

Abstract
This study presents an investigation of pacemaker mechanisms underlying lymphatic vasomotion. We tested the hypothesis that active inositol 1,4,5-trisphosphate receptor (IP3R)-operated Ca2+ stores interact as coupled oscillators to produce near-synchronous Ca2+ release events and associated pacemaker potentials, this driving action potentials and constrictions of lymphatic smooth muscle. Application of endothelin 1 (ET-1), an agonist known to enhance synthesis of IP3, to quiescent lymphatic smooth muscle syncytia first enhanced spontaneous Ca2+ transients and/or intracellular Ca2+ waves. Larger near-synchronous Ca2+ transients then occurred leading to global synchronous Ca2+ transients associated with action potentials and resultant vasomotion. In contrast, blockade of L-type Ca2+ channels with nifedipine prevented ET-1 from inducing near-synchronous Ca2+ transients and resultant action potentials, leaving only asynchronous Ca2+ transients and local Ca2+ waves. These data were well simulated by a model of lymphatic smooth muscle with: 1), oscillatory Ca2+ release from IP3R-operated Ca2+ stores, which causes depolarization; 2), L-type Ca2+ channels; and 3), gap junctions between cells. Stimulation of the stores caused global pacemaker activity through coupled oscillator-based entrainment of the stores. Membrane potential changes and positive feedback by L-type Ca2+ channels to produce more store activity were fundamental to this process providing long-range electrochemical coupling between the Ca2+ store oscillators. We conclude that lymphatic pacemaking is mediated by coupled oscillator-based interactions between active Ca2+ stores. These are weakly coupled by inter- and intracellular diffusion of store activators and strongly coupled by membrane potential. Ca2+ store-based pacemaking is predicted for cellular systems where: 1), oscillatory Ca2+ release induces depolarization; 2), membrane depolarization provides positive feedback to induce further store Ca2+ release; and 3), cells are interconnected. These conditions are met in a surprisingly large number of cellular systems including gastrointestinal, lymphatic, urethral, and vascular tissues, and in heart pacemaker cells.
INTRODUCTION
Lymphatic and various blood vessels display vasomotion, a rhythmic constriction-dilation cycle of smooth muscle in the vessel walls (1,2). In blood vessels vasomotion modulates local vascular resistance and blood flow (see Haddock et al. (3)) and possibly improves tissue oxygenation (see Nilsson and Aalkjaer (4)). In contrast, vasomotion in lymphatic vessels propels lymph fluid through frequently occurring unidirectional valves that divide the collecting lymphatic vessels into multiple chambers (2,5,6). The aims of this study were to investigate the pacemaker mechanisms that underlie lymphatic vasomotion.
Two models have been proposed to explain lymphatic pacemaking: a classical cardiac-like and a Ca2+ store-controlled pacemaker model. Both depend on Ca2+ action potentials initiating constriction of the smooth muscle (7–10). The classical model for heart pacemaking is based on membrane pacemaker currents including a hyperpolarization-activated inward current, known as Ifunny (If) that depolarizes the pacemaker cell membrane at the termination of an action potential and continues the cycle (11). Thus in this model the pacemaker clock is set by activation of ion channels in the cell membrane. Significantly, an If-like current has been reported in bovine and sheep lymphatic vessels (12,13), blocking of which has been shown to slow down lymphatic pacemaking (13).
In the second model pacemaking of lymphatic smooth muscle is controlled by cyclical release-refill of intracellular IP3R-operated Ca2+ stores (10,14–18). Ca2+ released from intracellular Ca2+ stores activates spontaneous transient inward currents (STICs) (19,20), these generating excitatory depolarizations termed spontaneous transient depolarizations (STDs). Vasomotion in guinea pig mesenteric lymphatics involves summation of STDs, resulting in pacemaker potentials that trigger the action potential and smooth muscle constriction (10,14). Thus in this store pacemaker model, the pacemaker clock is intracellular, set by the release-refill cycle of Ca2+ stores. Importantly, this mechanism may play a role in a much wider range of tissues. For example, recent evidence obtained from studies on isolated heart pacemaker cells opens up the possibility of a major role for store pacemaking in the heart (see Lakatta et al. (21)).
The focus of this study is to determine how Ca2+ stores achieve synchrony to produce sufficient current to drive pacemaking in both small and large lymphatic vessels. Conducting Ca2+ waves (22,23), events that result through Ca2+-induced Ca2+ release (CICR) along arrays of stores (24,25), are unlikely to explain this synchrony. This is because conducting Ca2+ waves propagate slowly (typically <0.1 mm/s; see Berridge (26)) and are unlikely to activate sufficient stores at any one time to drive pacemaking in lymphatic tissues (see van Helden et al. (14)). In contrast, a pacemaker mechanism, based on Ca2+ stores interacting as coupled oscillators provides a powerful alternative, one that could readily underlie lymphatic vasomotion (15,27).
Coupled oscillator-based interactions have been proposed to underlie diverse biological systems (28), such as heart pacemaking (29,30), intestinal peristalsis (31–33), and signaling in astrocytes (34). Importantly, studies on bovine lymphatic vessels indicate that lymphatic vasomotion also arises through coupled oscillator-based interactions, as central disruption of constrictions by a putative gap junction blocker left vasomotion in the two vessel ends but the rhythmic constrictions were no longer in phase (35). The authors suggested that electrical coupling along the length of the lymphatic duct allowed these sections to entrain their constrictions to a mutual compromise frequency.
To understand coupled oscillator interactions between Ca2+ stores it is useful to consider a row of pendulums interconnected by springs. Random activation of the pendulums causes the pendulums to interact, each advancing or retarding the phase of others, until there is global entrainment. Such entrainment can lead to local synchrony of the pendulums. However, because the coupling between pendulums is imperfect, phase delays establish which can lead to phase waves along the row of pendulums, each pendulum swinging at the same frequency, but starting at a slightly different phase to its neighbor (see van Helden and Imtiaz (36)).
In the case of stores, the pendulums are used to depict the store oscillators, though strictly speaking the stores are relaxation oscillators (i.e., rapidly releasing their contents and slowly refilling the cycle then repeating), and the springs represent the coupling mechanisms between the oscillators. Stores exhibit two primary coupling mechanisms, chemical and electrochemical. An example of the former is diffusion of Ca2+, which couples the stores by CICR. However, such coupling can only result in local synchrony because of limited effective diffusion of Ca2+ (e.g., ∼5 μm; (37)), with this type of coupling being too weak to successfully synchronize the Ca2+ store oscillations over large cellular syncytia (27). In contrast, electrochemical coupling has a 100–1000 times greater range than that for Ca2+ diffusion-based coupling. This is because electrical signals act in the millimeter range in smooth muscle syncytia (e.g., the length constant of vascular smooth muscle is ∼2mm; (38)). Therefore electrochemical coupling can act as a long-range “spring” interconnecting multiple oscillators. Such interactions have been hypothesized to underlie lymphatic vasomotion (15), vasomotion in mesenteric arteries (39–42), and slow wave pacemaking in a gastrointestinal smooth muscle (27), where electrochemical coupling is mediated by interaction between membrane voltage and Ca2+ stores linked by either voltage-dependent production of IP3 and/or Ca2+ entry.
Three elements are essential for effective long-range coupling between stores: 1), oscillatory Ca2+ release-induced depolarization; 2), membrane depolarization providing positive feedback to induce further store Ca2+ release; and 3), connectivity between cells (e.g., gap junctions). Lymphatic smooth muscle exhibits all these elements, because it has oscillatory Ca2+ release that causes depolarization, L-type Ca2+ channels that cause store Ca2+ release, and gap junctions between cells, and hence a coupled oscillator-based store pacemaker mechanism is predicted as we confirm here. Some of the results of this study have been presented previously in abstract form (43).
METHODS
Experimental methods
Tissue preparation
Young guinea pigs (<10 days) of either sex were killed by overexposure to the inhalation anesthetic isoflurane (5–10% in air) followed by decapitation, a protocol approved by the University of Newcastle Animal Care and Ethics Committee. The small intestine and attached mesentery were rapidly removed and placed in a physiological saline solution of the following composition (in mM): CaCl2, 2.5; KCl, 5; MgCl2, 2; NaCl, 120; NaHCO3, 25; NaHPO4, 1; and glucose 10. The pH was maintained near 7.4 by constant bubbling with a 95% O2 and 5% CO2 gas mixture. Lymphatic vessels of diameter typically <300 μm were selected and isolated in situ in the mesothelium together with any nearby arteries and/or veins. The mesothelium was either pinned flat onto the sylgard-coated (Dow Corning, Midland, MI) base of a small organ bath (volume 0.2–1 ml) or held flat against the coverslip base of a bath (volume 0.5–1 ml) using a fine stainless steel frame. Alternatively, single lymphatic chambers were mounted on a wire myograph, which was used to record constrictions and hold the vessel chamber flat against the coverslip. Care was taken to minimally stretch the lymphatic vessels in all cases. Preparations were superfused with the physiological saline heated to 34–36°C at a rate of 6 ml/min. Vessel chambers formed by adjacent valves (i.e., lymphangions; (5)) were isolated by cutting just inside the valves (length 300–500 μm and diameter 200–350 μm) for electrophysiological recording. Vessels were not internally perfused during recording. Tissues were normally used within 1–4 h of isolation and were stored at 4°C in physiological saline until use.
Recording techniques
Intracellular microelectrode recording was performed by impaling the smooth muscle of vessel segments. Microelectrodes were filled with 1 mM KCl and typically had resistances of 100–150 MΩ. Electrical data were recorded and stored digitally with analysis performed using Axograph (Axon Instruments, Foster City, CA).
Ca2+ imaging experiments were made using a Bio-Rad 1000 confocal laser system (Cambridge, MA) connected to an inverted microscope (Nikon TE200, Tokyo, Japan). Tissues were viewed directly through the coverslip that formed the base of the bath with a water immersion objective (magnification ×60, numerical aperture 1.2) with the confocal aperture set to provide a depth resolution of ∼2 μm. Tissues were loaded by two procedures. The first was to luminally perfuse vessels for 30 min at ∼35°C with a physiological saline in which the CaCl2 concentration was reduced to 1 mM and that contained 2 μM Oregon green/AM (Molecular Probes, Eugene, OR). This was followed by a 5-min perfusion using the normal low-calcium (1 mM) saline to wash out extra dye in the vessel lumen. Loading of the smooth muscle by this procedure was only successful in endothelium-denuded vessels, a condition that was achieved by briefly passing bubbles of air through the vessel lumen (see Gao et al. (44)). Vasomotion in these vessels occurs independent of the endothelium with endothelial factors only subserving a modulatory role (45). The second method was to externally load the smooth muscle by adding 2 μM Oregon green/AM to the normal physiological saline for 30 min at 35°C. This was used for loading lymphatic smooth muscle in tissues where the mesothelium had been largely removed. The Oregon green-loaded lymphatic smooth muscle was excited with light of wavelength 488 nm using an argon ion laser. Emission fluorescence was collected through a 510-nm dichroic mirror and 515-nm bandpass filter. Ca2+ transients were deemed measurable and appropriate for analysis if the associated transient increase in fluorescence of the Oregon green was at least twice the baseline noise.
