

Abstract
A new type of self-assembling peptide (sapeptide) scaffolds that serve as substrates for neurite outgrowth and synapse formation is described. These peptide-based scaffolds are amenable to molecular design by using chemical or biotechnological syntheses. They can be tailored to a variety of applications. The sapeptide scaffolds are formed through the spontaneous assembly of ionic self-complementary β-sheet oligopeptides under physiological conditions, producing a hydrogel material. The scaffolds can support neuronal cell attachment and differentiation as well as extensive neurite outgrowth. Furthermore, they are permissive substrates for functional synapse formation between the attached neurons. That primary rat neurons form active synapses on such scaffold surfaces in situ suggests these scaffolds could be useful for tissue engineering applications. The buoyant sapeptide scaffolds with attached cells in culture can be transported readily from one environment to another. Furthermore, these peptides did not elicit a measurable immune response or tissue inflammation when introduced into animals. These biological materials created through molecular design and self assembly may be developed as a biologically compatible scaffold for tissue repair and tissue engineering.
Keywords: biological materials, cell attachment, molecular material design, primary neurons, PC12 cells
The development of new biological materials, particularly those biologically compatible scaffolds that serve as permissive substrates for cell growth, differentiation, and biological function, is a key area for advancing medical technology. Recently, attempts have been made to develop new biologically compatible scaffolds for controlled drug release, tissue repair, and tissue engineering (1–3). These biomaterials have altered our concept of medical treatment and health care technology (4–6). Because many diseases cannot be treated solely by small-molecule drugs, cell-based therapy is emerging as an alternative approach. Several types of collagen-based biological scaffolds, their derivatives, and other biocompatible polymers showed great promise in this area (6–8). The ideal biologically compatible scaffold for supporting cell attachment and growth should meet several criteria: (i) components should be derived from biological sources; (ii) basic units should be amenable to design and modification to achieve specific needs; (iii) the scaffold should exhibit a controlled rate of biodegradation of the material; (iv) it should have no cytotoxicity; (v) it should possess properties that promote cell-substrate interactions; (vi) it should elicit minimal immune responses and inflammation; (vii) it should have easy and scaleable material production, purification, and processing; (viii) it should be readily transportable; and (ix) it should be chemically compatible with aqueous solutions and physiological conditions.
We have described previously a class of biomaterials that are made through spontaneous assembly of ionic self-complementary oligopeptides (9, 10). These biological material scaffolds are hydrogels that consist of greater than 99% water content (peptide content 1–10 mg/ml). They formed hydrogels when the sapeptide solution was exposed to physiological media or salt solution (9, 10). The constituents of the hydrogel scaffold are amphiphilic oligopeptides that have alternatively repeating units of positively charged lysine or arginine and negatively charged aspartate and glutamate. These peptides are designated as type I self-assembling peptides (sapeptides) because they contain amino acid sequences that facilitate the formation of a hydrogel scaffold through spontaneous molecular self assembly. The scaffolds consist of β-sheet ionic sapeptides that contain 50% charged residues (9, 10). These sapeptides are characterized by their periodic repeats of alternating ionic hydrophilic and hydrophobic amino acids. Thus, the β-sheets have distinct polar and nonpolar surfaces (9–11). The first molecule of this type was found as a segment in a yeast protein, zuotin. Zuotin was originally characterized by binding to left-handed Z-DNA (12). A number of additional type I sapeptides have been designed, synthesized, and characterized for salt-facilitated matrix formation (9, 10). Several sapeptide scaffolds support cell attachment of a variety of mammalian and avian primary and tissue culture cells (10).
