

Abstract
Perlecan, a heparan sulfate proteoglycan, has been suggested to be critical for regulation of vascular repair. We generated clones of endothelial cells expressing an antisense vector targeting domain III of perlecan. Transfected cells produced significantly less perlecan than parent cells and showed a reduced ability to inhibit the binding and mitogenic activity of fibroblast growth factor-2 in vascular smooth muscle cells. Endothelial cells were seeded onto three-dimensional polymeric matrices and implanted adjacent to porcine carotid arteries subjected to deep injury. Although the parent endothelial cells prevented occlusive thrombosis, perlecan-deficient cells were completely ineffective. The ability of endothelial cells to inhibit intimal hyperplasia, however, was abrogated only in part by perlecan suppression. The differential regulation by perlecan of these different aspects of vascular repair may explain why control of clinical clot formation does not lead to full control of intimal hyperplasia. Thus the use of genetically modified tissue-engineered cells provides a new approach for dissecting the role of specific factors within the complex environment of the blood vessel wall.
The vascular endothelium plays a critical role in regulating blood vessel wall homeostasis and repair (1). Compromised endothelial cell function is a hallmark of vascular disease, and endothelial injury has been implicated in contributing to virtually every aspect of the generation and propagation of native and accelerated forms of atherosclerosis (2–4). Endothelial disease impacts on thrombosis, inflammation, and the migration and proliferation of smooth muscle cells (5–10). Endothelial injury and accelerated arteriopathies (restenosis) lead to failure of 30–50% of vascular interventions such as angioplasty, vascular bypass grafts, and organ transplantation (11, 12). The role of the endothelium in normal maintenance of vascular wall integrity is complex. As a continuous monolayer, the endothelium provides structural boundaries to the circulating blood in the lumen and the vascular wall and ensures continued fluid flow by serving as a selectively permeable thromboresistant surface. The biochemical control established by the endothelium is not an isolated surface phenomenon; it pervades the blood vessel wall. Endothelial cells produce factors that regulate virtually every aspect of vascular biology. What has not been clear to date is whether the chemical control of each of the different biological elements is linked, in lock step, or potentially independently regulated.
We have previously used tissue engineering to investigate the role of endothelial cells as biochemical regulators of vascular injury (13–15). Endothelial implants placed into the perivascular space reduced intimal thickening and thrombotic occlusion after controlled mural injury of rat and pig carotid arteries (14, 15). In this system, the endothelial cells are far from the lumen, allowing their biochemical regulatory role to be dissociated from their boundary properties. Thus perivascular transplantation combined with the ability to genetically modify cultured endothelial cells can provide a powerful tool to dissect the roles of various endothelial cell products in controlling the vascular response to injury.
The heparan sulfate proteoglycan perlecan stands out as an especially important endothelial cell-derived regulator of vascular homeostasis (16–20). Perlecan is a multidomain proteoglycan that can contain heparan sulfate and chondroitin sulfate chains and is found prominently in vascular basement membranes (16, 17). Perlecan represents the major proteoglycan secreted by cultured endothelial cells, and it is a potent inhibitor of smooth muscle cell proliferation in vitro (18–20). Furthermore, the growth inhibitory activity of endothelial cell-conditioned media is lost when media are first treated with heparinase, demonstrating that heparan sulfate is required (9, 10, 19, 20). Perlecan has been proposed to bind to heparin-binding mitogens, such as fibroblast growth factor-2 (FGF-2), and prevent them from stimulating smooth muscle cells (10, 19). Thus, endothelial cell-secreted perlecan likely plays a critical role in the regulation of vascular injury in vivo, yet no direct test of its role within this system has been conducted.
To test directly the relative importance of perlecan within the complex environment of the blood vessel wall and to determine which of the phases of the vascular injury response are regulated by perlecan, we generated stably transfected clones of endothelial cells that express high levels of an antisense vector targeting a large conserved region (domain III) of perlecan. Transfected cells produced significantly less perlecan in vitro and showed a reduced ability to inhibit FGF-2 binding to and mitogenic activity in cultured smooth muscle cells. When three-dimensional polymeric matrices containing these transfected endothelial cells were implanted adjacent to injured porcine carotid arteries, protection against occlusive thrombosis was lost in full, but control of neointimal proliferation was abrogated only in part. Thus, we show that perlecan plays a critical role in regulating the vascular response to injury in vivo. Perlecan was absolutely required for the prevention of thrombosis after deep vascular injury but was only partially responsible for inhibition of the resultant tissue hyperplasia. The differential regulation by perlecan of these different aspects of vascular repair may explain the divergent results seen when promising animal experiments cannot be extended to higher animal species or humans and why control of clinical clot formation does not lead to full control of intimal hyperplasia. Genetically modified tissue-engineered cells may enhance our understanding of the complexity of vascular biology and the response to injury.