Internal perfusion
Some experiments were made using internal perfusion to activate lymphatic chambers. In these cases a glass cannula (tip diameter ∼50 μm was loosely inserted into the upstream end of a lymphatic vessel with the perfusion rate gradually increased until the lymphatic chambers in the vessel became active (final flow rate ∼2.5 μl/min). The perfusing solution used was a modified physiological saline differing only in that the CaCl2 was decreased to 0.3 mM. Constrictions in the vessels were then recorded videoscopically viewed through an inverted microscope with a ×4 objective. Constriction frequencies and propagation velocities were determined using an edge detection algorithm (46), with up to 10 vessel edges tracked simultaneously. Some experiments were made on large single chambers isolated from vessels. These were isolated by cutting the adjacent chambers near the valves at both ends of the chamber leaving enough remnant upstream chamber for insertion of the cannula, which was positioned into the upstream valve.
Statistics
All values are presented as the mean ± SE with n the number of samples used, where n samples are derived from a minimum of five different animals contributing at least one sample each. Any variation is specifically stated. Significance was determined using a two-tailed Student's t-test, with p < 0.05 considered significant.
Chemicals
The chemicals used were endothelin 1 (ET-1), nifedipine, wortmannin, and U46619 (9,11-dideoxy-9α,11α-methanoepoxy prostaglandin F2α) from Sigma (St. Louis, MO) and Oregon green/acetoxymethyl (AM) from Molecular Probes. Stock solutions were made up in either distilled water or dimethylsulphoxide (DMSO) at concentrations of 10−4–10−1 M and stored at −20°C. The concentration of DMSO was always <0.1%, which had no significant effect on lymphatic pumping.
Modeling: single lymphatic smooth muscle cell
Experimental background
We base our model of the lymphatic smooth muscle on previously published findings (10,15,18,35,47) and data presented in this study. Schematic in Fig. 1 and Table 1 summarize the essential events and pathways underlying Ca2+ and membrane potential dynamics of a single lymphatic smooth muscle cell. Cyclical release and refill of Ca2+ stores through activation of IP3 receptors (events 2–6) occur due to cytosolic IP3, elevated in response to application of agonists such as ET-1 (event 1). Cyclical oscillations in cytosolic Ca2+ are transformed into membrane potential oscillations through activation of an inward Cl− current (events A, B). When depolarization is superthreshold, it triggers an L-type Ca2+ channel-mediated action potential and resultant Ca2+ entry (events C, D). This influx of Ca2+ further reinforces store Ca2+ cycling (event E). Current flow to adjacent cells through gap junctions occur (event F) and provide a coupling mechanism between cycling stores across cells.
FIGURE 1.

Schematic summary of dynamics in a single lymphatic cell. This schematic shows pathways and feedback loops underlying Ca2+ and electrical oscillations in a single lymphatic cell. See text and Table 1 for further details.
TABLE 1.
Summary of experimental data underlying model construction
| Event sequence | Label on schematic | Reference | Notes | |
|---|---|---|---|---|
| Ca2+ dynamics | ET-1 causes synthesis of IP3 | 1 | (18) | Events 1–6 repeat in cyclical manner, constituting a store-cytosolic oscillator (29,14,30,50) |
| IP3 causes release of Ca2+ from Ca2+ stores through IP3Rs | 2, 3 | (10,14–19) | ||
| Ca2+ release from a store can induce further release of Ca2+ through IP3Rs (CICR) | 4, 5 | (24,25) | ||
| Ca2+ is pumped into the ER through a Ca2+-ATPase | 6 | (10,14–19) | ||
| Increased cytosolic Ca2+ can release SR Ca2+ through activation of RyRs | 4, 7 | (18) | Release from RyR does not play a significant role in guinea pig lymphatic pacemaking (see Zhao and van Helden (18)) | |
| Electrical dynamics | Ca2+ released from stores cause an inward Cl− current, depolarizing the membrane (+ΔV) | A, B | (14,20–24) | This transforms store Ca2+ oscillations into membrane potential oscillations |
| A superthreshold +ΔV causes an action potential primarily by activating L-type voltage operated Ca2+ channels on the plasma membrane causing influx of Ca2+ | C, D | (10,14,18) | Events C, D, and E provide feedback from plasmalemma oscillator to store Ca2+ oscillator | |
| Ca2+ flux through L-type Ca2+ channels causes increase in [Ca2+]c inducing further store refill/release | E | (18), and results in this article | ||
| Inward (i.e., depolarizing) currents at any location invade other cells through gap junctions and cause depolarization | F | (15,27), and results in this article | This mechanism provides coupling between store Ca2+ oscillations across cells |
Other experimental findings that relate to our simulations include the following. i), Application of ryanodine (1 and 20 μM) does not significantly alter lymphatic vasomotion (18). Thus we have not included ryanodine receptors in this model of cytosolic-store excitability. ii), Data presented in this and previous studies (18) have shown that influx of Ca2+ through voltage-gated channels is not a prerequisite for oscillatory Ca2+ release from stores. iii), Previous studies have shown that guinea pig mesenteric lymphatic vessels cut to short lengths or chambers of length <∼500 μm have a smooth muscle syncytium that is electrically short (10). This is because the smooth muscle layer exhibits a length constant of ∼1 mm (48). As such, potentials recorded from any smooth muscle cell reasonably reflect activity at any site in the vessel segment. iv), The endothelium modulates vasomotion but does not have a role in lymphatic pacemaking per se (18); therefore we only include a smooth muscle layer in our model. v), Simulations of the smooth muscle layer do not exclude the possibility that mixed in this layer and integrally coupled to the smooth muscle is a population of cells paralleling Interstitial Cells of Cajal (ICCs). ICCs are known to have a fundamental pacemaker role in gastric and other smooth muscles. However, despite some effort we do not have conclusive evidence for or against the presence of such cells in guinea pig lymphatic smooth muscle, because they have yet to be revealed by antibodies known to label gastric and some other ICCs (e.g., antibodies against cKit). Importantly, our model is based on Ca2+ stores operating in a network of cells that are electrically coupled, and hence the model should be applicable whether or not lymphatic vasomotion is paced by ICCs.
In summary, there are two main components of lymphatic smooth muscle excitability: a), cytosolic-store Ca2+ excitability, and b), an excitable effect of voltage-dependent L-type Ca2+ channels. We now formulate the model using these two components with reference to experimental data.
Ca2+ excitability
A previous experimental study (18) has shown that ET-1 induced vasomotion occurs through Ca2+ release from intracellular Ca2+ stores through IP3 receptors. ET-1 activates ETA receptors, which stimulates phospholipase C production by a Pertussis toxin-insensitive G-protein leading to IP3 production (18). Sustained levels of IP3 result in cyclical release-refill of intracellular IP3R-operated Ca2+ stores (26) (also see (14,27,49)). Ca2+ stores are refilled by a thapsigargin-sensitive Ca2+-ATPase pump after each release (18). The excitability of this cytosolic-store Ca2+ system, as controlled by the intracellular levels of IP3, is represented by the following set of equations:
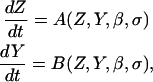 |
(1) |
where the functions A and B describe the dynamics of the cytosolic-store Ca2+ system. Z and Y are the Ca2+ concentration in the cytosol ([Ca2+]c) and in the lumen of the IP3-sensitive intracellular Ca2+ store ([Ca2+]s), respectively. The IP3 concentration in the cytosol ([IP3]c) is denoted by the variable β. The parameter σ defines the sensitivity of the cell to IP3. This parameter is used as a cell population heterogeneity parameter (see below).
The single pool model of Dupont and Goldbeter (50) adequately encapsulates the above given cytosolic-store Ca2+ excitability. Thus we use their model (50) to describe the functions A(Z,Y,β,σ) and B(Z,Y,β,σ). Further details of the cytosolic-store Ca2+ excitability system are given in Appendix I and Table 2.
TABLE 2.
Parameters related to Ca2+ dynamics given by Eqs. A1–A5
| Parameter | Description | Value |
|---|---|---|
| V0 | Constant Ca2+ influx from extracellular space into cytosol | 3.4 μM/min |
| V1 | IP3 dependent Ca2+ influx from extracellular space into cytosol | 3.4 μM/min |
| kf | Rate constant for Ca2+ leak from store to cytosol | 1 min−1 |
| K | Rate constant for Ca2+ extrusion from the cytosol | 10 min−1 |
| VM2 | Maximal value for Ca2+ pump into the store | 50 μM/min |
| n | Hill coefficient | 2 |
| K2 | Cytosolic Ca2+ dependent threshold constant for V2 | 1 μM |
| VM3 | Maximal value for Ca2+ release from the store | 650 μM/min |
| w | Hill coefficient | 4 |
| ka | Cytosolic Ca2+ dependent threshold constant for V3 | 0.9 μM |
| kr | Store Ca2+ dependent threshold constant for V3 | 2 μM |
| u | Hill coefficient | 2 |
| σ | IP3 dependent threshold constant for V3 | Population mean 0.48 μM |
| o | Hill coefficient | 4 |
Ca2+-induced membrane oscillations
Ca2+ increases in the cytosol result in depolarization. This transformation of Ca2+ release into membrane depolarization occurs through a Ca2+-activated inward current, most likely carried by Cl− ions (14,51). This Ca2+-induced inward current underlies cytosolic Ca2+-voltage coupling. The Ca2+-induced conductance, Gca, for this channel is defined by the following equation:
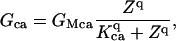 |
(2) |
where GMca is the maximum value for this conductance and Kca is the [Ca2+]c at half-maximal value of this conductance.
Current and voltage dynamics, including the Ca2+-induced current, is described by:
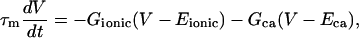 |
(3) |
where τm is the time constant of the membrane. A “leakage” current passing through passive ionic channels is represented by the first right-hand side term. The second term gives the Ca2+-induced current. Voltage-gated channels have not yet been introduced. Thus, at this stage of the model formulation all conductances in Eq. 3, except GCa, are taken as constant. A summary of parameters for the system defined by Eqs. 2 and 3 is given in Table 3.