We have investigated cell attachment and differentiation, neurite outgrowth, and the formation of functional synapses of primary and cultured neuronal cells on sapeptide scaffolds. Neurite outgrowth generally requires the attachment of neurons to a permissive substrate. Several extracellular matrix (ECM) proteins, such as laminin, fibronectin, and collagen, that support neurite outgrowth in situ have been identified (13–17). These ECM proteins contain specific motifs that are particularly favorable for cell attachment and neurite outgrowth (16, 17). ECM molecules and their adhesion domain fragments, either conjugated with polymers or alone, have also been used to coat surfaces (i.e., glass, polystyrene plastic) that otherwise provide poor support for neurite outgrowth (16, 17). Other coating materials such as poly-l-lysine and Matrigel (Becton Dickinson) also provide good support for neurite outgrowth (18–20). A number of in vitro preparations have been used to examine the interactions between neurons and individual protein-derived materials as well as their effect on neurite outgrowth (17, 20, 21). However, such in vitro systems present serious limitations for certain applications, such as tissue repair and tissue engineering. Once neurons attach to coated surfaces, they cannot be transported readily to tissues without the incorporation of nonbiological materials, such as polymer fibers.
Materials and Methods
Peptide Design and Synthesis.
Peptides of Arginine–Alanine–Aspartate (RAD)16-I and RAD16-II have the sequences:
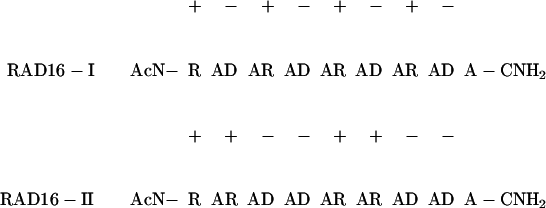 |
RAD16-I has a modulus of one based on the formula of (RADA)n, whereas RAD16-II has a modulus of two based on the formula (RARADADA)n. These sequences were derived from a segment found in a yeast protein, zuotin, EAK16-II (n-AEAEAKAKAEAEAKAK-c; modulus two). In this study, the N and C termini were blocked by acetylation and amidation to prevent rapid degradation. The motif RAD was incorporated to mimic the known cell adhesion motif RGD that is found in many ECM proteins (17). These sapeptides were synthesized by using an Applied Biosystems Model 430A solid-state-phase peptide synthesizer, were HPLC purified, and were analyzed by MS as described previously (9). The sapeptide scaffolds were processed by cannula injection of 1–10 mg/ml of water-dissolved sapeptide into PBS (5 mM Na-PO4/150 mM NaCl) or RPMI 1640 cell-culture medium (GIBCO/Life Technologies, Gaithersburg, MD). For visualization, the matrices were stained with 5 μg/ml Congo red solution (Sigma).
PC12 Cell Culture and Scanning Confocal Microscopy.
Rat PC12 cells (22) were maintained in RPMI 1640 medium supplemented with 10% heat-inactivated horse serum and 5% FBS in tissue culture flasks coated with Matrigel. For some experiments, the cells were preprimed with nerve growth factor (NGF 2.5S, 50 ng/ml medium, 10 days, Becton Dickinson). Cells were dissociated by trypsin treatment and introduced to the sapeptide scaffolds formed in the medium. The cell-bearing matrices were transferred to 24-well plates containing fresh medium (with or without 50 ng/ml NGF) 24 h after plating. The cells were labeled with the fluorescent vital dye Cell Tracker-CMFDA (Molecular Probes). The sapeptide scaffolds were moved from cell-culture wells to glass coverslips for scanning confocal microscopy image collection. Cell morphology and neurite outgrowth were assessed by scanning laser confocal microscopy (MRC-600, Bio-Rad).
Primary Cell Culture.
The cerebellum and hippocampus from 7-day-old mice (maintained in accordance with National Institutes of Health guidelines) (Charles River Breeding Laboratories) were dissected, removing the meninges, and mechanically dissociated in serum-free medium after incubation in an EDTA-PBS buffer (5 min at 37°C, Versene 1/5,000, GIBCO/Life Technologies). Cells (approximately 1.4–1.5 × 106 cells/ml) were introduced to sapeptide scaffolds formed in the medium in culture dishes (6-, 12-, or 24-well culture clusters; Falcon) by modification of the methods described in refs. 23–25. To prevent glial proliferation, 10 μM cytosine arabinoside was added to the medium 48 h after plating. Embryonic chick dorsal root ganglion cells were prepared by methods described previously (18, 20).