Materials and Methods
Materials.
Human recombinant FGF-2 was from Scios-Nova (Mountain View, CA). 125I-FGF-2 was prepared by a modification of the Bolton–Hunter procedure (21). Heparinase III, Chondroitinase ABC, heparan sulfate from bovine kidney, gelatin, Hepes, and trypsin were from Sigma. Dimethylmethylene blue was obtained from Aldrich. The Bio-Rad Protein Assay was used to determine protein concentrations based on BSA standards. Antiheparan sulfate proteoglycan (Upstate Biotechnology) served as the primary perlecan antibody, whereas anti-rat IgG, horseradish peroxidase-linked whole antibody from sheep (Amersham) served as the secondary antibody for Western blots (19).
Cell Cultures.
Bovine aortic endothelial cells (BAE) were used between passages 1–7 and bovine aortic vascular smooth muscle cells (VSMC) were used between passages 3–9. First passage BAE cells were purchased from Clonetics (Walkersville, MD). VSMC were isolated from calf aorta as described (22). All cells were maintained in DMEM (low glucose, GIBCO), supplemented with penicillin (100 units/ml), streptomycin (100 μg/ml), glutamine (2 mM), and 10% calf serum (HyClone).
Antisense Vector and Generation of Stably Transfected Clones.
A human antisense construct comprising ≈1 kb (bp 3120–4120) of perlecan domain III (23, 24) ligated into the eukaryotic expression vector pcDNA3 (Invitrogen) driven by the potent cytomegalovirus promoter was used as the antiperlecan vector (AP). Vector without the perlecan antisense sequence was used as a control (NEO). Approximately 5 × 105 BAE cells (passage 2) were transfected with 10 μg of plasmid DNA (AP or NEO) by using LipofectAMINE (GIBCO) and grown for 48 h in nonselective medium, which allowed for the expression of the transfected gene. The cells were then passaged and selected in the presence of G418 (800 μg/ml). Clones were isolated by ring cloning and expanded and maintained in medium containing G418 (400 μg/ml). Isolated transfected BAE cells were used for all experiments between passages 5 and 7.
Conditioned Medium.
Confluent BAE cells (2.5–5 × 106 cells/100 mm dish) were established in DMEM containing 10% calf serum and maintained in culture for 2 days before conditioning. Conditioned medium was prepared by washing the cells for 1 h in serum-free DMEM at 37°C then incubating the cells for 24 h in serum-free DMEM at 37°C. Medium was collected and centrifuged (30 min at 1,800 × g, 4°C). Anionic exchange chromatography (Q-Sepharose) was used to purify the proteoglycan fraction from endothelial-conditioned medium (19). The glycosaminoglycan (GAG) containing fraction was eluted with Tris-buffered saline, 1 M urea, 1.5 M NaCl, and GAG content determined by using the dimethylmethylene blue dye-binding assay with a heparan sulfate standard (25). Samples were dialyzed and concentrated to a final volume of 1 ml/1 liter of initial conditioned media by partial lyophylization.
Cell Engraftment.
BAEs transfected with the perlecan antisense vector (BAE-AP3) or with the empty vector (BAE-NEO1) were seeded in Gelfoam matrices to confluency and then implanted (15). Sterile Gelfoam was supplied by Pharmacia & Upjohn (Kalamazoo, MI) and cut into 2.5 × 1.0 × 0.3-cm blocks. The Gelfoam blocks were hydrated in PBS and placed in 35 × 10-mm tissue culture dishes. BAE-AP3 or BAE-NEO1 (1 × 105) cells were added directly to hydrated Gelfoam sponges in a total volume of 0.1 ml. The cells were placed at 37°C in a humidified 5% CO2, 95% air incubator and allowed to adhere for 2 h. The cell-loaded Gelfoam blocks were subsequently placed in 17 × 100-mm polypropylene tubes containing 2 ml media and incubated for up to 2 weeks at a 45° angle. Growth medium was changed every 72 h.
Animals.
We assessed the ability of the endothelial cell Gelfoam grafts to reduce intimal hyperplasia and thrombosis when wrapped around balloon-injured porcine carotid arteries (15). This study conformed to the National Institutes of Health “Guide for Care and Use of Laboratory Animals” and was approved by the Institutional Animal Care and Use Committee of the Veterans Association Medical Center (West Roxbury, MA). Twelve male domestic pigs, 34.7 kg ± 0.9 kg, were obtained from Animal Biotech (Danboro, PA). Anesthesia was induced with intramuscular ketamine (1,000 mg), xylazine (150 mg), and atropine (0.6 mg) and maintained with inhaled isofluorane (0.5–1.5%) via an endotracheal tube. All animals also received i.v. cefazolin (500 mg, pre- and postsurgery) to prevent infection. The intraarterial pressure and electrocardiogram were monitored continuously throughout the procedure.
