified into 16 geometrical groups, which we found were sufficient to characterize most of the reconstituted particles imaged by STEM (Fig. 6 c). A few larger particles that lack definitive morphology were excluded in our analysis, and we speculate that they were fused vesicles.
The statistics indicate that the two nanogolds in a C2 symmetry (Fig. 6 c, group #5) occurs most frequently among the 16 geometrical groups. Following that are the groups of two nanogolds in a nonsymmetrical assembly (#7), three nanogolds with C3 symmetry (#9), and three nanogolds in a nonsymmetrical assembly (#10). It should be noted that incomplete nanogold labeling is possible in our preparation. Therefore, some particles in group #7 may in fact have three proteins in a C3 symmetry. Similarly, some particles in group #10 may have four proteins in a C4 symmetry. Since incomplete labeling should occur equally in all geometrical groups, the fact that only a few particles have a single nanogold label suggests that labeling is close to complete. Based on the ratio between particles with a single gold label and particles with two gold labels, we believe that incomplete labeling is ∼10% in our preparation (Fig. 6, a).
Size of the reconstituted DMPC particles
Since the image intensity is a measure of sample density, STEM provides a direct measurement of the molecular weight for each reconstituted particle. The average molecular weight of a Ni-NTA-Nanogold is 25.7 ± 9.0 kDa, as measured from 207 nanogolds in 10 independent images. The molecular weight of each reconstituted particle is calculated from the measured molecular weight excluding the molecular weight of the nanogold, i.e., the number of nanogolds times 25.7 kDa.
The average molecular weight can be compared among particles with different numbers of observed nanogold labels (Fig. 7 a). A statistically significant increase (p < 0.01) in molecular weight is observed with an increasing number of nanogold labels from 2 to 4, the three groups that constitute 93% of all analyzed particles. Since the number of nanogolds corresponds to the number of proteins per particle, this increase in molecular weight is expected. Rather surprisingly, the increase is smaller than 56 kDa, the molecular weight of B6.4-17. The increase of the average molecular weight for particles with two nanogold labels to three nanogold labels is 28 kDa and from three to four is 39 kDa (Fig. 7 a). In other words, current data do not suggest a correlation between the increase in the size of the DMPC core and the increase of the number of proteins on the particle. Rather, the addition of the third protein results in a particle with fewer lipid molecules.
FIGURE 7.

Molecular weights of nanogold-labeled particles. (a) The average molecular weight of the reconstituted particle when different numbers of nanogold labels are present. The molecular weight of each reconstituted particle was measured with PCMass28 (30). The mass that corresponds to the nanogold was excluded during statistical analysis. The error bar corresponds to the standard deviation of the calculated molecular weight at each stoichiometry. The number of particles in each group was labeled on the top of each dataset. Datasets with an asterisk have a P-value <0.01 among each other, as calculated by analysis of variance in Origin 7.5 (Microcal). (b,c) The frequency of occurrence of the molecular weight for particles with a C2 symmetry (b) or a C3 symmetry (c).
Subgroups #5 (C2 symmetry) and #9 (C3 symmetry) represent the most commonly observed geometrical orientations (Fig. 6 c). The molecular weight distributions of the reconstituted particles in these two subgroups indicate a slight, but discernable increase in the molecular weight (Fig. 7, b and c). The average molecular weight of particles in the C2 and C3 symmetry is 263 ± 77 kDa and 289 ± 81 kDa, respectively. Based on these values, the average number of protein and DMPC molecules in each particle is: C2, two proteins and 222 DMPC molecules; and C3, three proteins and 181 DMPC molecules. These values correspond to the L/P weight ratios of 1.34 and 0.72. Applying similar calculations on all geometric groups, the average L/P weight ratio for all 1315 particles is 1.17. Notably, when using previously described biochemical methods to measure the protein and phospholipid concentrations separately, we obtain an L/P weight ratio of 1.24 for the purified minimal-sized B6.4-17/DMPC particles (24). Therefore, the measurements made from the single particle STEM analysis is consistent with those obtained from gross biochemical analysis. Moreover, this single-particle-based analysis presents geometrical and statistical information that is unavailable in gross biochemical analysis.