TABLE 3.
| Parameter | Description | Value |
|---|---|---|
| τm | Membrane time constant | 0.0017 min |
| Gionic | Lumped conductance of passive ionic channels | 1.12 mS |
| Eionic | Lumped reversal/null potential of passive ionic channels | −67.2 mV |
| Eca | Reversal/null potential for the Ca2+ modulated current(s) | −20 mV |
| GMca | Maximal value of Gca | 4 mS |
| kca | Half-saturation constant for GCa | 1.4 μM |
| q | Hill coefficient | 4 |
Voltage-dependent Ca2+ channels
Previous studies (18) and experimental observations presented here show that the lymphatic smooth muscle contain L-type Ca2+ channels. This channel is voltage dependent and activates at voltage thresholds approximately +15 mV above resting membrane potential. The current through the L-type Ca2+ channels is given as,
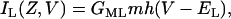 |
(4) |
where IL(Z,V), the current through the L-type Ca2+ channels is dependent on Z the [Ca2+]c, and V the membrane potential. The maximum conductance through the L-type Ca2+ channels is given by GML and EL is the reversal potential of this current. The current IL(Z,V) activates through a voltage threshold controlled by the gating variable m and is deactivated by an intracellular Ca2+ concentration threshold controlled by the gating variable h.
The activation of IL(Z,V) is very fast and here is assumed to be instantaneous. Thus the activation gating variable m is given as,
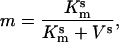 |
(5) |
where Km is the half-saturation value of the gate with a Hill coefficient s.
The deactivation of IL(Z,V) is dependent on the concentration of Ca2+ in the cytosol and is controlled by the gating variable h as follows,
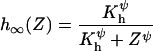 |
(6) |
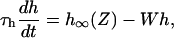 |
(7) |
where h∞(Z) is the steady-state response of the deactivation gate h with half-saturation value Kh and Hill coefficient ψ. Equation 7 gives the dynamics of the gate h with a time constant τh and degradation variable W.
The current IL(Z,V) is due to influx of Ca2+ through the L-type Ca2+ channels, therefore it contributes to the intracellular concentration of Ca2+ in the cytosol. The change in Ca2+ concentration in the cytosol due to IL(Z,V) is given as,
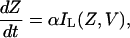 |
(8) |
where α is a scaling variable that defines the change in concentration of Ca2+ ions caused by the current IL(Z,V). Table 4 summarizes the parameter values used in Eqs. 4–8.
TABLE 4.
| Parameter | Description | Value |
|---|---|---|
| GML | Lumped conductance of L-type Ca2+ channels | 5 mS |
| EL | Reversal/null potential of L-type Ca2+ channels | 20 mV |
| Km | Half saturation constant for activation gate m | −42 mV |
| S | Hill coefficient | 20 |
| Kh | Half saturation constant for inactivation gate h | 0.7 μM/min |
| Ψ | Hill coefficient | 20 |
| τh | Time constant for inactivation gate h | 0.02 min |
| W | Degradation rate constant for inactivation gate h | 1 |
| α | Current to Ca2+ scaling factor | −1.33 |
Modeling: lymphatic smooth muscle syncytium
The dynamics of a single lymphatic smooth muscle isopotential unit j (with adjacent units i and k) that includes the excitable cytosolic-store Ca2+ system and excitability of the membrane due to L-type Ca2+ channels, is given as follows.
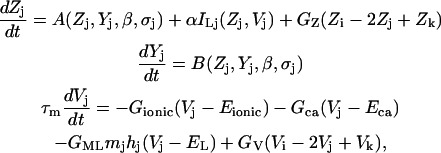 |
(9) |
where GZ and GV are the Ca2+ and current coupling coefficients, respectively. Zj and Vj are the cytosolic Ca2+ and voltage in unit j. In this model [IP3]c is considered to be dependent on the external stimulus β (e.g., level of ET-1) and is considered uniform across the whole cell population. Thus, there is no flux of IP3 through the gap junctions or within cells.
A typical vascular smooth muscle cell is elongated, fusiform with tapering ends and has average diameter and length of 3–6 μm and 150–200 μm, respectively (52,53). Here we assume each smooth muscle cell to be 100 μm, and model it with a one-dimensional array of 10 isopotential units described by Eq. 9.
The smooth muscle in each lymphatic chamber is a tissue syncytium, with cells interconnected by gap junctions. The smooth muscle syncytium is now modeled by making a two-dimensional network of gap junction connected cells. Gap junctions were placed with coupling in the transverse direction 10 times lower in accordance to observed experimental data (27). The electrical length constant of lymphatic smooth muscle has been reported to be between 0.8–2 mm (54). Various simulations were run with longitudinal electrical length constant between 0.8 and 2 mm and Ca2+ diffusion of 1.2 μm2/min, with coupling of units within cells 300 times that of gap junctions. “No flux” boundary conditions were used for all simulations.
Large lymphatic tissue (2 mm) comprised a one-dimensional array of 200 coupled units. Each unit representing 100 μm isopotential tissue length was composed of a Ca2+ store and plasmalemma oscillator.
Cell heterogeneity
The lymphatic smooth muscle syncytium is composed of a collection of cells. The inference from experimental data shows that the cells in the population are heterogeneous in their response to IP3. This heterogeneity could arise from many factors, such as heterogeneous expression of IP3 receptors on the Ca2+ stores, heterogeneous populations of Ca2+ stores within cells, cell size, or even cell type. Furthermore, variation in IP3 concentration in different regions of the tissue would also result in heterogeneous response of the cells.
Although the heterogeneity of the cell population could arise from various sources, the outcome relevant to our study of synchronization is the effect of these factors on the excitability and oscillatory frequency of the cells in response to agonist. Thus the heterogeneity of the cell arising out of various parameters can be lumped in our study by considering them to arise from a single source, i.e., the response to IP3. The response of the cell to IP3 is given by the parameter σ in Eq. 9 describing the calcium excitability of the cell. This parameter defines the sensitivity of a cell to IP3 and controls the response of the cell to the level of agonist available to the syncytium. Although the stimulus level β is same for the syncytium, each cell responds according to its sensitivity to IP3.
Parameter values
The model presented here is qualitative, i.e., it encapsulates the essential features of the experimental data and interrelationships of observed variables. Membrane potential records were used as a basic comparison variable for the experimental and simulated data. All other variables and parameters were adjusted using this criterion. We note that simulated time was scaled to match duty cycle of the experimental action potentials.
Numerical simulation and analysis were performed using MATLAB/SIMULINK (Mathworks, Natick, MA) on an IBM-compatible desktop computer.
RESULTS
Membrane potential recordings were made from isolated lymphatic chambers (length <500 μm, diameter <300 μm). The preparations were generally quiescent under unstimulated control conditions with relatively few chambers spontaneously active (∼10% from over 100). Intracellular microelectrode recordings revealed a resting membrane potential of −55 ± 2 mV (mean ± SE, n = 100). The isolated vessel segments were electrically short allowing measurement of activity generated in any region of the smooth muscle (10).
Spontaneous pacemaker events
Intracellular voltage recordings and/or confocal Ca2+ imaging were made in the smooth muscle of spontaneously active chambers (Fig. 2). Shown are the regions of interest (ROIs) for two types of analysis (Fig. 2, A and B). This tissue exhibited action potentials, associated increases in [Ca2+]c and constrictions (Fig. 2, C–I). The recordings presented here (representative of nine experiments from six animals) were made on application of 5 μM wortmannin before it had become fully effective (Fig. 2, C–E) and during treatment after it fully stopped constrictions (Fig. 2, F–I). This agent inhibits myosin light chain kinase, thus preventing tissue constrictions and dislodgement of the electrode. Importantly, it did not inhibit pacemaking, consistent with studies on another phasic smooth muscle (55) and indicating that other known actions of wortmannin such as inhibition of PIP3 kinase (56) do not significantly interfere with lymphatic pacemaking. Action potentials and associated large synchronous global increases in [Ca2+]c (Fig. 2, C and D) exhibited constrictions, as indicated by the downward deflections of the ROI placed on the edge of the tissue (Fig. 2 C, ROI 1, green trace). Action potentials were >15 mV having a mean amplitude of 33 ± 3 mV and half-duration (duration at 50% of peak) of 1.2 ± 0.2 s, with associated Ca2+ transients having a half-duration of 1.5 ± 0.05 s (n = 9 tissues). Subthreshold pacemaker events were also observed consisting of depolarizations with associated near-synchronous increases in [Ca2+]c. However, unlike the action potentials they did not cause constrictions. Pacemaker potentials had a mean amplitude of 6.6 ± 0.3 mV and a half-duration of 2.8 ± 0.9 s (n = 10 events), with [Ca2+]c having a half-duration 106.8 ± 1.0% and amplitude 17.5 ± 0.5% of the action potential-associated [Ca2+]c transients (n = 6 events).
FIGURE 2.

Spontaneous pacemaker events. Membrane potential and associated [Ca2+]c transients recorded during application (C–E) and in 5 μM wortmannin (F–I). (A and B) Section of a lymphatic chamber showing Oregon green loaded smooth muscle with two types of regions of interest used for data analysis. (C) Membrane potential recordings (blue upper trace) and simultaneous record of [Ca2+]c for the nine ROIs marked in panel A, along with mean [Ca2+]c for the nine ROIs. ROI No. 1 was placed on the edge of the tissue so as to monitor tissue movements (green trace). Deflection of this trace below the dotted line shows tissue constriction, whereas upward movement above the dotted line is generated by actual increase in [Ca2+]c (verified by frame-by-frame analysis). Vertical dotted line shows that during a pacemaker event different regions of the tissue were synchronized without any associated movement. Inset in panel C shows expanded [Ca2+]c records for ROIs 2–9 with superimposed membrane potential recording to highlight correlation between these two types of recordings. The inset also indicates that another pacemaker event has occurred after PP1. (D) Space-time plot showing temporal changes in relative intensity for all the ROIs shown in panel B. This plot was produced by plotting [Ca2+]c transients within each ROI vertically down in the sequence shown in panel B as a function of time (i.e., ROIs 1–10 for row a, then ROIs 1–10 for row b, etc., plotted on the ordinate against time on the abscissa). (E) Mean difference image showing active regions during pacemaker event PP1. This image was produced by averaging five sequential images during peak of PP1, then subtracting average of five sequential images during baseline (taken at upward arrow in C). (F–H) Same as panels C–E but in the presence of 5 μM wortmannin, ROI 1 trace (green) shows that now there is no movement of the tissue. (I) Mean difference image showing active regions during an action potential (AP1 in F), produced in a similar manner to panels E and H. Color scale bar in B applies to A, representing 8-bit (0–255) grayscale nonratiometric fluorescence intensity. Scale bars in F apply to corresponding traces in C. Timescale bar in G applies to C, D, and F. Normalized intensity color scale bar in G applies to D. Intensity color bar in I applies to E and H.
An inspection of the [Ca2+]c transients showed that a pacemaker event (PP1) occurred synchronously throughout the tissue, Fig. 2, C and D. This synchronous increase in [Ca2+]c during the pacemaker event was not a result of tissue movement because ROI 1 (green trace) showed no tissue movement. This was further corroborated by a mean difference image, Fig. 2 E, which showed that widely dispersed regions within the field of view participated in the pacemaker event. Fig. 1, F–I, shows similar data but now after wortmannin fully prevented tissue constrictions. Once again the pacemaker event PP2 was found to be synchronous throughout the imaged region. These results show that synchronous Ca2+ events can occur in the absence of action potentials.