Surgical Procedure.
Porcine carotid arterial injury and matrix implantation were performed as described (15). Right femoral arterial access with an 8 French sheath was obtained via cut down, and an 8.0-mm-diameter angioplasty balloon (Cordis, Miami) was advanced to the common carotid artery under fluoroscopic guidance. Angiography was performed and recorded by cineradiography. The right and left carotid arteries were injured by 30-second balloon inflations at 8 atmospheres pressure (5 inflations per side, in overlapping segments). The balloon/artery ratio (1.39 ± 0.04) did not vary significantly between treatment groups. After final angiography to assess vessel patency, a mid-line neck incision exposed both common carotid arteries, which were isolated and gently wrapped with Gelfoam containing BAE-NEO1 (n = 6 arteries, 2 arteries per animal), BAE-AP3 (n = 10 arteries, 2 arteries per animal), or no cells (n = 8 arteries, 2 arteries per animal). The carotid sheath was closed to immobilize the device and sutured to facilitate the location of implants at sacrifice.
Tissue Processing.
Tissue was processed and sections analyzed as described (15). On the 28th postoperative day, animals were euthanized and the carotid arteries were fixed in situ by perfusion with 4% paraformaldehyde in 0.1 mol/liter PBS (pH 7.4). The arteries with attached implants were isolated and paraffin embedded. Five-μm sections were obtained and stained with Verhoeff's elastin stain. Morphometric analysis was performed on all segments.
Results
Decreased Perlecan Expression Reduces Endothelial Cell-Mediated Inhibition of FGF-2 Growth Stimulation in VSMC.
We have demonstrated previously that endothelial cell heparan sulfate proteoglycans inhibit FGF-2 binding and mitogenesis in VSMC (10, 19). To test the role of perlecan specifically, we generated stable transfected clones of primary BAE cells expressing an antisense cDNA targeting domain III of perlecan (Fig. 1A). Several antisense transfected clones (AP1–4) showed reduced perlecan core protein expression as compared with the parent cells or cells transfected with the vector alone (NEO1–4) (Fig. 1B). The ability of conditioned media from these clones to inhibit FGF-2 binding to VSMC was analyzed as a functional activity. All of the clones that showed significantly reduced perlecan levels also showed reduced ability to inhibit FGF-2 binding. Likewise, clones that did not show reduced perlecan expression (AP5 and 6) retained the ability to inhibit FGF-2 binding (Fig. 1C).
Figure 1.

Expression of perlecan antisense leads to reduced perlecan secretion and loss of FGF-2 binding inhibition in endothelial cell-conditioned media. (A) Perlecan antisense construct. (B) Western blot of conditioned media from untransfected BAE cells, and clones transfected with the antiperlecan sequences (BAE-AP1–6) or the vector alone (BAE-NEO1–4). All samples were normalized for cell number (1 × 104 cells) and subjected to heparinase treatment before gel electrophoresis. (C) FGF-2 binding to smooth muscle cells is inhibited by conditioned media from BAE-NEO cells to a greater extent than by that from BAE-AP cells 125I-FGF-2 binding (2 ng/ml) to VSMCs was conducted as described (10). VSMCs were plated (5 × 104 cells/well) in 24-well culture plates (Costar) in DMEM, 10% calf serum. When the cells reached confluence (day 3), the medium was removed, and the monolayers were washed with cold (0°C) binding buffer (DMEM/25 mM Hepes/0.05% gelatin), and binding of 125I-FGF-2 was conducted in the presence of conditioned media (equivalent to the media conditioned by 3.5 × 104 cells) for 2.5 h at 4°C. The data shown are the averages ± SEM of triplicate determinations. Control binding represented the amount of 125I-FGF-2 bound to cells in the absence of any conditioned media (100%).
BAE cell clones transfected with the perlecan antisense vector (BAE-AP3), and the empty vector (BAE-NEO1) were studied in detail. Proteoglycan secretion by BAE-NEO1 and BAE-AP3 was quantified after partial purification (Fig. 2A). Although total protein production was similar in the two clones, secretion of proteoglycan into the media was reduced by more than 95% in the BAE-AP3 cells compared with the controls (0.1 vs. 2.6 μg/106 cells/24h for BAE-AP3 and BAE-NEO1, respectively). This was paralleled by a ≈90% decrease in the amount of perlecan core protein (Fig. 2B). No changes in the levels of other heparan sulfate proteoglycan core proteins were noted in the AP cells compared with controls, as determined by using an antibody that recognized desaturated uronic acid residues on protein cores after heparinase III digestion (3G10, Seikagaku Kogyo, Tokyo) (26) (data not shown). The ability of these cells to inhibit FGF-2-induced VSMC mitogenesis in a coculture system was also analyzed as a measure of the biological activity of these cells. FGF-2 significantly stimulated VSMC proliferation in the absence of BAE cells; however, FGF-2-mediated mitogenesis was dramatically reduced in the presence of BAE or BAE-NEO1 cells. In the presence of BAE-AP3 cells, FGF-2 stimulated VSMC mitogenesis to nearly the same extent as that observed in the absence of cells.