DISCUSSION
After three decades of extensive study, the mechanism that underlies the formation of lipoproteins remains elusive. The difficulty in obtaining high resolution structures for apolipoproteins, especially in the lipid-bound form, prevents our understanding of this vital process at a molecular level. Presumably, one of the major difficulties derives from the conformational flexibilities of apolipoproteins, which may be critical for their physiological functions, but imposes enormous technical complications in structural studies. A structurally homogenous sample is the prerequisite for most structural methods, including crystallography, NMR, and electron microscopic reconstruction. On the other hand, apolipoproteins are structurally simpler, because they often have characteristic secondary structural elements, i.e., amphipathic α-helices or β-sheets. Computational modeling incorporated with constraints obtained from biophysical methods and direct imaging can be a valid strategy to obtain structural insights into the function of these proteins.
Here we introduce a novel approach to obtain such structural constraints for the N-terminal domain of apoB in a reconstituted lipoprotein particle. Direct imaging using cryoelectron microscopy and pattern characterization has been successfully utilized by van Antwerpen and co-workers to unveil the heterogeneity in serum derived lipoproteins (36). Our method is based on a similar strategy, but involves the use of Ni-NTA-Nanogold labeling and scanning transmission electron microscopy (STEM). This method allows an easier and more precise evaluation of individual particles in a sample that is heterogeneous, but not random. The geometrical assembly and molecular weight information obtained from this study reveals the range of the heterogeneity of these particles and provides insights into the possible structural basis behind such heterogeneity. This information is not accessible with average-based biochemical analyses of these particles. It should be noted that such nonrandom heterogeneity may not only exist in the B6.4-17/DMPC complex, but also among other naturally present lipoproteins or lipoprotein intermediates. Behind these heterogeneous observations lie the biophysical properties of the apolipoproteins, their domain structures and interactions.
Combined with limited proteolysis, biophysical characterization and comparative modeling, we propose a working model of B6.4-17 in a reconstituted DMPC complex. In this model, three structural elements in B6.4-17 are involved in DMPC binding: the N-terminal half of the α-helical domain, the β-sheets in the C-sheet domain, and two proposed amphipathic α-helices in the middle of the C-sheet domain that are missing in the lipovitellin-based model (Fig. 8, a and b). These three structural elements shield the exposed hydrocarbon chains on the circumference of the DMPC bilayer and stabilize the particle (Fig. 8, b and c).
FIGURE 8.

Models of reconstituted B6.4-17/DMPC particles. (a) Domain diagram of B6.4-17. The N-terminal half of the α-helical domain, green; C-terminal half of the α-helical domain, cyan; β-sheets in the C-sheet domain, red; proposed α-helical region in the C-sheet domain missing in the lipovitellin crystal structure, magenta. (b) Proposed domain interactions in lipid-bound B6.4-17. The black sphere indicates a nanogold label. (c) A three-dimensional cartoon of B6.4-17 in a discoidal particle. The orange disks represent a DMPC bilayer. The lipid core and B6.4-17 domains are shown approximately to scale. (d–f) Proposed protein assemblies in the B6.4-17/DMPC particle. (d) Two proteins in a head-to-tail assembly. (e) Two proteins in a head-to-head assembly. (f) Three proteins in a symmetric assembly. The expected STEM views are shown on the right corner of each model.
The N-terminal half of the α-helical domain contains the phospholipid remodeling activity, while the C-terminal half does not (24). When the protein binds a phospholipid bilayer, some structural adjustments in N-terminal subdomain may occur. Such adjustment may involve a hinge motion in the middle of the α-helical domain. Some opening of the α-helical bundles can contribute to the change of the orientation of the N-terminal subdomain (green) compared to its position in the lipovitellin-based model (Fig. 8 c). This hypothesis is in agreement with the molten globule nature of this subdomain as identified by circular dichroism. Moreover, the limited proteolysis study also identified small changes of the trypsin accessibility in the very N-terminus of B6.4-17.
The C-sheet domain (B13-17), particularly its N-terminal half (B13-15), has a strong phospholipid binding activity (24). Furthermore, the protection of a fragment corresponding to the predicted C-sheet is significantly enhanced in the trypsin digestion of B6.4-17/DMPC particles, also indicating that the C-sheet region in B6.4-17 is directly involved in DMPC binding. However, it should be mentioned that the identified trypsin cleavage sites (Lys614 and Lys627) in the C-sheet domain are both located in the middle of the predicted β-sheets in the B17 model (Table 1), which suggests potential inaccuracies in the current homology model.