Sympathetic neuronal cultures were prepared from neonatal rats by modification of methods described in refs. 20, 21, and 26. The superior cervical ganglion was dissected from these neonatal Fisher 344 rats and treated with 1.5 mg/ml collagenase (Type I, Worthington) and 5 mg/ml dispase (Life Technologies) for 1 h at 37°C. Schwann cell and fibroblast density was reduced by preplating. Sympathetic neurons were introduced to the sapeptide scaffolds formed in 24-well plates with wells containing 1 ml of cell culture medium. The medium contained L15CO2, 10% FBS, 6 μg/ml dextrose, 2 mM glutamine, 100 units/ml penicillin, 100 μg/ml streptomycin, 1 μg/ml 6,7, dimethyl-5, 6, 7, 8-tetrahydropterine (Calbiochem), 5 μg/ml glutathione (Sigma), 100 μg/ml l-ascorbic acid, 1 μM cytosine arabinofuranoside (Sigma), and 50 ng of 2.5S NGF (Collaborative Research).
Optical Labeling of Synapse Formation and Activity.
Standard procedures for culturing hippocampal neurons and FM1–43 dye staining were used (26). Briefly, FM1–43 dye incubation was performed at room temperature (23–25°C) from spindle-shaped pyramidal neurons kept in culture for 14–20 days. The small experimental chamber (0.25 ml) was continuously perfused (0.25 ml/min) with Tyrode solution containing 25 mM Hepes (pH 7.4 with NaOH), 124 mM NaCl, 5 mM KCl, 2 mM CaCl2, 1 mM MgCl2, and 30 mM glucose. Tetrodotoxin (1 μM), 5 μM d-carboxypiperazin-propyl-phosphonic acid, and 50 μM picrotoxin were added to block action potentials, the NMDA component of synaptic current, and Cl− channels mediating inhibitory synaptic currents. The hippocampal neurons were stained with 10 μM FM1–43 in 70 mM [K+] for 1 min, then washed for >5 min. The experimental recorded image was obtained through an inverted Olympus Fluo-view Personal Confocal Microscope, by using an Olympus ×40 planapochromat water-immersion lens (1.15 numerical aperture). Images were captured with software provided by Olympus.
Sapeptide in Vivo Injection.
Fisher 344 rats (male, 10 wk old, 135–142 g, n = 3), maintained in accordance with National Institutes of Health guidelines, were used in these experiments. Intramuscular peptide (1 mg/ml) injections were carried out by using 20 μl each in rat nos. 35 and 36 (RAD sapeptide in the right leg, sapeptide EAK in the left leg, both sapeptide solutions in hind limbs of the right and left legs). Two injections, 20 μl each, of RAD sapeptide in the right leg and 20 μl of EAK sapeptide in the left leg were given in rat no. 37. The time interval between injections and perfusion was 9 days for rat no. 35 and 5 wk for rats nos. 36 and 37. These rats did not exhibit any movement deficits within the 5 wk after the injections. The rats were then perfused transcardially under deep barbital anesthesia with 100 ml of PBS, followed by 400 ml of 4% paraformaldehyde and 0.2% picric acid in 0.1 M PBS (pH 7.5) followed by 150 ml of 20% sucrose. The leg muscle was dissected, and 10-μm tissue sections of the entire muscle were cut with a cryostat. The sections were stained with 0.1% thionin, Nissl or Congo red, and were examined under a high-power optical microscope for signs of mononuclear cell infiltration and/or muscle necrosis.
Peptides were tested for immunogenicity by coupling them to BSA, and serial injections were carried out in rabbits and goats.
Results
Sapeptide Matrix Formation.