Figure 2.

BAE-AP3 shows significantly reduced proteoglycan synthesis and is less able to inhibit FGF-2 stimulation of smooth muscle cell proliferation compared with BAE-NEO1. (A) Serum-free conditioned media was generated from BAE-NEO1 and BAE-AP3 cells, assayed for total protein concentration, and then subjected to ion exchange chromatography to isolate the proteoglycan fraction. The total amount of glycosaminoglycan and protein determined in BAE-NEO1 (shaded bars) and BAE-AP3 (filled bars) (see Materials and Methods). (B) Western blot for perlecan of conditioned media-derived proteoglycan from BAE-NEO1 (left lane) and BAE-AP3 (right lane). (C) Inhibition of FGF-2-mediated smooth muscle cell proliferation by BAE, BAE-NEO1, and BAE-AP2 cells when grown in coculture. VSMC proliferation was analyzed in coculture with BAE cells as described (10). Confluent monolayers of BAE cells were established in Falcon cell culture inserts (0.45 μm pore size, Cyclopore Membrane, Falcon). Sparse VSMC cultures (4,000 cells/well) were established in separate six-well plates (Falcon). The media were changed to DMEM, 5% calf serum in both the BAE and VSMC cultures, and the BAE-containing culture inserts were transferred on top of the underlying VSMC. FGF-2 was administered continuously by controlled release from an alginate microsphere system within the lower (VSMC containing) chamber (42). VSMC cell number was determined after 5 days by counting trypsin suspended cells with a Coulter Counter (Model Z2). The results represent the average ± SEM stimulation of cell growth by FGF-2 [FGF-2 stimulation = (cell no. with FGF-2/cell no. without FGF-2)]. For example, the VSMC number in wells with “no cells” in the coculture insert was 1.5 × 104 and 3.3 × 104 per well in the absence and presence of FGF-2, respectively.
BAE-AP3 cells were cultured within Gelfoam matrices (Fig. 3A). BAE-AP3 cells grew well within the three-dimensional Gelfoam matrices with cell doublings observed approximately every 36 h and a saturation density of ≈1 × 106 cell/cm3 Gelfoam. Cell viability remained at 95–100% during the 2-week culture course. The growth kinetics, saturation density, and viability are similar to those we have previously reported for BAE cells (14, 15). Moreover, BAE-AP3 cells cultured within Gelfoam matrices retained their ability to take up acetylated low-density lipoprotein (LDL) (Fig. 3B).
Figure 3.
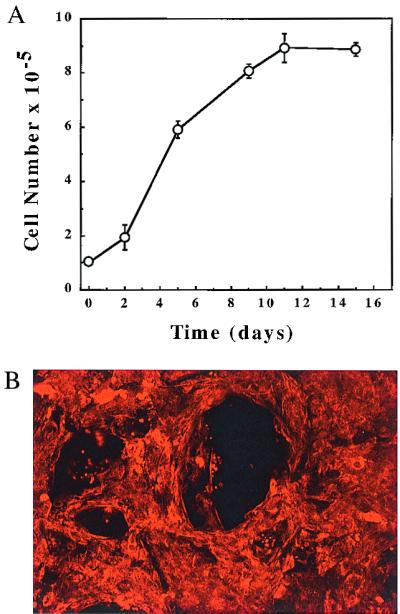
Characterization of BAE-AP3 cells. (A) BAE-AP3 cells cultured within Gelfoam matrices followed a growth pattern similar to normal BAEs (14, 15). The number of cells attached to the Gelfoam was determined with a hemacytometer after digestion with collagenase (1 mg/ml, Type I, Worthington). Cell viability was assessed by trypan blue exclusion. Cell growth reached a plateau after approximately 10 days in culture. (B) The preservation of endothelial cell identity was determined by uptake of acetylated LDL. Gelfoam matrices were incubated with DiI-Ac-LDL (Biomedical Technologies, Stoughton, MA) at a concentration of 10 μg/ml for 6 h at 37°C and fixed in 3% paraformaldehyde. Confocal laser-scanning microscopy (Bio-Rad MRC 600), using standard rhodamine excitation and emission filters, was used to visualize positive cells within the matrices after 2 weeks in culture. The computer was programmed to collect images from the surface at 25-μm intervals. The sections were then combined into a single image by using customized software. Red cells indicate positive Ac-LDL uptake. Empty control Gelfoam matrices showed no positive staining (not shown). (×200).