CD measurements of the truncated C-sheet (B13-17) suggest ∼20% β-sheet content and ∼30% α-helical content (15). Therefore, the term “C-sheet” may not accurately reflect the structure of this domain. Notably, a segment of 64 amino acids (B15.4-16.4) cannot be modeled due to the lack of electron density in the lipovitellin structure. This segment contains seven Lys residues, which have better protection than Lys614 or Lys627 in the trypsin digestion, indicating this segment may be folded and is likely to interact with DMPC. Indeed, some sequences in this segment have high scores for the prediction of being α-helical, which may explain the observed α-helical content in B13-17. Segrest and co-workers proposed that this sequence folds into two amphipathic helices, and stabilizes the lipoprotein particle containing B22 (17,37). Our observation is consistent with this hypothesis. We propose that these two helices constitute the third DMPC binding element in B6.4-17.
Ni-NTA-Nanogold labels and STEM imaging provides new insights into the molecular assembly of B6.4-17 in a DMPC particle. First, the exclusive circumferential location of the nanogold labels directly supports the model that these reconstituted particles adopt a discoidal shape, with proteins wrapping around and shielding the hydrophobic acyl chains of DMPC. Secondly, the reconstituted B6.4-17/DMPC complexes are structurally heterogeneous, and multiple forms of protein assembly are observed. Most DMPC complexes have two or three B6.4-17 molecules attached. Among these groups, the geometry of two proteins in a symmetric layout has the highest frequency. The fact that a symmetric distribution is more common than nonsymmetrical distributions suggests that a head-to-tail protein assembly is preferred. Another unexpected finding is that the size of the DMPC bilayer does not increase as more proteins are incorporated into a single particle. This observation suggests a high degree of flexibility in the way the proteins associate with DMPC bilayers. Notably, the sample analyzed here is prepared under conditions of excess protein, which may promote the attachment of more proteins onto the reconstituted particle. Indeed, in samples prepared in excess DMPC, the C2 geometric group has an even higher frequency (data not shown).
The heterogeneity of the reconstituted B6.4-17/DMPC particles comes from the different combinations of proteins assembled on the circumference of a DMPC bilayer (Fig. 8, d–f). A head-to-tail arrangement will give rise to a symmetric geometry of nanogold labels, such as C2 and C3 (Fig. 8, d and f). A head-to-head assembly will result in nonsymmetrical geometry, such as the one shown in Fig. 8 e. Another level of heterogeneity can arise from the fact that some subdomains in B6.4-17 may detach from DMPC. Wang and co-workers have demonstrated that amphipathic β-sheets bind hydrophobic surfaces irreversibly, while amphipathic α-helices may be expelled when the interfacial pressure reaches a critical point (38–40). Thus, the amphipathic β-sheets could outcompete the weaker binding helical regions at the hydrophobic edge of the particle. We suspect that not all lipid-binding elements in B6.4-17 are associated with DMPC when the number of proteins in the particle increases (Fig. 8 f). This adaptability in the protein can give rise to particles with different number of proteins, but a similar sized lipid core. This would also explain the occasional observation of reconstituted particles with two nanogold labels very close to each other (Fig. 6, #6 and #11).
The reconstituted B6.4-17/DMPC complexes appear morphologically very similar to premature HDL particles formed with apoA-I. However, our study indicates that the N-terminal α-helical and C-sheet domains in apoB behave differently in comparison with apoA-I. ApoA-I is one of the major protein components in HDL. Structurally, lipid-free apoA-I is believed to be a molten globule (41). The formation of HDL-like particles by apoA-I involves significant conformational changes, which are manifested by changes in the α-helical content (42), thermal (33,42,43) and chemical stability (43), proteolytic patterns (44), and antibody accessibilities (45). This conformational flexibility of apoA-I is also reflected by the two drastically different apoA-I crystal structures, showing either a fully extended pseudo-continuous α-helical belt structure or a much more compact six helices forming two helical bundles (46,47). In contrast to apoA-I, the formation of a B6.4-17/DMPC particle does not require significant changes in either the α-helical content or the chemical stability in the helical domain. A similar fragment corresponding to a largely untouched α-helical domain is observed during limited proteolysis of B6.4-17 in reconstituted DMPC complexes, which is different from the observation in apoA-I (44). Moreover, the minimal-sized B6.4-17/DMPC complex has an average dimension of 152 Å in diameter, while apoA-I makes discoidal particles as small as 60 Å in diameter (24,33). This difference may derive from the rigidity of the B6.4-17 protein, which is a result of tighter tertiary folding compared to the putative extended helix belt model of apoA-I. Indeed, the molecular surface of two B6.4-17 as shown in our model with minimal conformational adjustment already provides sufficient hydrophobic interface to cover the hydrocarbon chains in the DMPC complex. In short, a completely extended helical belt model, which has been proposed for apoA-I when the protein binds phospholipid, does not fit the observation made in B6.4-17.