Isobuoyant macroscopic scaffolds were processed from the sapeptide RAD16-II in the presence of millimolar monovalent salts. The sapeptide scaffolds were fabricated into various geometric forms with relatively even thickness, either as tapes (Fig. 1A), strings (Fig. 1B), or sheets (Fig. 1C). The geometry and dimensions of the macroscopic matrices are governed by the concentration of sapeptide, amount of sapeptide solution applied, concentration of monovalent salt, and dimensions of the processing apparatus.
Figure 1.

Fabrication of sapeptide scaffolds. The sapeptide dissolved in water was injected through a device (two pieces of thin wire spaced 5 mm apart are sandwiched between two glass slides) into PBS to produce a tape-shaped macroscopic sapeptide scaffold that was stained with Congo red (A). The dimensions of the tape shown in A are approximately 8 cm in length, 0.5 cm in width, and 0.3 mm in thickness. The sapeptide scaffold was fabricated as a rope under similar conditions when the aqueous sapeptide solution was introduced into PBS using a 3-ml syringe (B). The dimensions of the sapeptide scaffold string shown are 18 cm in length and 2 mm in diameter. Sapeptide scaffold was fabricated as a sheet (C). Part of the sheet was removed and used for cell attachment experiments. The scale in centimeters is shown below the sheet structure.
Sapeptide scaffolds are formed as sheets up to several square centimeters and strings greater than 20 centimeters in length. These β-sheet ionic self-complementary sapeptides rapidly self assemble into macroscopic scaffolds in the presence of millimolar concentrations of monovalent salts (Fig. 1), including NaCl and KCl, at levels that are found in physiological solutions (9, 10).
Sapeptide Scaffold Examined by High-Resolution Scanning Electron Microscopy (SEM).
When the RAD16-I and RAD16-II sapeptide scaffolds were examined by using SEM, the high-resolution structure of the materials was revealed. The RAD sapeptide scaffolds consist of individual interwoven fibers (Fig. 2), similar to the previously observed matrix of EAK16-II (9, 10), EFK8, and several other sapeptide matrices. The individual fibers are approximately 10–20 nm in diameter. The fiber density correlates with the concentration of sapeptide solution that is used to produce the materials (data not shown).
Figure 2.

Determination of the sapeptide scaffold structure by scanning electron microscopy. Sapeptide scaffolds were subjected to serial dehydration by using 5–100% ethanol immersion followed by liquid CO2 immersion for scanning electron microscopy analysis. Six photographs are shown (a–f) at increasing magnification (×2,000–60,000). At lower magnification, the scaffold has a felt-like appearance. Higher magnification reveals interwoven sapeptide fibers that are approximately 10–20 nm in diameter. The scale bar on each image is included: 10 μm (a), 1 μm (b–e) to 100 nm (f). Molecular models of the ionic self-complementary peptide RAD16-II in β-strand form with distinct polar and nonpolar sides are shown (g). The proposed interpeptide interaction is shown (h).
How can such short peptides, approximately 5 nm in length, form macroscopic materials? We have proposed a molecular model based on our experimental observations to explain the formation of a macroscopic sapeptide scaffold from β-sheet ionic self-complementary peptides (9, 10). The formation and stability of macroscopic matrices is facilitated by: (i) intermolecular hydrogen bonding between peptide backbones in conventional β-sheets, (ii) formation of intermolecular ionic bonds from the side chains on one side of the β-sheet between regularly repeating units, with positively and negatively charged residues, (iii) formation of hydrophobic interactions between the alanine methyl groups on the other side of the sheet, (iv) formation of overlap interactions among the individual peptides, and (v) coordination by salt ions between intermolecular ionic groups. In addition, peptide length contributes positively to macroscopic matrix formation and stability (9, 10). These features are summarized in an updated molecular model for the self assembly of macroscopic sapeptide scaffolds from the β-sheet ionic self-complementary sapeptide RAD16-II (Fig. 2 g and h). In contrast to the well-ordered sapeptide scaffolds formed from Type I sapeptides, peptides composed exclusively of alternating positively charged residues lysine, arginine (or exclusively negatively charged glutamate or aspartate), and hydrophobic residues such as valine, phenylalanine, or tyrosine do not self assemble to form macroscopic peptide matrices. Instead, these peptides form disordered precipitates in the presence of salts or pH changes (30–32).