BAE-AP3 Cells Show Reduced Inhibition of Intimal Thickening and Thrombosis.
Pigs were randomly selected to receive BAE-AP3/Gelfoam, BAE-NEO1/Gelfoam, or Gelfoam implants without cells after balloon injury (15). All 12 pigs recovered well from the surgical procedure and gained weight during the 28-day postoperative period. Host immune responses to the endothelial implants were similar to that previously reported for BAE implants (15). The bovine cell grafts displayed infiltration of leukocytes 28 days after implantation. Moreover, the host response was similar when animals were implanted with either BAE-NEO1 or BAE-AP3 (data not shown).
Morphometric analysis of each artery showed no significant differences in the injury index between the three experimental groups (Table 1). A significant portion (22%) of the carotid arteries treated with BAE-AP3 implants showed extensive occlusive organized thrombus at the site of injury, which is comparable to that observed for arteries treated with empty Gelfoam matrices (Fig. 4A) (15). Occlusive organized thrombus was not observed in any of the arteries that received BAE-NEO1 implants. Differences were also noted in the extent of intimal thickening among the three groups (Fig. 4B, Table 1). The restenosis index of the control animals treated with Gelfoam containing no cells (1.34 ± 0.16) was similar to that previously reported for both balloon injury alone and balloon injury followed by treatment with empty Gelfoam matrices (15). Arteries treated with BAE-NEO1 implants reduced the restenosis index by 42% (0.78 ± 0.05, P < 0.05) compared with empty Gelfoam controls (Fig. 4C), which was similar to that previously observed with unmodified BAE cells [49% (15)]. In contrast, the restenosis index of the arteries treated with BAE-AP3 implants (0.99 ± 0.05) was not significantly reduced (P = not significant) compared with control (Fig. 4C). Thus, BAE-NEO1 implants showed a significantly greater reduction in intimal thickening than the BAE-AP3 implants (P < 0.05). These results suggest that endothelial cell-secreted perlecan plays an important role in regulating vascular restenosis and thrombosis after balloon injury but regulates the various aspects of the vascular response to injury differentially.
Table 1.
Histopathological characteristics of porcine carotid arteries after balloon injury
| Characteristics | Empty Gelfoam | BAE-AP3 | BAE-NEO1 |
|---|---|---|---|
| EEL area, mm2 | 9.15 ± 0.90 | 8.86 ± 0.71 | 9.90 ± 0.39 |
| IEL length, mm | 6.51 ± 0.62 | 6.58 ± 0.41 | 7.20 ± 0.36 |
| Fracture length, mm | 1.36 ± 0.15 | 1.27 ± 0.09 | 1.40 ± 0.24 |
| Intima area, mm2 | 1.50 ± 0.2 | 1.0 ± 0.13 | 0.78 ± 0.14‡ |
| Media area, mm2 | 4.10 ± 0.35 | 4.08 ± 0.25 | 4.20 ± 0.20 |
| Lumen area, mm2 | 2.59 ± 0.49 | 2.58 ± 0.32 | 3.56 ± 0.45 |
| Residual lumen, ratio | 0.65 ± 0.04 | 0.73 ± 0.02 | 0.82 ± 0.04‡ |
| Injury index* | 0.20 ± 0.01 | 0.19 ± 0.01 | 0.20 ± 0.03 |
| Restenosis index† | 1.34 ± 0.16 | 0.99 ± 0.05 | 0.78 ± 0.05‡§ |
The intimal (I), medial (M), lumen (L), and external elastic lamina (EEL) areas as well as the internal elastic lamina (IEL) circumference and IEL fracture length (F) were measured by using computerized digital planimetry with a video microscope and customized software (15). Morphometric measurements were performed by an observer blinded to the treatment groups. The extent of injury was represented by the fracture length of the IEL, normalized for the size of the artery by the circumference of the IEL: injury index = F/IEL. Intimal hyperplasia was also normalized by the total artery wall area: I/(I+M). A restenosis index (43) was established taking into account the degree of injury: restenosis index = [I/(I+M)]/(F/IEL). Arteries with an intact IEL were excluded from all analyses. The residual lumen was also measured, which reflected the change in vessel geometry after injury and repair (44). The residual lumen was defined as L/(L+I). Arteries that were occluded with organized thrombus were not included in the intimal thickening analyses.
Injury index = IEL fracture length/IEL circumference.
Restenosis index = [I/(I+M)]/(F/IEL).
P < 0.05 compared to control arteries.
P < 0.05 compared to BAE-AP3 arteries.