Agonist-induced emergence of pacemaking
Experiments recording changes in membrane potential and/or [Ca2+]c were also made on quiescent chambers, now observing the effects of agonists such as ET-1, which are known to increase synthesis of IP3. Application of ET-1 (1–3 nM) first caused an enhancement in the amplitude of measurable STDs (i.e., >0.5 mV), these increasing by 156 ± 14% of control (p = .0007; n = 10), as measured for 30-s periods after ET-1 application. There was also a tendency for the frequency of these STDs to increase (166 ± 32% of control), though this was not significant (p = 0.052). This initial enhancement in activity usually led to generation of action potentials and associated constrictions (Fig. 3 A). Action potentials persisted long after removal of the ET-1. For example, 1–2 min application of 1–3 nM ET-1 induced vasomotion that gradually subsided over the next 5–30 min after return to control solution (n = 9; data not shown). The above sequence of events was also observed for application of the thromboxane A2 mimetic U46619 (n = 5, data not shown), an agonist that is also known to enhance lymphatic vasomotion most likely through synthesis of IP3 (57,58).
FIGURE 3.

Effect of ET-1. (A) Membrane potential recordings (upper traces) and simultaneous vessel edge recordings (lower traces) upon application of 3 nM ET-1. Numbered arrows correspond to regions shown on an expanded timescale. (B) [Ca2+]c transients (upper record) averaged over all the imaged areas shown in C and associated smooth muscle constrictions (lower record) recorded from a lymphatic chamber mounted on a wire myograph. Labeled regions show record sections analyzed below in corresponding panels. (C) Section of a lymphatic chamber showing Oregon green loaded smooth muscle with regions of interest used for data analysis. (D) Records of changes in [Ca2+]c upon and during maintained application of 3 nM ET-1 (panel B, arrow D). Application of ET-1 caused the emergence of pacemaker events as first evidenced by the Ca2+ transients becoming near-synchronous in areas 1–3 (*). The following cycle showed larger near-synchronous Ca2+ transients across all sites except area 6. (E) The synchrony and magnitude of the activity improved over time resulting in large, rhythmically occurring Ca2+ transients. Force scale represents relative force.
The data presented in Fig. 3, B–E, shows measurements of relative [Ca2+]c (representative of 15 experiments from 11 animals), and in this case simultaneous constriction measurements. Fig. 3 B presents the summed fluorescence of the Ca2+ sensing fluorophore for the entire imaged area of the lymphatic smooth muscle and the corresponding record of constriction. Analysis of intensity changes used the regions of interest depicted in Fig. 3 C. ET-1 caused the emergence of vasomotion (Fig. 3 B). Specifically, application of 3 nM ET-1 caused the asynchronous Ca2+ transients present under control conditions to synchronize with synchrony first beginning in more localized regions (e.g., see *, Fig. 3 D). Such emergence of synchronicity appeared as near-synchronous events distributed in local regions over multiple cells. However, even after the onset of more global synchrony some regions did not show significant increases in [Ca2+]c (e.g., Fig. 3 D, ROI 6). Over time all areas showed near-synchronous increases in [Ca2+]c (Fig. 3 E), the Ca2+ transients and constrictions now being larger and occurring at a higher frequency. Local Ca2+ waves conducting with low velocities (39 ± 9 μm/s; n = 5) were observed within but not between smooth muscle cells during quiescent periods between tissue constrictions.
Inhibition of pacemaking by blockade of L-type Ca2+ channels
Nifedipine blocks L-type Ca2+ channels and hence constriction. However, in addition to this it was found that nifedipine also inhibited synchronization of Ca2+ store release events. This was investigated by firstly measuring membrane potential. Nifedipine (1 μM) was applied to lymphatic chambers that were either spontaneously active, or in which activity was induced by prior application of 1–3 nM ET-1. Nifedipine first blocked action potentials revealing the underlying pacemaker potentials, with these then diminishing to leave apparently unsynchronized activity (Fig. 4 A). These findings were observed in all tissues tested (representative of nine experiments from nine animals).
FIGURE 4.

Effect of nifedipine. (A) Membrane potential recordings (upper traces) and simultaneous vessel edge recordings (lower traces) upon application of 1 μM nifedipine. L-NAME (30 μM), which like other NO synthase inhibitors (45) does not itself inhibit vasomotion, was included to inhibit NO release, because nifedipine has been reported to release NO from the endothelium (79). This figure is continued from Fig. 3 A where vasomotion had been initiated using 3 nM ET-1. Numbered arrows correspond to regions shown on an expanded timescale. (B) Membrane potential recordings (blue upper trace) and simultaneous record of [Ca2+]c for the seven ROIs marked in C, along with mean [Ca2+]c for the seven ROIs. ROI No. 1 was placed on the edge of the tissue so as to monitor tissue movements (green trace). Deflection of this trace below the dotted line shows tissue constriction, whereas upward movement above the dotted line is generated by actual increase in [Ca2+]c (verified by frame-by-frame analysis). (D and E) Superimposed ROIs (both axes scaled ×5) at arrow D (marked PP indicating pacemaker event) and E (Fig. 4 B).
Fig. 4 B shows simultaneous measurement of membrane potential and [Ca2+]c during application of nifedipine to a spontaneously active chamber. Fig. 4 B presents membrane potential (blue trace), fluorescence of the Ca2+ sensing fluorophore for regions of interest depicted in Fig. 4 C, and mean fluorescence of all ROIs. All regions showed simultaneous increases in [Ca2+]c during action potentials (Fig. 4 B). As nifedipine became effective, action potentials were blocked exposing underlying pacemaker potentials (Fig. 4, B and D), which then themselves diminished as synchrony between regions was lost. Finally, all synchrony was lost with ROIs now showing only asynchronous increases in [Ca2+]c (Fig. 4, B and E).
KCl was applied when nifedipine had blocked synchronous oscillations and was found to cause no significant increase in baseline [Ca2+]c (101 ± 1% n = 4) or initiate vasomotion (data not shown). Whereas, KCl caused increase in frequency of vasomotion (150 ± 10%, n = 5) and then vasospasm when applied in control (data not shown). This indicates that L-type Ca2+ channels are a major pathway for increasing [Ca2+]c.
Effect of agonist during blockade of L-type Ca2+ channels
The role of L-type Ca2+ channels in facilitating synchrony of Ca2+ release was examined by addition of the agonist ET-1 to tissues in which L-type Ca2+ channels had been blocked by nifedipine. An example of 10 experiments from 10 animals is presented in Fig. 5 A. This figure shows that addition of ET-1 (3 nM) caused small depolarization but no synchronization. Further insight into the action of ET-1 was provided from Ca2+ imaging studies and an example is presented in Fig. 5, B–E. Application of 1 μM nifedipine abolished large constriction-associated Ca2+ transients (Fig. 5 B). Plots of the changes in [Ca2+]c for the ROIs shown in Fig. 5 C demonstrated asynchronous spontaneous Ca2+ transients (Fig. 5 D). Application of 3 nM ET-1 in the presence of nifedipine (Fig. 5 B, arrow E) did not overcome the nifedipine-associated blockade of pacemaker potentials, however, there was an increase in the frequency of asynchronous Ca2+ transients (Fig. 5 E). These data indicate that L-type Ca2+ channels are critical for global synchrony of Ca2+ release and vasomotion.
FIGURE 5.

Effect of ET-1 in the presence of nifedipine. (A) Membrane potential recordings (upper traces) and simultaneous vessel edge recordings (lower traces) upon application 3 nM ET-1 in the presence of 1 μM nifedipine. This figure is continued from Fig. 4 (L-NAME (30 μM) also added with nifedipine). Numbered arrows correspond to regions shown on an expanded timescale. (B) [Ca2+]c transients (upper record) averaged over all the imaged areas shown in C and associated smooth muscle constrictions (lower record) recorded from the lymphatic chamber of Fig. 3, B–E. Labeled regions show record sections analyzed below in corresponding panels. (C) Section of a lymphatic chamber showing Oregon green loaded smooth muscle with regions of interest used for data analysis. (D and E) Plots of relative [Ca2+]c as indicated on panel B (arrows D and E) obtained in the presence of 1 μM nifedipine before (D), and during application of 3 nM ET-1 (E). Force scale represents relative force.
Modeling
Simulations using the model set out in the Methods section were made. The key feature of such simulations is that the outcomes are in no way predetermined, rather they simply reflect physical interactions that arise when Ca2+ stores in an array of coupled cells are stimulated, each cell imbued with the known properties of a lymphatic smooth muscle cell. The essential features of this model are: 1), a two-dimensional array of cells interconnected by gap junctions; 2), each cell exhibits oscillatory release of Ca2+ from IP3-receptor operated Ca2+ stores; 3), activation of a Ca2+ store can activate other stores through Ca2+-induced Ca2+ release; 4), store-induced increases in [Ca2+]c generate inward current and resultant membrane depolarization; 5), there is voltage-dependent Ca2+ influx through L-type Ca2+ channels, this in turn causing store release through CICR; and 6), stores interact through chemical coupling (i.e., diffusion of Ca2+ between stores and CICR) and electrochemical coupling (i.e., store release > membrane depolarization > voltage-dependent Ca2+ entry > store release). Increasing the concentration of IP3 in the model simulates the action of IP3 enhancing agonists such as ET-1. The increase in IP3 activates stores that interact as a system of coupled relaxation oscillators within and across the cellular syncytium. Most significantly, the outcome of these interactions closely parallels the observed emergence of lymphatic pacemaking.
Response of syncytium model to low levels of IP3
The dynamic response of the model syncytium was studied by increasing stimulus β (i.e., [IP3]c) at a fixed rate (Fig. 6). During stimulation with low levels of IP3, [Ca2+]c levels become noisy. Each cell has a Ca2+ store population with a heterogeneous IP3 sensitivity distribution (s) (Methods). Thus, within each cell stores that are most sensitive to IP3 (i.e., low σ) respond to increasing IP3 first. The sequence in response can also be due to variation in IP3 concentration in different regions of the tissue, as noted in the Methods section. This can be seen in Fig. 6 B (1a), which shows the Ca2+ response of all the stores in a single cell (this corresponds to cell number (3,3) of Fig. 6 B (1b). In this cell each store responds according to its IP3 sensitivity to increasing stimulus β (i.e., [IP3]).
FIGURE 6.