Sapeptide Scaffolds Support Extensive Neurite Outgrowth.
We demonstrated previously that a number of mammalian cell types could attach to sapeptide scaffolds (10). Here we demonstrate that the sapeptide scaffolds support extensive neurite outgrowth. Experiments were conducted by using several types of neuronal cells, including transformed cell lines and freshly isolated primary cells. In one set of experiments, NGF preprimed PC12 rat pheochromocytoma cells (22) were seeded on RAD16-II sapeptide scaffolds. Optical sections were collected by using scanning laser confocal microscopy. Individual images do not show the entire neurite arbors because the neurite processes move in and out of the focal plane as they follow the irregular contours of the sapeptide scaffolds. These individual images were merged in register to show the full extent of neurite outgrowth along the contours of the sapeptide scaffolds. Using phase contrast microscope, we observed phase-bright cells growing on the sapeptide scaffolds 18 h after attachment. The attached PC12 cells had a cuboidal appearance, suggesting that the soft matrix wraps around the cells, as compared with the appearance of cells grown on rigid culture plastic surfaces. This cuboidal appearance is consistent with the appearance of cells grown on other malleable substrates (19). In another set of experiments, cell-laden sapeptide scaffolds were transferred to other wells. NGF (50 ng/ml) was added to half of the cultures, and the other half served as NGF-minus controls. Neurite outgrowth on the sapeptide scaffolds was observed for the NGF-treated cells 24 h after the treatment. The NGF-treated cells projected extensive neurites that follow the contours of the sapeptide scaffolds (Fig. 3A). In contrast, control cells without NGF treatment did not project neurites on the sapeptide scaffolds (Fig. 3B). The neurite-bearing PC12 cell cultures grown on the sapeptide scaffolds have been maintained for up to 3 wk after NGF treatment. Similar extensive PC12 cell neurite outgrowth on sapeptide scaffolds was observed by using cells that were not preprimed with NGF, except that the neurites were not observed until 3 to 4 days after NGF treatment (Table 1). The pattern of neurite trajectory of NGF-treated PC12 cells on the malleable sapeptide scaffolds resembles the neurite arbors of nerve cells in vivo, in contrast to the linear appearance of neurites projecting from nerve cells plated on rigid culture plastic surfaces. This difference in neurite arbor appearance may reflect the contour differences between the malleable sapeptide scaffolds and the rigid plastic surface.
Figure 3.

PC12 cell neurite outgrowth and differentiation on sapeptide scaffolds with and without NGF. All images are merged stacks of multiple confocal optical sections. (A) NGF preprimed PC12 cells attached to sapeptide scaffolds. The cells projected extensive neurites that follow the contour of the scaffold after NGF treatment. The black spots on the image are holes in the scaffold. (B) Untreated PC12 cells attach to peptide scaffolds, but do not project neurites. Clusters of cell bodies appear as large white areas. All confocal micrographs were collected 10 days after the cells were introduced to the peptide scaffolds. Cell-laden sapeptide scaffolds were moved from cell-culture wells to glass slides for image collection. The micrographs are representative of at least four experiments for each condition. Bar = 50 μm for both A and B.
Table 1.
Neurite outgrowth from neuronal cells on self-assembling peptide scaffold
| Cell type | Neurite length, μm | Cell source†† |
|---|---|---|
| NGF-treated Rat PC12 | 400–500 | Cultured cell line |
| NGF-preprimed PC12 | 400–500 | Cultured cell line |
| Human SY5Y neuroblastoma | 400–500 | Cultured cell line |
| Mouse cerebellar granule neurons | 200–300 | ‡‡Primary cells |
| Mouse hippocampal neurons | 100–200 | ‡‡Primary cells |
| Rat hippocampal neurons | 200–300 | §§Primary cells |
†† Cells were seeded onto sapeptide scaffolds. The cell-bearing sapeptide scaffolds were transferred to dishes with fresh medium. Maximum neurite length was estimated visually with scale bars 3–7 days after cell attachment for primary cultures and 10–14 days after matrix attachment for the cultured cell lines.