The maintenance of a higher order tertiary structure during phospholipid binding may correlate with the function of this N-terminal apoB domain during lipoprotein assembly. Based on the lipovitellin structure, we speculate that the N-terminal half of the α-helical domain interacts with sequences between B17-20.5, which fold into another extended β-sheet, termed the “A-sheet” in lipovitellin (48). The phospholipid incorporation may be a cotranslational process, and the N-terminal half of the α-helical domain may assist the stabilization of a nascent initiation particle, when the proposed A-sheet is absent. When the A-sheet is folded, it may find its position on the initiation particle through the interaction with the N-terminal subdomain. Moreover, in the endoplasmic reticulum, MTP, which is approximately the size of B17, is also homologous to lipovitellin (11). MTP is proposed to be a triglyceride-shuttling protein (19). Its less efficient phospholipid transferring activity was recently suggested to be sufficient for the assembly and secretion of apoB-containing lipoproteins (21). However, no distinction between phospholipid remodeling versus phospholipid transferring activity has been made experimentally. Since MTP is homologous to B17, it may also have similar phospholipid remodeling and binding properties. It is possible that the predominantly dimeric assembly of B6.4-17 on a DMPC particle observed in vitro is replaced by a similar apoB/MTP interaction during lipoprotein assembly in vivo. Thus, MTP may play a structural role and stabilize a phospholipid-enriched initiating particle when the nascent apoB polypeptide is incompetent to fully encircle the trapped lipid moieties.
To fully elucidate the mechanism that initiates the formation of apoB-containing lipoproteins requires the establishment of in vitro systems containing apoB, MTP, and the lipid components. The heterogeneous nature of such model systems significantly complicates the application of most structural methods. With certain modifications, the direct imaging method described here provides a viable strategy to examine the structure of single lipoprotein particles at the molecular level, and should facilitate the ultimate goal of understanding the in vivo assembly of lipoproteins.
Acknowledgments
We thank Beth Y. Lin for all the STEM specimen preparation and Drs. Donald Small, David Atkinson, Esther Bullitt, Haya Herscovitz, Margaretha Carraway, and Libo Wang for their insightful discussions.
This work was supported by grants from the National Institutes of Health (No. HL-26335) and the American Heart Association (grant No. 0625943T). The Brookhaven National Laboratory STEM is a National Institutes of Health Supported Resource Center (grant No. 5 P41 EB2181) with additional support provided by the Department of Energy, Office of Biological and Environmental Research.