Model simulations. (A) Cellular syncytium comprising a two-dimensional 5 × 4 array of 20 coupled cells with each cell having 10 Ca2+ stores. (A) Average Ca2+ (upper trace) and membrane potential changes in the model syncytium and corresponding space-time plot. This space-time plot shows the Ca2+ store activity as reflected by changes in cytosolic Ca2+concentration in all (10) regions of each cell, plotted vertically down in the sequence shown in panel B (1b) as a function of time (i.e., stores 1–10 for cell (1,1) then cell (1,2), etc., plotted on the ordinate against time on the abscissa). The boxed inset shows an action potential and associated Ca2+ transient on an expanded (10×) timescale. (B) Regions marked with numbered bars in panel A shown in expanded format. (B (1a)) Ca2+ changes in all stores within a single cell (cell (3,3) marked with arrowhead in B (1b)). Numbers on the left indicate location of the store within the model cell (see Methods). (B (1b) Average Ca2+ changes in each cell of the syncytium. Numbers on the left indicate location of the cell within the syncytium (see Methods). These traces show asynchronous Ca2+ transients in the syncytium during low levels of IP3. (B (2a) Ca2+ wave arising through sequential activation of the array of stores in a single cell before the emergence of pacemaking (cell (3,3) marked with arrowhead in B (2b)). (B (2b)) Corresponding space-time plot for all stores showing asynchronous Ca2+ transients and Ca2+ waves within the cellular syncytium (this is plotted in the same format as for the space-time plot in A but on expanded scales). (B (3a)) Traces showing synchronous store-associated Ca2+ transients in a single cell (cell (1,3) marked with arrowhead in B (3b)) during a pacemaker event. (B (3b)) corresponding space-time plot showing that loosely synchronous Ca2+ release across the model syncytium results in a pacemaker event. (B (4a)) Traces showing store-associated Ca2+ changes in a single cell during action potentials (cell (3,3) marked with arrowhead in B (4b)). (B (4b)) Corresponding space-time plot showing that Ca2+ transients are highly synchronous once pacemaker activity becomes superthreshold and action potentials are generated. Local waves continue to occur during the interval between pacemaker-induced action potentials. Timescale bar in panel A applies to all traces and space-time plot. Color bar applies to all space-time plots. Ca2+ scale bar in B (4a) applies to all traces in panel B. Timescale bar in B (4a) applies to all traces in B except B (1a) and B (1b). Stimulation (i.e., [IP3]) was increased at a fixed rate of 0.0058 μM/min from 0.24 to 0.8 μM. All voltage and Ca2+ scale bars represent normalized values.
The response of a cell is the averaged response of all the Ca2+ stores within it. Fig. 6 B (1b) shows the Ca2+ response of all cells during low levels of IP3. It can be seen that cells respond in a heterogeneous manner to changing IP3; those that have most sensitive Ca2+ stores begin oscillating first, with the oscillation frequency of each cell dependent on its store population. This results in asynchronous Ca2+ oscillations across the model syncytium. Store Ca2+ release from a cell generates inward current causing depolarization in this and other electrically connected cells, dependent on the size of the current and the passive electrical properties of the cellular syncytium. This induces subthreshold Ca2+ influx and resultant Ca2+ release in these cells. This interaction provides the fundamental coupling mechanism in the cell network. In this manner cells interact and influence the dynamics of other cells, but due to the Gaussian distribution of frequencies, the overall syncytium response at first appears random (Fig. 6 A). These Ca2+ release events show close parallels to asynchronous Ca2+ transients observed in the lymphatic smooth muscle during resting conditions.
Ca2+ released from a store can induce Ca2+ release from adjacent stores through CICR; this occasionally continues and results in a propagating Ca2+ wave conducting with a low velocity within a cell (Fig. 6 B (2a)). These waves can arise when a more sensitive store becomes active, its Ca2+ release activating neighboring stores by CICR, with this process continuing along the array of stores. Gap junctions present significant discontinuity to these waves due to the low permeability of gap junctions to Ca2+ (see Hofer et al. (59)). Therefore a diffusive wave is impeded in crossing this barrier (Fig. 6 B (2b)), consistent with our observations that such waves are conducted within and not between lymphatic smooth muscle cells.
Emergence of synchronicity
Initially Ca2+ release events in cells were asynchronous. However with increasing IP3 more cells became excitable and oscillatory, and at a critical level of IP3 (β ≈ 0.48 μM) the overall response of the syncytium became more coordinated and a loose global synchrony appeared (Fig. 6 A, space-time plot). This caused the noisy baseline to be interspersed with aggregated larger amplitude Ca2+ release events and associated membrane potential oscillations (Fig. 6 A). This pacemaker activity parallels that observed in lymphatic smooth muscle in response to ET-1 (Fig. 2). Ca2+ oscillations within single cells became more synchronous during such pacemaker events (Fig. 6 B (3a)), and a loose synchrony appeared across the syncytium, where most, but not all, cells (20–40% of store population) participated in the event (Fig. 6 B (3b)).
Asynchronous Ca2+ transients and pacemaker-like oscillations persisted with increasing IP3, but eventually action potentials and associated large Ca2+ transients appeared (Fig. 6 A). Ca2+ transients within cells became highly synchronous during such events (Fig. 6 B (4a)), and also across the syncytium (Fig. 6 B (4b)). These simulation outcomes parallel the synchronous global Ca2+ transients in lymphatic smooth muscle (Fig. 3). The local intracellular Ca2+ waves that occurred under low levels of stimulation, or between action potentials when the simulated [IP3]c was higher, also parallel the experimental observations. The Ca2+ wave velocity was dependent on the amount of stimulation, wave velocity increasing with higher stimulus over a range of 20–37 μm/s for β = 0.24–0.8 μM. The frequency of action potentials in the model syncytium increased with increasing IP3. However, pacemaking was abolished at very high [IP3]c with the cells entering a state of persistent elevated [Ca2+]c and membrane depolarization (data not shown). This outcome predicts vasospasm, a behavior exhibited by lymphatic smooth muscle during stimulation with high concentrations of ET-1 (18).
Role of L-type Ca2+ channels in model syncytium
A block of L-type Ca2+ channels was achieved by blocking the associated conductance (GML = 0) in Eq. 9. Ramp application of IP3 (i.e., stimulus β) induced asynchronous Ca2+ release events and associated membrane depolarization. Increasing IP3 increased the frequency and amplitude of asynchronous Ca2+ transients and associated STDs, however, synchronous Ca2+ oscillations and associated depolarizations did not emerge (Fig. 7, A and B). Local Ca2+ waves continued to occur within cells (Fig. 7 B (1b)). This behavior of the model syncytium in the presence of L-type Ca2+ channel blockade accords with experimental observations (Figs. 4 and 5).
FIGURE 7.

Role of L-type Ca2+ channels in model simulations. (A) Average Ca2+ (upper trace) and membrane potential response (lower trace) to increasing IP3 in the same simulation as for Fig. 6, with L-type Ca2+ channels blocked. (B) Regions marked with numbered bars in panel A shown in further detail. (B (1a)) Average Ca2+ changes in each cell shows asynchronous Ca2+ transients across the syncytium. (B (1b)) Space-time plot shows asynchronous Ca2+ transients and local Ca2+ waves within cells. (B (2a)) With increasing stimulation (i.e., [IP3]), the frequency of Ca2+ transients is increased but synchronous pacemaker Ca2+ transients do not arise. (B (2b)) Space-time plot shows further details of the store Ca2+ transients. Timescale bar in B (2b) applies to all traces and space-time plots in panel B. Ca2+ scale bar applies to all traces. All voltage and Ca2+ scale bars represent normalized values.
Propagation in large lymphatic chambers
The behavior of guinea-pig mesenteric lymphatics is somewhat different under perfusion. For example constrictions are generally more rhythmic during perfusion than when nonperfused vessels are agonist stimulated. Experiments were made on 11 large perfused lymphatic chambers (mean length 1835 ± 77 μm, range 1500–2300 μm, mean diameter 248 ± 14 μm, range 175–350 μm). Video analysis (Methods) at four approximately evenly spaced sites along the chambers demonstrated waves of constriction. The main direction of propagation of constriction was found to vary between chambers with five of the chambers mainly propagating constriction in the retrograde direction, three chambers mainly in the orthograde direction, and the remaining three chambers exhibited no dominant direction of propagation. Constrictions propagated approximately uniformly in seven of the chambers with other four showing differences in propagation between the tracking sites and in some cases occurring synchronously across local regions. For example in one chamber propagation velocity between recording sites 1–2 (separation 445 μm), 2–3 (separation 396 μm), and 3–4 (separation 314 μm) was 5.0 ± 0.4 mm/s (n = 61 events), 4.6 ± 0.3 mm/s (n = 62 events), and 1.9 ± 0.3 mm/s (n = 64 events), respectively. The mean propagation velocity for the 11 chambers taken between sites 1 and 4 was 2.9 ± 0.3 mm/s.
The modeling results presented so far have studied electrically short syncytia, as would be observed in experiments within the field of imaging. We now use the model to predict the Ca2+ dynamics underlying the experimentally observed behavior of smooth muscle tissue in large lymphatic chambers, where Ca2+ imaging experiments cannot be readily made. Ca2+ dynamics consequent to agonist stimulation was examined in a one-dimensional array of 200 coupled cells, representing a lymphatic chamber of length 2 mm. The IP3 sensitivity of the cells were randomly assigned from a Gaussian distribution. The data of Fig. 8 A, taken once global rhythmic activity had emerged, shows the average membrane potential and [Ca2+]c recorded at three sites along the chamber during a rhythmic depolarization. Conduction delays occur along the length of the model tissue. In the example shown, depolarization first appears in region 1 and then with some delay occurs in region 2 and 3 (Fig. 8 A). Importantly, were each region examined in isolation (i.e., decoupled from the other regions), then each region would exhibit rhythmic activity with its own intrinsic frequency, due to the variability in IP3 sensitivity of the cells. Coupled oscillator-based interaction between these oscillators result in an organized pattern (Fig. 8 B) where a wave appears to be conducting from region 1 toward regions 2 and 3. This is not necessarily a conducting wave but a phase wave, which is a consequence of phase locking between the oscillators. The delays in initiation of action potentials in different regions of the model tissue are similar to constriction delays observed in large lymphatic chambers (∼3–4 mm/s).
FIGURE 8.

Simulation of a large lymphatic chamber. (A) Cellular syncytium comprising a one-dimensional array of 200 coupled units, each 100 μm in length, was used to simulate a lymphatic chamber of 2-mm length. Each unit was composed of a Ca2+ store and plasmalemma oscillator (see Methods). (A) Average membrane potential and [Ca2+]c in three different regions of the model syncytium as shown in the schematic inset with corresponding color code and labels. [Ca2+]c changes all units within each corresponding region are shown below the averages. (B) Space-time plot showing [Ca2+]c changes in all units for the corresponding traces in A. Color-coded and numbered bar on the left shows spatial scale. Timescale bar in panel B applies to all traces. All voltage and Ca2+ scale bars represent normalized values.
Taken together, the modeling suggests that the experimentally observed constriction waves arise due to phase locking of local Ca2+ oscillators that generate global Ca2+ phase waves.