‡‡Seven-day-old mouse.
§§One-day-old rat.
Primary Neuron Neurite Outgrowth on Sapeptide Scaffolds.
Neurite outgrowth from primary neuronal cultures was also tested by using several nerve cell types, including primary dissociated neurons from the mouse cerebellum and rat hippocampus (20). Cerebellar granule neurons undergo postnatal development and are morphologically distinguishable from other cerebellar cells (20, 25). Sapeptide scaffolds support extensive neurite outgrowth from cerebellar granule neurons prepared from 7-day-old mice (Fig. 4). The neurites were readily visualized in two different focal planes (Fig. 4), suggesting that the neurites closely follow the contours of the matrices. The primary cerebellar neuronal cultures on the sapeptide scaffolds were maintained for up to 4 wk. Dissociated mouse and rat hippocampal neurons also attach and project neurites on sapeptide scaffold (Table 1).
Figure 4.

Extensive neurite growth of primary neurons on sapeptide scaffolds. Primary mouse cerebellar granule neurons project extensive neurites on scaffolds. The nonconfocal images show two planes of focus in the same field (A and B), indicating that neurite outgrowth follows the contour of the sapeptide scaffold. A phase-bright neurite varicosity is indicated at the arrows. Comparison of the two photographs reveals that some segments of neurites are in focus in one plane and not in the other, whereas for other segments, it is vice versa. The micrographs are representative of four experiments. Bar = 100 μm.
Synapse Formation on the Sapeptide Surface.
Cultured neurons are capable of forming active synaptic connections with other neurons (20, 21, 28). The location of the active synapses can be labeled by optical imaging by using the fluorescent lipophilic probe FM1–43. As FM1–43 can selectively trace synaptic vesicle turnover during the process of synaptic transmission (26), individual FM1–43 dye spots after staining provide information about the locations of active presynaptic terminals. Furthermore, our previous data indicated that the postsynaptic membrane under most FM1–43 dye spots contains a cluster of functional glutamate receptors (26, 27). As shown in Fig. 5A, cultured hippocampal neurons can form extensive neurites on the sapeptide surface. Some of these neurites made contact with adjacent cells and other neurites. After staining with FM1–43, bright punctuate fluorescent signals were readily detectable at many of the primary neurite contact sites of cells on the sapeptide surface, suggesting that the sapeptide surface can support not only the growth of hippocampal neurons but also the formation of functional synaptic connections. For control experiments, the coating material Matrigel was used as the substrate. Similar levels of synapse formation and synaptic activities were observed for the neurons grown on the sapeptide and Matrigel. The number of active synaptic sites between the sapeptide scaffolds and the Matrigel was indistinguishable. These results show that the sapeptide scaffold is a permissive substrate for primary neuronal synapse formation. This is an important property for further development of the sapeptide scaffolds for neurorepair, neuroengineering, and other general applications for tissue engineering.
Figure 5.