References
- 1.Mahley, R. W., T. L. Innerarity, S. C. Rall, Jr., and K. H. Weisgraber. 1984. Plasma lipoproteins: apolipoprotein structure and function. J. Lipid Res. 25:1277–1294. [PubMed] [Google Scholar]
- 2.Segrest, J. P., H. De Loof, J. G. Dohlman, C. G. Brouillette, and G. M. Anantharamaiah. 1990. Amphipathic helix motif: classes and properties. Proteins. 8:103–117. [DOI] [PubMed] [Google Scholar]
- 3.Segrest, J. P., M. K. Jones, V. K. Mishra, G. M. Anantharamaiah, and D. W. Garber. 1994. ApoB-100 has a pentapartite structure composed of three amphipathic α-helical domains alternating with two amphipathic β-strand domains. Detection by the computer program LOCATE. Arterioscler. Thromb. 14:1674–1685. [DOI] [PubMed] [Google Scholar]
- 4.Young, S. G. 1990. Recent progress in understanding apolipoprotein B. Circulation. 82:1574–1594. [DOI] [PubMed] [Google Scholar]
- 5.Yang, C. Y., S. H. Chen, S. H. Gianturco, W. A. Bradley, J. T. Sparrow, M. Tanimura, W. H. Li, D. A. Sparrow, H. DeLoof, M. Rosseneu, and others. 1986. Sequence, structure, receptor-binding domains and internal repeats of human apolipoprotein B-100. Nature. 323:738–742. [DOI] [PubMed] [Google Scholar]
- 6.Walsh, M. T., and D. Atkinson. 1983. Solubilization of low-density lipoprotein with sodium deoxycholate and recombination of apoprotein B with dimyristoylphosphatidylcholine. Biochemistry. 22:3170–3178. [DOI] [PubMed] [Google Scholar]
- 7.Orlova, E. V., M. B. Sherman, W. Chiu, H. Mowri, L. C. Smith, and A. M. Gotto, Jr. 1999. Three-dimensional structure of low density lipoproteins by electron cryomicroscopy. Proc. Natl. Acad. Sci. USA. 96:8420–8425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Spin, J. M., and D. Atkinson. 1995. Cryoelectron microscopy of low density lipoprotein in vitreous ice. Biophys. J. 68:2115–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Poulos, G. 2001. The three-dimensional structure of low-density lipoprotein via cryoelectron microscopy. Thesis. Boston University, School of Medicine, Boston, MA.
- 10.Lunin, V. Y., N. L. Lunina, S. Ritter, I. Frey, A. Berg, K. Diederichs, A. D. Podjarny, A. Urzhumtsev, and M. W. Baumstark. 2001. Low-resolution data analysis for low-density lipoprotein particle. Acta Crystallogr. D Biol. Crystallogr. 57:108–121. [DOI] [PubMed] [Google Scholar]
- 11.Mann, C. J., T. A. Anderson, J. Read, S. A. Chester, G. B. Harrison, S. Kochl, P. J. Ritchie, P. Bradbury, F. S. Hussain, J. Amey, B. Vanloo, M. Rosseneu, R. Infante, J. M. Hancock, D. G. Levitt, L. J. Banaszak, J. Scott, and C. C. Shoulders. 1999. The structure of vitellogenin provides a molecular model for the assembly and secretion of atherogenic lipoproteins. J. Mol. Biol. 285:391–408. [DOI] [PubMed] [Google Scholar]
- 12.Raag, R., K. Appelt, N. H. Xuong, and L. Banaszak. 1988. Structure of the lamprey yolk lipid-protein complex lipovitellin-phosvitin at 2.8 Å resolution. J. Mol. Biol. 200:553–569. [DOI] [PubMed] [Google Scholar]
- 13.Anderson, T. A., D. G. Levitt, and L. J. Banaszak. 1998. The structural basis of lipid interactions in lipovitellin, a soluble lipoprotein. Structure. 6:895–909. [DOI] [PubMed] [Google Scholar]
- 14.Segrest, J. P., M. K. Jones, and N. Dashti. 1999. N-terminal domain of apolipoprotein B has structural homology to lipovitellin and microsomal triglyceride transfer protein: a “lipid pocket” model for self-assembly of apoB-containing lipoprotein particles. J. Lipid Res. 40:1401–1416. [PubMed] [Google Scholar]
- 15.Jiang, Z. G., M. Carraway, and C. J. McKnight. 2005. Limited proteolysis and biophysical characterization of the lipovitellin homology region in apolipoprotein B. Biochemistry. 44:1163–1173. [DOI] [PubMed] [Google Scholar]
- 16.Shelness, G. S., L. Hou, A. S. Ledford, J. S. Parks, and R. B. Weinberg. 2003. Identification of the lipoprotein initiating domain of apolipoprotein B. J. Biol. Chem. 278:44702–44707. [DOI] [PubMed] [Google Scholar]