DISCUSSION
Emergence of pacemaker potentials
The experimental data presented have investigated spontaneous and ET-1 induced lymphatic pacemaking. Measurement of relative [Ca2+]c showed loose global synchronization of asynchronous and locally produced spontaneous Ca2+ transients. Measurement of membrane potential in isolated chambers showed corresponding enhancement of spontaneous transient depolarizations and loose synchronization of these events to form pacemaker potentials, which when superthreshold triggered action potentials and vasomotion. Importantly, blockade of L-type Ca2+ channels, while not preventing ET-1 action to enhance the activity of asynchronous Ca2+ transients, prevented the emergence of global synchrony between Ca2+ stores. The fundamental dependence of pacemaking on Ca2+ entry through a voltage-dependent Ca2+ channel pathway indicates that this is the key chemical link in electrochemical coupling between stores. In guinea pig mesenteric lymphatics, the release of Ca2+ from stores is mediated by IP3Rs and not ryanodine receptors (17,18).
The experimental findings were predicted by simulations based on a two-dimensional array of gap-junction connected cells with each cell having an array of stores and L-type Ca2+ channels. In the example provided, the array consisted of 5 × 4 cells, simulating an electrically short smooth muscle syncytia. Gradually increasing [IP3]c across the array caused asynchronous oscillatory Ca2+ release from stores and associated spontaneous transient depolarizations, the level of this activity increasing as more stores were recruited. Global synchrony then emerged leading to formation of Ca2+ transients and associated pacemaker potentials, which when superthreshold triggered action potentials. Importantly, blockade of L-type Ca2+ channels in the model prevented the emergence of pacemaking.
The close parallel between the experimental and simulated emergence of pacemaking consequent to exposure to elevated levels of IP3, provides considerable insight into this process. In the simulation, pacemaking was induced by gradually increasing IP3 levels. At low levels, Ca2+ release events were asynchronous, as relatively few stores were active and hence the interaction between stores was weak, each store causing minimal depolarization. As the [IP3]c was increased more stores became active resulting in local coupled oscillator interactions between Ca2+ stores through both diffusion of store-released Ca2+ and through the now larger Ca2+ transient-induced depolarization causing some voltage-dependent entry of Ca2+. The Ca2+ entry, like Ca2+ released from stores (but more globally), acts to advance or retard store release-refill cycles or to activate new stores. This together with further increase in the number of active stores through the increase in [IP3]c led to more global synchrony and the emergence of pacemaker Ca2+ release and associated pacemaker potentials. Thus, both IP3 and Ca2+ play an essential role in store coupling and synchrony. Importantly, although pacemaker Ca2+ release simulated in Fig. 6 appears to arise near-synchronously it is in effect a Ca2+ phase wave. It appears near-synchronous because of the small array of stores used to simulate the naturally occurring short lymphatic chambers present in the guinea pig mesentery (60). The effect of phase delays and resultant Ca2+ phase waves become apparent in larger size tissues, as the simulation shown in Fig. 8 predicts. Such phase waves are likely to explain the conduction delays observed during constrictions of large lymphatic chambers.
The simulations also provide insight into other observations including local slow conducting Ca2+ waves and “failed” pacemaker events. The former event probably results when a more sensitive IP3R-operated Ca2+ store becomes activated, the resultant high local [Ca2+] diffusing to nearby stores within the same cell sequentially activating these by CICR. This process continues along the array of stores within the cell to generate a Ca2+ wave. Weak coupled oscillator interactions through diffusion of Ca2+ may also underlie some of these local Ca2+ waves (34). The existence of local Ca2+ waves increases the duration of Ca2+ release during subthreshold pacemaker events (Fig. 6 B (3b)). However, when action potentials are triggered there is rapid global Ca2+ influx through the L-type Ca2+ channels. This induces further release of Ca2+from the stores within cells through CICR from IP3 receptors (ryanodine receptors do not seem to be involved in this response (18)). This sequence of events now markedly shortens the underlying pacemaker event (Fig. 6 B (4b)). Another characteristic of lymphatic pacemaking is the occurrence of “failed” pacemaker events (Fig. 6 A), and such activity is also demonstrated experimentally in Fig. 2. These occur when there is a wide spread in natural frequencies of the individual stores leading to imperfect event-to-event entrainment. Thus, slower stores may not be able to refill every cycle leading to variability in synchronization of Ca2+ release and hence the magnitude of the pacemaker potential. Furthermore, stores that are oscillating at very high frequency may not be able to lock in with the majority of the store population. Such rapidly cycling stores contribute to noise levels during the resting state between action potentials. With increasing IP3 the slower stores become more excitable and oscillate at higher frequency (see Berridge (26)) and the chances of “failed” pacemaker events are reduced.
Role of L-type Ca2+ channels
Nifedipine first blocks action potentials and associated synchronized Ca2+ transients. Initially, when action potentials are blocked, subthreshold pacemaker activity is often revealed (Fig. 4) but such activity is rapidly lost leaving only unsynchronized activity, as we have observed many times by either voltage recording or Ca2+ imaging. We interpret this on the basis that initially during nifedipine blockade there are still sufficient L-type Ca2+ channels opened at potentials just above the resting potential to mediate coupled oscillator-based interactions and hence the generation of pacemaker potentials. We consider the data of Figs. 2 and 3 in the same way, namely that the voltage-dependent Ca2+ channels show a spectrum of voltage sensitivities and that low threshold channels are in operation just above the resting membrane potential. The result is generation of pacemaker signals that do not necessarily trigger action potentials (e.g., the “subthreshold” pacemaker signals of Fig. 2). The fact that all synchronicity is lost after establishment of nifedipine block indicates that the channels responsible are L-type Ca2+ channels and precludes a major role for other voltage-dependent Ca2+ channels, voltage-dependent production of IP3 (27), or diffusion of Ca2+ (41). Our simulations support this interpretation as “subthreshold” pacemaker signals occur during simulations (Fig. 6) and all synchronized pacemaker activity is lost when L-type Ca2+ channels are blocked (Fig. 7).
Synchrony is enhanced if stores are brought into their excitable/oscillatory domain (41,61,62). It is possible that Ca2+ entry through the L-type Ca2+ channels “primes” the Ca2+ stores and brings them into their oscillatory domain so that they may synchronize through diffusion of Ca2+; i.e., L-type Ca2+ channels may play only a secondary role in synchronization of stores. However, our experiments in lymphatic smooth muscle show that L-type Ca2+ channels are the primary coupling mechanism. We deduce this because application of ET-1 in the presence of nifedipine brings stores into oscillatory domain (increased frequency of Ca2+ oscillations; Fig. 5), and yet no global synchrony emerges whereas frequency of asynchronous Ca2+ oscillations increase due to increase in frequency and number of oscillatory cells. Thus membrane potential acting through L-type Ca2+ channels is likely the primary source of coupling rather than diffusion of Ca2+ or IP3. These observations are supported by our simulations where increasing store stimulation (i.e., bringing stores into oscillatory domain) while blocking L-type Ca2+ channels does not result in global synchrony (Fig. 7).
Interstitial cells of Cajal
Specialized pacemaker cells, known as interstitial cells of Cajal (ICC) have been identified as pacemaker cells in tissues such as gastrointestinal smooth muscle (63,64). ICC-like cells have also been found in sheep lymphatic vessels (65). However, despite considerable effort, we have yet to demonstrate the existence of these cells in guinea-pig mesenteric lymphatics because the studies made with antibodies such as c-Kit have so far been inconclusive despite readily labeling ICCs in gastric muscle taken from the same animals. Thus, although it remains possible that ICCs have a key role in pacemaking, this role has yet to be confirmed. Should such a role eventuate then these cells would be expected to exhibit dominant pacemaker Ca2+ release and/or enhance coupling between smooth muscle cells. Cells that exhibit dominant pacemaker Ca2+ release would be expected to show rhythmicity even at low concentrations of IP3. In this regard, it is to be noted that we occasionally observed regions where there were oscillations in [Ca2+]c under control conditions where [IP3]c would be relatively low. Significantly, the model presented here implicitly incorporates the possibility of pacemaker ICCs by including a wide heterogeneous sensitivity of the cell population to IP3.
Parallels to slow waves in a gastric smooth muscle
The theoretical basis of this study relates closely to an earlier study of Ca2+ store-based pacemaking shown to generate slow waves in a gastric smooth muscle (27). The gastric slow wave was found to occur through coupled oscillator-based synchronization of oscillatory Ca2+ release from IP3-sensitive Ca2+. Here, entrainment of active Ca2+ stores caused summation of spontaneous transient depolarizations and the production of pacemaker potentials. However, unlike pacemaking in lymphatic smooth muscle, L-type Ca2+ channels are not essential for the generation of the slow waves (49,66). Rather, gastric pacemaking is dependent on depolarization-induced IP3R-mediated Ca2+ release (49,61,66,67). Either voltage-dependent Ca2+ entry or synthesis of IP3 can provide the “chemical” link in electrochemical coupling of Ca2+ stores. Thus pacemaking in gastric and lymphatic smooth muscle are fundamentally similar, both involving coupled oscillator-based synchronization of Ca2+ stores and resultant Ca2+ phase waves, but differing in the mechanisms of long-range coupling (i.e., the springs) by which Ca2+ store oscillators are strongly coupled.
Parallels to vasomotion in blood vessels
Two types of vasomotion have been described in blood vessels. The first, voltage-dependent vasomotion has been reported in rat basilar and mesenteric arteries (39,40,68,69) and the other, voltage-independent vasomotion has been reported in rat irideal arterioles (3,70). Both have been associated with oscillatory Ca2+ release and membrane potential changes (3,39,40,69–72), findings that have resulted in proposal of a hypothesis for the pacemaker mechanism underlying voltage-dependent vasomotion in blood vessels (40). In their model, initiation of pacemaking occurs when “sufficient number of cells become active at the same moment” (40). Here the authors formulated a model where simultaneous activation of store Ca2+ release caused a Ca2+-activated inward current and depolarization, this activating influx of Ca2+ through L-type Ca2+ channels. The voltage-dependent Ca2+ entry was considered to cause near-synchronous activation of Ca2+ stores, which then remained entrained, to act as a pacemaker for vasomotion. The coupled oscillator-based pacemaker mechanism that we present here shares similarities to both the lymphatic model (15) where electrochemical-based interaction of store oscillators was proposed, and the blood vessel model (40) as discussed above. The model we present here also takes into account store populations with heterogeneous responses to IP3, which predicts variation in pacemaker amplitude. Another recent work (41) has investigated the role of electrical, Ca2+, and IP3 (including phospholipase C-δ dependent IP3 synthesis) coupling in synchronization of Ca2+ oscillators. The model was validated using published data on rat mesenteric arterial segments. The authors concluded that coupling through Ca2+, but not IP3 or electrical current is capable of synchronizing Ca2+ oscillations. These results are in contrast to the studies performed on the lymphatic smooth muscle, where electrical coupling is essential for global synchrony. Similar results were obtained in our previous study of the gastric smooth muscle where electrical, but not coupling through diffusion of Ca2+ or IP3 was found to be essential for global synchrony (see van Helden and Imtiaz (27)).