Figure 4.

Effects of BAE-NEO1 and BAE-AP3 on thrombosis and intimal thickening of balloon-injured porcine carotid arteries. (A) Bar graph shows a decrease in occlusive thrombosis for arteries treated with BAE-NEO1 (P < 0.05) and no effect for arteries treated with the anti-perlecan transfected cells, compared with control. (B) Photomicrographs of cross sections of arteries show the effects of perivascular endothelial cell implants on neointimal formation 28 days after balloon injury of porcine carotid arteries [Verhoeff's elastin stain (×100)]. Control Gelfoam (no cells) implants (Top), BAE-AP3 cell implants (Middle), and BAE-NEO1 cell implants (Bottom). I = Intima, M = Media. (C) Bar graph shows that BAE-NEO1 significantly reduced the restenosis index when compared with BAE-AP3 and control Gelfoam treated arteries (P < 0.05). BAE-AP3-treated arteries did not significantly reduce the restenosis index when compared with control. Statistical analysis comparing treatment groups used a nonpaired t test. Values of P < 0.05 (two-tailed analysis) were considered significant.
Discussion
Endothelial cell dysfunction is considered a decisive factor in the generation and propagation of atherosclerosis and the accelerated arterial obstructions that follow mechanical vascular interventions (1–4). Recent evidence indicates that endothelial secreted compounds might mediate vascular repair (9, 10, 27–29). Among the various candidate compounds, considerable attention has been focused on the heparan sulfate proteoglycan perlecan. The antiproliferative effects of heparin on smooth muscle cells have long been considered a reflection of the potential role of endothelial cell-produced heparan sulfate (18, 20, 30–32). Indeed, endothelial cell-derived heparan sulfate proteoglycans are potent inhibitors of smooth muscle cell proliferation in vitro (9). Furthermore, isolated perlecan regulates the extracellular transport and cell surface binding of a potent smooth muscle cell mitogen, FGF-2 (19, 33). The antithrombotic effect of heparin is also likely to reflect an important function of endothelial heparan sulfate in modulating vascular repair. The present study set out to evaluate the hypothesis that the secretion of perlecan is required for endothelial cells to maintain biochemical control of vascular injury. To test this hypothesis, genetically modified endothelial cells were generated to express reduced amounts of perlecan. Cells with reduced perlecan expression were less able to inhibit FGF-2 binding and activity in cultured VSMC when compared with the nontransfected (parental) cells (Fig. 1).
BAE cells were cultured within Gelfoam matrices and placed adjacent to balloon-injured porcine arteries so that their effects on the vascular response to injury could be evaluated. The swine model of vascular injury offered advantages over other animal models for the present study. Its complexity and time course of response provide a series of specific reactions that can be quantified in a manner reflective of the histologic and tissue reaction to human vascular injury (34–36). An interesting divergence of biological effect of the modified BAE cell implants was observed. Endothelial-cell implants with reduced perlecan expression were less effective than implants containing parental cells at inhibiting experimental restenosis (Fig. 4) yet showed a complete loss of the antithrombotic effect observed with the parental-cell implants (Fig. 4). It is interesting to note that the effects of the endothelial cells are sensed within the lumen, at a distance from the implant. One possibility is that endothelial cell products, such as perlecan, are able to move throughout the blood vessel wall, or that these factors alter the bioavailability and distribution of endogenous vasoregulatory compounds such as growth factors (33, 37). The experimental model presented here should provide a useful method for systematically testing the roles of individual endothelial-derived components in the complex environment of the blood vessel wall.
These results not only implicate perlecan directly as an important regulator of intimal hyperplasia and occlusive thrombosis that follow vascular interventions but also show the complexity of the regulatory role of this compound. Perlecan is absolutely necessary to control the occlusive thrombotic reaction to deep vascular injury, but its contribution to the hyperplastic reaction is only a part of the overall regulation that the intact endothelium elicits. It is likely that endothelial control over intimal thickening results from a combination of perlecan and other secreted cell-based products such as nitric oxide, endothelins, prostaglandins, and a myriad of growth factors, cytokines, and vasoreactive agents (1, 38–41). By removing an important factor (perlecan) that directly interacts in these processes, the implanted cells displayed a decreased ability to reduce intimal thickening. Our data reflect that the cooperative action of endothelial cell factors is necessary to observe inhibition of intimal thickening. These observations could be clinically relevant and may explain why single endothelial-derived products, aimed at one of the cellular events thought to be involved in either thrombosis or smooth muscle cell proliferation, do not lead to full control of restenosis. This is not surprising when one considers the large number of cells and biological events involved in restenosis (2, 3, 8, 11, 12). Cell-based therapies offer new tools for added insight into vascular repair and, when coupled with molecular modification technology, may enable us to dissect further the biology of vascular injury.