- 17.Manchekar, M., P. E. Richardson, T. M. Forte, G. Datta, J. P. Segrest, and N. Dashti. 2004. Apolipoprotein B-containing lipoprotein particle assembly: lipid capacity of the nascent lipoprotein particle. J. Biol. Chem. 279:39757–39766. [DOI] [PubMed] [Google Scholar]
- 18.Hussain, M. M., A. Bakillah, N. Nayak, and G. S. Shelness. 1998. Amino acids 430–570 in apolipoprotein B are critical for its binding to microsomal triglyceride transfer protein. J. Biol. Chem. 273:25612–25615. [DOI] [PubMed] [Google Scholar]
- 19.Atzel, A., and J. R. Wetterau. 1993. Mechanism of microsomal triglyceride transfer protein catalyzed lipid transport. Biochemistry. 32:10444–10450. [DOI] [PubMed] [Google Scholar]
- 20.Dashti, N., M. Gandhi, X. Liu, X. Lin, and J. P. Segrest. 2002. The N-terminal 1000 residues of apolipoprotein B associate with microsomal triglyceride transfer protein to create a lipid transfer pocket required for lipoprotein assembly. Biochemistry. 41:6978–6987. [DOI] [PubMed] [Google Scholar]
- 21.Rava, P., G. K. Ojakian, G. S. Shelness, and M. M. Hussain. 2006. Phospholipid transfer activity of microsomal triglyceride transfer protein is sufficient for the assembly and secretion of apoB-lipoproteins. J. Biol. Chem. 281:11019–11027. [DOI] [PubMed] [Google Scholar]
- 22.Herscovitz, H., M. Hadzopoulou-Cladaras, M. T. Walsh, C. Cladaras, V. I. Zannis, and D. M. Small. 1991. Expression, secretion, and lipid-binding characterization of the N- terminal 17% of apolipoprotein B. Proc. Natl. Acad. Sci. USA. 88:7313–7317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.DeLozier, J. A., J. S. Parks, and G. S. Shelness. 2001. Vesicle-binding properties of wild-type and cysteine mutant forms of α1 domain of apolipoprotein B. J. Lipid Res. 42:399–406. [PubMed] [Google Scholar]
- 24.Jiang, Z. G., D. Gantz, E. Bullitt, and C. J. McKnight. 2006. Defining lipid-interacting domains in the N-terminal region of apolipoprotein B. Biochemistry. 45:11799–11808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Edelhoch, H. 1967. Spectroscopic determination of tryptophan and tyrosine in proteins. Biochemistry. 6:1948–1954. [DOI] [PubMed] [Google Scholar]
- 26.Schagger, H., and G. von Jagow. 1987. Tricine-sodium dodecyl sulfate-polyacrylamide gel electrophoresis for the separation of proteins in the range from 1 to 100 kDa. Anal. Biochem. 166:368–379. [DOI] [PubMed] [Google Scholar]
- 27.Harris, J. R., and R. W. Horne. 1991. Negative staining. In Electron Microscopy in Biology. J. R. Harris, editor. IRL Press, Oxford, UK.
- 28.Wall, J. S., J. F. Hainfeld, and M. N. Simon. 1998. Scanning transmission electron microscopy of nuclear structures. Methods Cell Biol. 53:139–164. [DOI] [PubMed] [Google Scholar]
- 29.Wall, J. S., and M. N. Simon. 2001. Scanning transmission electron microscopy of DNA-protein complexes. Methods Mol. Biol. 148:589–601. [DOI] [PubMed] [Google Scholar]
- 30.Wall, J. S., and J. F. Hainfeld. 1986. Mass mapping with the scanning transmission electron microscope. Annu. Rev. Biophys. Biophys. Chem. 15:355–376. [DOI] [PubMed] [Google Scholar]
- 31.Koradi, R., M. Billeter, and K. Wuthrich. 1996. MOLMOL: a program for display and analysis of macromolecular structures. J. Mol. Graph. 14:29–55. [DOI] [PubMed] [Google Scholar]
- 32.Atkinson, D., and D. M. Small. 1986. Recombinant lipoproteins: implications for structure and assembly of native lipoproteins. Annu. Rev. Biophys. Biophys. Chem. 15:403–456. [DOI] [PubMed] [Google Scholar]
- 33.Tall, A. R., D. M. Small, R. J. Deckelbaum, and G. G. Shipley. 1977. Structure and thermodynamic properties of high density lipoprotein recombinants. J. Biol. Chem. 252:4701–4711. [PubMed] [Google Scholar]