The lymphatic pacemaker model may also predict pacemaking in circumstances where L-type Ca2+ channels activate nonregeneratively. For example, voltage-dependent vasomotion in resistance vessels while dependent on L-type Ca2+ channels can occur with or without regenerative action potentials. The latter circumstance would differ from lymphatics, which generally shows an associated action potential. However, the underlying pacemaker mechanisms would be no different except that the L-type Ca2+ channels associated with the pacemaker depolarization would not lead to regenerative activation possibly because of shunting by the conductance underlying the pacemaker potential (e.g., ClCa channels). Store-based pacemaking is also likely to underlie voltage-independent vasomotion (3). However, such pacemaking would be predicted to be weak unless there were large numbers of active (i.e., pacemaker) stores and/or there were other coupling factors including strong gap junction coupling between cells. Coupling might also be strengthened if cells were chemically coupled extracellularly. For example, it is known that cellular release of ATP, can activate Ca2+ release in other cells, this underlying some Ca2+ waves (73).
CONCLUSIONS
This article has presented experimental data and model simulations to study pacemaking in lymphatic smooth muscle. The results indicate that pacemaking occurs through distributed coupled oscillator-based interactions of intracellular IP3R-operated Ca2+ stores across the smooth muscle syncytium. Positive feedback by voltage-dependent Ca2+ entry provides long-range electrochemical coupling between the Ca2+ store oscillators and produces global entrainment of pacemaker Ca2+ stores. It is likely that this pacemaker mechanism may have a key role in many cellular rhythms where these elements are expressed. Significantly, single cell studies have shown that these elements are also present in the heart, and are proposed to play a role in pacemaking (74–77). Similarly, studies on isolated human lymphatics show they exhibit behavior that parallels that of guinea-pig mesenteric lymphatics. For example, when isolated they are generally quiescent but exhibit rhythmic constrictions in response to agonists such as norepinephrine (e.g., see Sjoberg and Steen (78)).
Acknowledgments
We thank Peter Dosen for technical assistance and Chris Katnik for fruitful discussion.
The work was supported by the National Health and Medical Research Council of Australia (NHMRC grant No. 141771), the Australian Research Council (ARC LIEF grant No. LE0453630), the Brawn Bequest, and the Hunter Medical Research Institute.
APPENDIX I: CALCIUM DYNAMICS
We follow the original notation of the single pool model of Dupont and Goldbeter (50) for Ca2+ concentration in the cytosol ([Ca2+]c) and IP3-sensitive intracellular Ca2+ store ([Ca2+]s) and denote them by the state variables Z and Y, respectively. We denote IP3 concentration in the cytosol ([IP3]c) by the variable β. The functions describing the Ca2+ dynamics are:
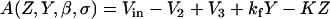 |
(A1) |
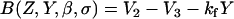 |
(A2) |
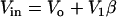 |
(A3) |
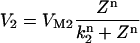 |
(A4) |
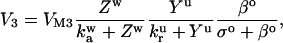 |
(A5) |
where Vin is the Ca2+ influx from extracellular space, (composed of an IP3 independent component (V0), and an IP3 dependent component (V1β)). Ca2+ is expelled from the cytosol into the extracellular space by a [Ca2+]c dependent pump, represented by the term KZ. V2 is the Ca2+ flux into the intracellular Ca2+ store. V3 is the Ca2+ and IP3 dependent release of calcium from the intracellular Ca2+ store. The term kfY represents the leakage from the store dependent on the [Ca2+]s. Parameters for the system defined by Eqs. A1–A5 are summarized in Table 2.
We note that an oscillating IP3 concentration is not required to generate oscillations in this model. Thus, synchrony in our model appears without the need of oscillating IP3 concentration. A bifurcation diagram of Ca2+ with stimulus β as the bifurcation parameter is given in Fig. 9, which shows the overall response of the single Ca2+ oscillator.
FIGURE 9.

Response of the single Ca2+ oscillator to stimulus. A bifurcation diagram of Ca2+ with stimulus β as the bifurcation parameter showing the overall response of the single Ca2+ oscillator. The thick solid lines give envelop of maximum and minimum [Ca2+]c values during oscillations with respect to stimulus. The system is nonoscillatory outside this envelop.
References
- 1.Florey, H. W. 1927. Observations on the contractility of lacteals. Part 1. J. Physiol. (Lond.). 62:267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson, P. C. 1980. The myogenic response. In Handbook of Physiology: The Cardiovascular System. S. R. Geiger, D. F. Bohr, A. P. Somlyo, and H. V. Sparks, editors. American Physiological Society, Bethesda, MD. 400–442.
- 3.Haddock, R. E., G. D. Hirst, and C. E. Hill. 2002. Voltage independence of vasomotion in isolated irideal arterioles of the rat. J. Physiol. 540:219–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nilsson, H., and C. Aalkjaer. 2003. Vasomotion: mechanisms and physiological importance. Mol. Interv. 3:79–89. [DOI] [PubMed] [Google Scholar]
- 5.Mislin, H. 1983. The lymphangion. In Lymphangiology. M. Foldi, and J. R. Casley-Smith, editors. F. K. Schattauer Verlag, New York. 165–175.
- 6.McHale, N. G. 1990. Lymphatic innervation. Blood Vessels. 27:127–136. [DOI] [PubMed] [Google Scholar]
- 7.Kirkpatrick, C. T., and N. G. McHale. 1977. Electrical and mechanical activity of isolated lymphatic vessels. J. Physiol. 272:33P–34P. [PubMed] [Google Scholar]
- 8.Ohhashi, T., T. Azuma, and M. Sakaguchi. 1978. Transmembrane potentials in bovine lymphatic smooth muscle. Proc. Soc. Exp. Biol. Med. 159:350–352. [DOI] [PubMed] [Google Scholar]
- 9.Ward, S. M., K. M. Sanders, K. D. Thornbury, and N. G. McHale. 1991. Spontaneous electrical activity in isolated bovine lymphatics recorded by intracellular microelectrodes. J. Physiol. 438:168P. [Google Scholar]
- 10.van Helden, D. F. 1993. Pacemaker potentials in lymphatic smooth muscle of the guinea-pig mesentery. J. Physiol. 471:465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.DiFrancesco, D. 1991. The contribution of the ‘pacemaker’ current (if) to generation of spontaneous activity in rabbit sino-atrial node myocytes. J. Physiol. 434:23–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Allen, J. M., and N. G. McHale. 1988. The effect of known K+-channel blockers on the electrical activity of bovine lymphatic smooth muscle. Pflugers Arch. 411:167–172. [DOI] [PubMed] [Google Scholar]
- 13.McCloskey, K. D., H. M. Toland, M. A. Hollywood, K. D. Thornbury, and N. G. McHale. 1999. Hyperpolarisation-activated inward current in isolated sheep mesenteric lymphatic smooth muscle. J. Physiol. 521:201–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.van Helden, D. F., P.-Y. von der Weid, and M. J. Crowe. 1996. Intracellular Ca2++ release: a basis for electrical pacemaking in lymphatic smooth muscle. In Smooth Muscle Excitation. T. B. Bolton and T. Tomita, editors. Academic Press, London. 355–373.
- 15.van Helden, D. F., and J. Zhao. 2000. Lymphatic vasomotion. Clin. Exp. Physiol. Pharmacol. 27:1014–1018. [DOI] [PubMed] [Google Scholar]
- 16.Ferrusi, I., J. Zhao, D. van Helden, and P. Y. von der Weid. 2004. Cyclopiazonic acid decreases spontaneous transient depolarizations in guinea pig mesenteric lymphatic vessels in endothelium-dependent and -independent manners. Am. J. Physiol. Heart Circ. Physiol. 286:H2287–H2295. [DOI] [PubMed] [Google Scholar]
- 17.Zhao, J., and D. F. van Helden. 2002. ATP-induced endothelium-independent enhancement of lymphatic vasomotion in guinea-pig mesentery involves P2X and P2Y receptors. Br. J. Pharmacol. 137:477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhao, J., and D. F. van Helden. 2003. ET-1-associated vasomotion and vasospasm in lymphatic vessels of the guinea-pig mesentery. Br. J. Pharmacol. 140:1399–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.van Helden, D. F. 1991. Spontaneous and noradrenaline-induced transient depolarizations in the smooth muscle of guinea-pig mesenteric vein. J. Physiol. 437:511–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wang, Q., R. C. Hogg, and W. A. Large. 1992. Properties of spontaneous inward currents recorded in smooth muscle cells isolated from the rabbit portal vein. J. Physiol. 451:525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lakatta, E. G., T. Vinogradova, A. Lyashkov, S. Sirenko, W. Zhu, A. Ruknudin, and V. A. Maltsev. 2006. The integration of spontaneous intracellular Ca2+ cycling and surface membrane ion channel activation entrains normal automaticity in cells of the heart's pacemaker. Ann. N. Y. Acad. Sci. 1080:178–206. [DOI] [PubMed] [Google Scholar]
- 22.Keizer, J., Y. X. Li, S. Stojilkovic, and J. Rinzel. 1995. InsP3-induced Ca2+ excitability of the endoplasmic reticulum. Mol. Biol. Cell. 6:945–951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Parker, I., and Y. Yao. 1991. Regenerative release of calcium from functionally discrete subcellular stores by inositol trisphosphate. Proc Roy Soc Ser B. 246:269–274. [DOI] [PubMed] [Google Scholar]
- 24.Fabiato, A. 1983. Calcium-induced release of calcium from the cardiac sarcoplasmic reticulum. Am. J. Physiol. 245:C1–14. [DOI] [PubMed] [Google Scholar]
- 25.Yao, Y., J. Choi, and I. Parker. 1995. Quantal puffs of intracellular Ca2+ evoked by inositol trisphosphate in Xenopus oocytes. J. Physiol. 482:533–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Berridge, M. J. 1993. Inositol trisphosphate and calcium signalling. Nature. 361:315–325. [DOI] [PubMed] [Google Scholar]
- 27.van Helden, D. F., and M. S. Imtiaz. 2003. Ca2+ phase waves: a basis for cellular pacemaking and long-range synchronicity in the guinea-pig gastric pylorus. J. Physiol. 548:271–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Strogatz, S. H., and I. Stewart. 1993. Coupled oscillators and biological synchronization. Sci. Am. 269:102–109. [DOI] [PubMed] [Google Scholar]
- 29.van der Pol, B., and J. van der Mark. 1926. The heartbeat considered as a relaxation oscillation, and an electrical model of the heart. Phil Magnus Suppl. 6:763–775. [Google Scholar]
- 30.Jalife, J. 1984. Mutual entrainment and electrical coupling as mechanisms for synchronous firing of rabbit sino-atrial pace-maker cells. J. Physiol. 356:221–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nelsen, T. S., and J. C. Becker. 1968. Simulation of the electrical and mechanical gradient of the small intestine. Am. J. Physiol. 214:749–757. [DOI] [PubMed] [Google Scholar]
- 32.Sarna, S. K., E. E. Daniel, and Y. J. Kingma. 1971. Simulation of slow-wave electrical activity of small intestine. Am. J. Physiol. 221:166–175. [DOI] [PubMed] [Google Scholar]
- 33.Daniel, E. E., B. L. Bardakjian, J. D. Huizinga, and N. E. Diamant. 1994. Relaxation oscillator and core conductor models are needed for understanding of GI electrical activities. Am. J. Physiol. 266:G339–G349. [DOI] [PubMed] [Google Scholar]
- 34.Roth, B. J., S. V. Yagodin, L. Holtzclaw, and J. T. Russell. 1995. A mathematical model of agonist-induced propagation of calcium waves in astrocytes. Cell Calcium. 17:53–64. [DOI] [PubMed] [Google Scholar]
- 35.McHale, N. G., and M. K. Meharg. 1992. Co-ordination of pumping in isolated bovine lymphatic vessels. J. Physiol. 450:503–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.van Helden, D. F., and M. S. Imtiaz. 2003. Ca2+ phase waves emerge. Physiology News. 52:7–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Allbritton, N. L., T. Meyer, and L. Stryer. 1992. Range of messenger action of calcium ion and inositol 1,4,5-trisphosphate. Science. 258:1812–1815. [DOI] [PubMed] [Google Scholar]
- 38.Hirst, G. D., and T. O. Neild. 1978. An analysis of excitatory junctional potentials recorded from arterioles. J. Physiol. (Lond.). 280:87–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mauban, J. R., C. Lamont, C. W. Balke, and W. G. Wier. 2001. Adrenergic stimulation of rat resistance arteries affects Ca2+ sparks, Ca2+ waves, and Ca(2+) oscillations. Am. J. Physiol. Heart Circ. Physiol. 280:H2399–H2405. [DOI] [PubMed] [Google Scholar]
- 40.Peng, H., V. Matchkov, A. Ivarsen, C. Aalkjaer, and H. Nilsson. 2001. Hypothesis for the initiation of vasomotion. Circ. Res. 88:810–815. [DOI] [PubMed] [Google Scholar]
- 41.Koenigsberger, M., R. Sauser, M. Lamboley, J. L. Beny, and J. J. Meister. 2004. Ca2+ dynamics in a population of smooth muscle cells: modeling the recruitment and synchronization. Biophys. J. 87:92–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Haddock, R. E., and C. E. Hill. 2005. Rhythmicity in arterial smooth muscle. J. Physiol. 566:645–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Imtiaz, M. S., J. Zhao, and D. F. van Helden. 2002. A theoretical study of Ca2+ oscillations and pacemaker potentials underlying vasomotion in guinea-pig lymphatic smooth muscle. Proceedings of the Australian Health and Medical Research Congress, Melbourne, Australia. 1148. (Abstr.).