Acknowledgments
We acknowledge National Institutes of Health grants (HL56200, HL 60407, and GM/HL49039) and Reprogenesis, Inc. for financial support. We thank Danielle Bornstein, Philip Seifert, and Cynthia Richmond for expert technical assistance. We thank Gabriel Belfort, Dr. Gail Sonenshein, and members of her laboratory for assistance.
Abbreviations
- FGF-2
fibroblast growth factor-2
- BAE
bovine aortic endothelial cells
- LDL
low density lipoprotein
- VSMC
vascular smooth muscle cells
- AP
antiperlecan vector
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
References
- 1.Rubanyi G M. J Cardiovasc Pharmacol. 1993;22:S1–S14. doi: 10.1097/00005344-199322004-00002. [DOI] [PubMed] [Google Scholar]
- 2.Fischell T A, Derby G, Tse T M, Stadius M L. Circulation. 1988;78:1323–1334. doi: 10.1161/01.cir.78.6.1323. [DOI] [PubMed] [Google Scholar]
- 3.el-Tamimi H, Davies G J, Hackett D, Sritara P, Bertrand O, Crea F, Maseri A. Circulation. 1991;84:1198–1202. doi: 10.1161/01.cir.84.3.1198. [DOI] [PubMed] [Google Scholar]
- 4.Castaneda-Zuniga W R, Formanek A, Tadavarthy M, Vlodaver Z, Edwards J E, Zollikofer C, Amplatz K. Radiology. 1980;135:565–571. doi: 10.1148/radiology.135.3.7384437. [DOI] [PubMed] [Google Scholar]
- 5.Furchgott R F, Zawadzki J V. Nature (London) 1980;288:373–376. doi: 10.1038/288373a0. [DOI] [PubMed] [Google Scholar]
- 6.Autio I, Malo-Ranta U, Kallioniemi O P, Nikkari T. Artery. 1989;16:72–83. [PubMed] [Google Scholar]
- 7.Cybulsky M I, Gimbrone M A., Jr Science. 1991;251:788–791. doi: 10.1126/science.1990440. [DOI] [PubMed] [Google Scholar]
- 8.Casscells W. Circulation. 1992;86:723–729. doi: 10.1161/01.cir.86.3.723. [DOI] [PubMed] [Google Scholar]
- 9.Castellot J J, Addonizio M L, Rosenberg R D, Karnovsky M J. J Cell Biol. 1981;90:372–379. doi: 10.1083/jcb.90.2.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nugent M A, Karnovsky M J, Edelman E R. Circ Res. 1993;73:1051–1060. doi: 10.1161/01.res.73.6.1051. [DOI] [PubMed] [Google Scholar]
- 11.O'Keefe J H, Jr, Hartzler G O. J Invest Cardiol. 1989;1:109–122. [Google Scholar]
- 12.McBride W, Lange R A, Hillis L D. N Engl J Med. 1988;318:1734–1738. doi: 10.1056/NEJM198806303182606. [DOI] [PubMed] [Google Scholar]
- 13.Langer R, Vacanti J P. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 14.Nathan A, Nugent M A, Edelman E R. Proc Natl Acad Sci USA. 1995;92:8130–8134. doi: 10.1073/pnas.92.18.8130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nugent H M, Rogers C, Edelman E R. Circ Res. 1999;84:384–391. doi: 10.1161/01.res.84.4.384. [DOI] [PubMed] [Google Scholar]
- 16.Noonan D M, Fulle A, Valente P, Cai S, Horigan E, Sasaki M, Yamada Y, Hassell J R. J Biol Chem. 1991;266:22939–47. [PubMed] [Google Scholar]
- 17.Iozzo R V. Annu Rev Biochem. 1998;67:609–652. doi: 10.1146/annurev.biochem.67.1.609. [DOI] [PubMed] [Google Scholar]
- 18.Kojima T, Leone C W, Marchildon G A, Marcum J A, Rosenberg R D. J Biol Chem. 1992;267:4859–4869. [PubMed] [Google Scholar]
- 19.Forsten K E, Courant N A, Nugent M A. J Cell Physiol. 1997;172:209–220. doi: 10.1002/(SICI)1097-4652(199708)172:2<209::AID-JCP8>3.0.CO;2-S. [DOI] [PubMed] [Google Scholar]
- 20.Benitz W E, Kelley R T, Anderson C M, Lorant D E, Bernfield M. Am J Respir Cell Mol Biol. 1990;2:13–24. doi: 10.1165/ajrcmb/2.1.13. [DOI] [PubMed] [Google Scholar]
- 21.Nugent M A, Edelman E R. Biochemistry. 1992;31:8876–8883. doi: 10.1021/bi00152a026. [DOI] [PubMed] [Google Scholar]