- 34.Hainfeld, J. F., W. Liu, C. M. Halsey, P. Freimuth, and R. D. Powell. 1999. Ni-NTA-gold clusters target His-tagged proteins. J. Struct. Biol. 127:185–198. [DOI] [PubMed] [Google Scholar]
- 35.Woodcock, C. L., L. L. Frado, and J. S. Wall. 1980. Composition of native and reconstituted chromatin particles: direct mass determination by scanning transmission electron microscopy. Proc. Natl. Acad. Sci. USA. 77:4818–4822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Antwerpen, R., M. La Belle, E. Navratilova, and R. M. Krauss. 1999. Structural heterogeneity of apoB-containing serum lipoproteins visualized using cryo-electron microscopy. J. Lipid Res. 40:1827–1836. [PubMed] [Google Scholar]
- 37.Richardson, P. E., M. Manchekar, N. Dashti, M. K. Jones, A. Beigneux, S. G. Young, S. C. Harvey, and J. P. Segrest. 2005. Assembly of lipoprotein particles containing apolipoprotein-B: structural model for the nascent lipoprotein particle. Biophys. J. 88:2789–2800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wang, L., and D. M. Small. 2004. Interfacial properties of amphipathic β-strand consensus peptides of apolipoprotein B at oil/water interfaces. J. Lipid Res. 45:1704–1715. [DOI] [PubMed] [Google Scholar]
- 39.Wang, L., D. Atkinson, and D. M. Small. 2004. The interfacial properties of apoA-I and CSP—an amphipathic α-helix consensus peptide of exchangeable apolipoproteins at the triolein/water interface. J. Biol. Chem. 280:4154–4165. [DOI] [PubMed] [Google Scholar]
- 40.Wang, L., M. T. Walsh, and D. M. Small. 2006. Apolipoprotein B is conformationally flexible but anchored at a triolein/water interface: a possible model for lipoprotein surfaces. Proc. Natl. Acad. Sci. USA. 103:6871–6876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gursky, O., and D. Atkinson. 1996. Thermal unfolding of human high-density apolipoprotein A-1: implications for a lipid-free molten globular state. Proc. Natl. Acad. Sci. USA. 93:2991–2995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fang, Y., O. Gursky, and D. Atkinson. 2003. Lipid-binding studies of human apolipoprotein A-I and its terminally truncated mutants. Biochemistry. 42:13260–13268. [DOI] [PubMed] [Google Scholar]
- 43.Gorshkova, I. N., T. Liu, H. Y. Kan, A. Chroni, V. I. Zannis, and D. Atkinson. 2006. Structure and stability of apolipoprotein A-I in solution and in discoidal high-density lipoprotein probed by double charge ablation and deletion mutation. Biochemistry. 45:1242–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Roberts, L. M., M. J. Ray, T. W. Shih, E. Hayden, M. M. Reader, and C. G. Brouillette. 1997. Structural analysis of apolipoprotein A-I: limited proteolysis of methionine-reduced and -oxidized lipid-free and lipid-bound human Apo A-I. Biochemistry. 36:7615–7624. [DOI] [PubMed] [Google Scholar]
- 45.Bergeron, J., P. G. Frank, D. Scales, Q. H. Meng, G. Castro, and Y. L. Marcel. 1995. Apolipoprotein A-I conformation in reconstituted discoidal lipoproteins varying in phospholipid and cholesterol content. J. Biol. Chem. 270:27429–27438. [DOI] [PubMed] [Google Scholar]
- 46.Rogers, D. P., C. G. Brouillette, J. A. Engler, S. W. Tendian, L. Roberts, V. K. Mishra, G. M. Anantharamaiah, S. Lund-Katz, M. C. Phillips, and M. J. Ray. 1997. Truncation of the amino terminus of human apolipoprotein A-I substantially alters only the lipid-free conformation. Biochemistry. 36:288–300. [DOI] [PubMed] [Google Scholar]
- 47.Ajees, A. A., G. M. Anantharamaiah, V. K. Mishra, M. M. Hussain, and H. M. Murthy. 2006. Crystal structure of human apolipoprotein A-I: insights into its protective effect against cardiovascular diseases. Proc. Natl. Acad. Sci. USA. 103:2126–2131. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 48.Thompson, J. R., and L. J. Banaszak. 2002. Lipid-protein interactions in lipovitellin. Biochemistry. 41:9398–9409. [DOI] [PubMed] [Google Scholar]