- 44.Gao, J., J. Zhao, S. E. Rayner, and D. F. Van Helden. 1999. Evidence that the ATP-induced increase in vasomotion of guinea-pig mesenteric lymphatics involves an endothelium-dependent release of thromboxane A2. Br. J. Pharmacol. 127:1597–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.von der Weid, P. Y., M. J. Crowe, and D. F. Van Helden. 1996. Endothelium-dependent modulation of pacemaking in lymphatic vessels of the guinea-pig mesentery. J. Physiol. (Lond.). 493:563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Beresford-Smith, B., K. V. Nesbitt, and D. F. Van Helden. 1993. Edge detection at multiple locations using a ‘radar’ tracking algorithm as exemplified in isolated guinea-pig lymphatic vessels. J. Neurosci. Methods. 49:69–79. [DOI] [PubMed] [Google Scholar]
- 47.Toland, H. M., K. D. McCloskey, K. D. Thornbury, N. G. McHale, and M. A. Hollywood. 2000. Ca2+-activated Cl− current in sheep lymphatic smooth muscle. Am. J. Physiol. 279:C1327–C1335. [DOI] [PubMed] [Google Scholar]
- 48.Zhao, J. 2003. Lymphatic vasomotion. The University of Newcastle, Newcastle, Australia. 266.
- 49.van Helden, D. F., M. S. Imtiaz, K. Nurgaliyeva, P. von der Weid, and P. J. Dosen. 2000. Role of calcium stores and membrane voltage in the generation of slow wave action potentials in guinea-pig gastric pylorus. J. Physiol. 524:245–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dupont, G., and A. Goldbeter. 1993. One-pool model for Ca2+ oscillations involving Ca2+ and inositol 1,4,5-trisphosphate as co-agonists for Ca2+ release. Cell Calcium. 14:311–322. [DOI] [PubMed] [Google Scholar]
- 51.Large, W. A., and Q. Wang. 1996. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. Am. J. Physiol. 271:C435–C454. [DOI] [PubMed] [Google Scholar]
- 52.Somlyo, A. 1980. Ultrastructure of vascular smooth muscle. American Physiological Society, Bethesda, MD.
- 53.Somlyo, A., and A. Somlyo. 1992. The Heart and Cardiovascular System. Paven Press, New York.
- 54.Crowe, M. J. 1996. Control and Modulation of Constrictions in Lymphatic Vessels of the Guinea-Pig Mesentary. The University of Newcastle, Newcastle, Australia.
- 55.Burdyga, T. V., and S. Wray. 1998. The effect of inhibition of myosin light chain kinase by Wortmannin on intracellular [Ca2+], electrical activity and force in phasic smooth muscle. Pflugers Arch. 436:801–803. [DOI] [PubMed] [Google Scholar]
- 56.Ui, M., T. Okada, K. Hazeki, and O. Hazeki. 1995. Wortmannin as a unique probe for an intracellular signalling protein, phosphoinositide 3-kinase. Trends Biochem. Sci. 20:303–307. [DOI] [PubMed] [Google Scholar]
- 57.Rayner, S. E., and D. F. Van Helden. 1997. Evidence that the substance P-induced enhancement of pacemaking in lymphatics of the guinea-pig mesentery occurs through endothelial release of thromboxane A2. Br. J. Pharmacol. 121:1589–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.von der Weid, P. Y., J. Zhao, and D. F. Van Helden. 2001. Nitric oxide decreases pacemaker activity in lymphatic vessels of guinea pig mesentery. Am. J. Physiol. Heart Circ. Physiol. 280:H2707–H2716. [DOI] [PubMed] [Google Scholar]
- 59.Hofer, T., A. Politi, and H. Reinhert. 2001. Intercellular Ca2+ wave propagation through gap-junctional Ca2+ diffusion: a theoretical study. Biophys. J. 80:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Crowe, M. J., P. Y. von der Weid, J. A. Brock, and D. F. Van Helden. 1997. Co-ordination of contractile activity in guinea-pig mesenteric lymphatics. J. Physiol. (Lond.). 500:235–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Imtiaz, M. S., D. W. Smith, and D. F. Van Helden. 2002. A theoretical model of slow wave regulation using voltage-dependent synthesis of inositol 1,4,5-trisphosphate. Biophys. J. 83:1877–1890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Imtiaz, M. S., C. P. Katnik, D. W. Smith, and D. F. van Helden. 2006. Role of voltage-dependent modulation of store Ca2+ release in synchronization of Ca2+ oscillations. Biophys. J. 90:1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dickens, E. J., G. D. Hirst, and T. Tomita. 1999. Identification of rhythmically active cells in guinea-pig stomach. J. Physiol. 514:515–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ward, S. M., T. Ordog, S. D. Koh, S. Abu Baker, J. Y. Jun, G. Amberg, K. Monaghan, and K. M. Sanders. 2000. Pacemaking in interstitial cells of Cajal depends upon calcium handling by endoplasmic reticulum and mitochondria. J. Physiol. 525:355–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.McCloskey, K. D., M. A. Hollywood, K. D. Thornbury, S. M. Ward, and N. G. McHale. 2002. Kit-like immunopositive cells in sheep mesenteric lymphatic vessels. Cell Tissue Res. 310:77–84. [DOI] [PubMed] [Google Scholar]
- 66.Suzuki, H., and G. D. Hirst. 1999. Regenerative potentials evoked in circular smooth muscle of the antral region of guinea-pig stomach. J. Physiol. 517:563–573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hirst, G. D., and F. R. Edwards. 2001. Generation of slow waves in the antral region of guinea-pig stomach—a stochastic process. J. Physiol. 535:165–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fujii, K., D. D. Heistad, and F. M. Faraci. 1990. Ionic mechanisms in spontaneous vasomotion of the rat basilar artery in vivo. J. Physiol. 430:389–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Haddock, R. E., and C. E. Hill. 2002. Differential activation of ion channels by inositol 1,4,5-trisphosphate (IP(3))- and ryanodine-sensitive calcium stores in rat basilar artery vasomotion. J. Physiol. 545:615–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hill, C. E., J. Eade, and S. L. Sandow. 1999. Mechanisms underlying spontaneous rhythmical contractions in irideal arterioles of the rat. J. Physiol. 521:507–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Segal, S. S., and J. L. Beny. 1992. Intracellular recording and dye transfer in arterioles during blood flow control. Am. J. Physiol. 263:H1–H7. [DOI] [PubMed] [Google Scholar]
- 72.von der Weid, P. Y., and J. L. Beny. 1993. Simultaneous oscillations in the membrane potential of pig coronary artery endothelial and smooth muscle cells. J. Physiol. 471:13–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Jorgensen, N. R., S. T. Geist, R. Civitelli, and T. H. Steinberg. 1997. ATP- and gap junction-dependent intercellular calcium signaling in osteoblastic cells. J. Cell Biol. 139:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vinogradova, T. M., Y. Y. Zhou, V. Maltsev, A. Lyashkov, M. Stern, and E. G. Lakatta. 2004. Rhythmic ryanodine receptor Ca2+ releases during diastolic depolarization of sinoatrial pacemaker cells do not require membrane depolarization. Circ. Res. 94:802–809. [DOI] [PubMed] [Google Scholar]
- 75.Rigg, L., and D. A. Terrar. 1996. Possible role of calcium release from the sarcoplasmic reticulum in pacemaking in guinea-pig sino-atrial node. Exp. Physiol. 81:877–880. [DOI] [PubMed] [Google Scholar]
- 76.Ju, Y. K., and D. G. Allen. 1998. Intracellular calcium and Na+-Ca2+ exchange current in isolated toad pacemaker cells. J. Physiol. 508:153–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Lipsius, S. L., J. Huser, and L. A. Blatter. 2001. Intracellular Ca2+ release sparks atrial pacemaker activity. News Physiol. Sci. 16:101–106. [DOI] [PubMed] [Google Scholar]
- 78.Sjoberg, T., and S. Steen. 1991. In vitro effects of a thromboxane A2-analogue U-46619 and noradrenaline on contractions of the human thoracic duct. Lymphology. 24:113–115. [PubMed] [Google Scholar]
- 79.Dhein, S., A. Salameh, R. Berkels, and W. Klaus. 1999. Dual mode of action of dihydropyridine calcium antagonists: a role for nitric oxide. Drugs. 58:397–404. [DOI] [PubMed] [Google Scholar]