- 22.Ross R. J Cell Biol. 1971;50:172–186. doi: 10.1083/jcb.50.1.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Iozzo R V, Pillarisetti J, Sharma B, Murdoch A D, Danielson K G, Uitto J, Mauviel A. J Biol Chem. 1997;272:5219–5228. doi: 10.1074/jbc.272.8.5219. [DOI] [PubMed] [Google Scholar]
- 24.Sharma B, Handler M, Eichstetter I, Whitelock J M, Nugent M A, Iozzo R V. J Clin Invest. 1998;102:1599–1608. doi: 10.1172/JCI3793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Farndale R W, Buttle D J, Barrett A J. Biochim Biophys Acta. 1986;883:173–177. doi: 10.1016/0304-4165(86)90306-5. [DOI] [PubMed] [Google Scholar]
- 26.David G, Bai X M, Van der Schueren B, Cassiman J J, Van den Berghe H. J Cell Biol. 1992;119:961–975. doi: 10.1083/jcb.119.4.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ferns G, Reidy M, Ross R. Am J Pathol. 1990;137:403–413. [PMC free article] [PubMed] [Google Scholar]
- 28.Bjornsson T D, Dryjski M, Tluczek J, Mennie R, Ronan J, Mellin T N, Thomas K A. Proc Natl Acad Sci USA. 1991;88:8661–8665. doi: 10.1073/pnas.88.19.8651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fishman J A, Ryan G B, Karnovsky M J. Lab Invest. 1975;32:339–351. [PubMed] [Google Scholar]
- 30.Clowes A W, Karnovsky M J. Nature (London) 1977;265:625–626. doi: 10.1038/265625a0. [DOI] [PubMed] [Google Scholar]
- 31.Clowes A W, Karnovsky M J. J Surg Res. 1978;24:161–168. doi: 10.1016/0022-4804(78)90169-5. [DOI] [PubMed] [Google Scholar]
- 32.Fritze L, Reilly C, Rosenberg R. J Cell Biol. 1985;100:1041–1049. doi: 10.1083/jcb.100.4.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dowd C J, Cooney C L, Nugent M A. J Biol Chem. 1999;274:5236–5244. doi: 10.1074/jbc.274.8.5236. [DOI] [PubMed] [Google Scholar]
- 34.Leach C M, Thorburn G D. Prostaglandins. 1982;24:47–59. doi: 10.1016/0090-6980(82)90176-9. [DOI] [PubMed] [Google Scholar]
- 35.Steele P M, Chesebro J H, Stanson A W, Holmes D R, Jr, Dewanjee M K, Badimon L, Fuster V. Circ Res. 1985;57:105–112. doi: 10.1161/01.res.57.1.105. [DOI] [PubMed] [Google Scholar]
- 36.Karas S P, Gravanis M B, Santoian E C, Robinson K A, Anderberg K A, King S B d. J Am Coll Cardiol. 1992;20:467–474. doi: 10.1016/0735-1097(92)90119-8. [DOI] [PubMed] [Google Scholar]
- 37.Lovich M A, Philbrook M, Sawyer S, Weselcouch E, Edelman E R. Am J Physiol. 1998;275:H2236–H2242. doi: 10.1152/ajpheart.1998.275.6.H2236. [DOI] [PubMed] [Google Scholar]
- 38.Bobik A, Campbell J H. Pharmacol Rev. 1993;45:1–42. [PubMed] [Google Scholar]
- 39.Gertler J P, Abbott W M. J Surg Res. 1992;52:89–95. doi: 10.1016/0022-4804(92)90284-7. [DOI] [PubMed] [Google Scholar]
- 40.Furchgott R F, Vanhoutte P M. FASEB J. 1989;3:2007–2018. [PubMed] [Google Scholar]
- 41.Nabel E G. Am J Cardiol. 1991;68:6C–8C. doi: 10.1016/0002-9149(91)90217-9. [DOI] [PubMed] [Google Scholar]
- 42.Edelman E R, Mathiowitz E, Langer R, Klagsbrun M. Biomaterials. 1991;12:619–626. doi: 10.1016/0142-9612(91)90107-l. [DOI] [PubMed] [Google Scholar]
- 43.Bonan R, Paiement P, Scortichini D, Cloutier M J, Leung T K. Am Heart J. 1993;126:1334–1340. doi: 10.1016/0002-8703(93)90531-d. [DOI] [PubMed] [Google Scholar]
- 44.Schneider J E, Berk B C, Gravanis M B, Santoian E C, Cipolla G D, Tarazona N, Lassegue B, King S B d. Circulation. 1993;88:628–637. doi: 10.1161/01.cir.88.2.628. [DOI] [PubMed] [Google Scholar]