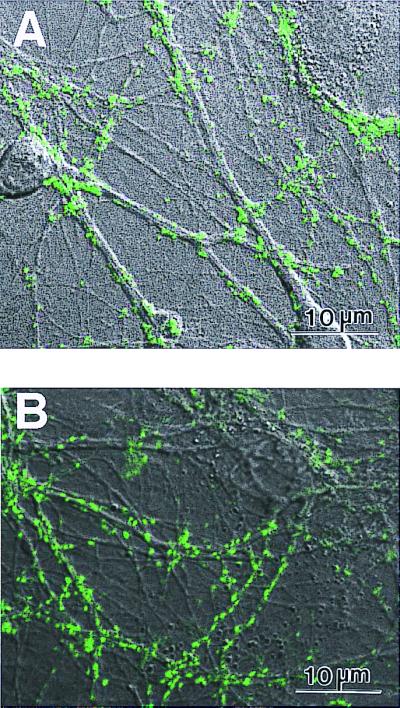
Primary rat hippocampal neurons form active synapses on peptide scaffolds. The confocal images show bright discrete green labeling indicative of synaptically active membranes after FM1–43 incubation of neurons. (A) Active synapses on the peptide surface. (B) Active synapses on Matrigel. The active synapses on these different materials are not readily distinguishable, indicating that the peptide scaffold is a permissible substrate for synapse formation. Bar = 10 μm.
Sapeptide Scaffolds Are Tolerated in Vivo.
The EAK16 and RAD16 oligopeptide scaffolds did not cause detectable toxic effects in animals. This was determined by injection of RAD16 peptides into the leg muscles of Fisher 344 rats. The time interval between injections and perfusion was 9 days for rat No. 35 and 5 wk for rats Nos. 36 and 37. The entire muscle was dissected and sectioned. The muscle sections were then stained with Nissl and thionin and examined by using light microscopy for mononuclear cell infiltration and/or muscle fiber necrosis for signs of inflammation and/or necrosis. There were no apparent signs of muscle pathology or any structural abnormalities. It was very difficult to find the site of the injection because of the lack of tissue damage. There was no observable evidence of muscle necrosis or inflammation at the site of injection 3 wk after sapeptide microinjection, nor was there observable evidence for motor impairment (data not shown).
Discussion
Molecular Self Assembly to Form Biological Sapeptide Scaffolds.
We previously reported the discovery of a class of ionic self-complementary sapeptides that form scaffolds in the presence of solutions that contain monovalent salts (9). EAK16-II, a 16-aa sapeptide, is a member of this class of peptides that was derived from a repetitive segment found in the yeast protein zuotin (12). We subsequently designed and synthesized the EAK16-II derivatives of including RAD16-I and RAD16-II, in which arginine and aspartate residues substitute lysine and glutamate. These sapeptides are highly soluble and form stable β-sheet structures in water because 50% residues are charged amino acids (9). Stable macroscopic matrix structures can be fabricated by the spontaneous self assembly of aqueous sapeptide solutions introduced to physiological salt-containing solutions. The self assembly of other sapeptide and protein materials has been described (9–11, 33–39). One peptide gel was reported by Aggeli et al., who found a fragment of a membrane protein that forms very stable β-sheet and can be processed into tape and other novel materials structures (33, 34). Several polymer-based peptide and protein self-assembly systems have also been developed by other investigators. These protein-based polymers were also designed de novo and produced through recombinant DNA biotechnology (35–39). Discovery and understanding of the nature of peptide- and protein-based biological materials is an emerging field. It is believed that the discovery and development of natural and biologically inspired materials will play an important role in bioengineering in the coming years.
Nerve Cell Attachment and Synapses.
The molecular mechanism for cell attachment and neurite outgrowth on the sapeptide scaffolds is not clear at this time. Two sapeptides, RAD16-I and RAD16-II, which assembled into sapeptide scaffolds, were used. They contain a tripeptide arginine–alanine–aspartate (RAD) sequence (40), which is similar to the RGD motif found in many ECM proteins and which serves as a binding site for some cell adhesion receptors (16, 17). It is believed that neuronal cell attachment and neurite outgrowth on the sapeptide scaffold may be two independent events. The neurite outgrowth on the sapeptide scaffold could take place through an integrin-dependent mechanism. For example, neurite outgrowth may be facilitated through binding to ECM proteins that are secreted by cells themselves (17). Synapse formation on Matrigel or on the peptide surface is indistinguishable under our experimental conditions. Commercial Matrigel contains a collection of laminin, collagen, and other ECM proteins as well as some growth factors. In contrast, the sapeptides contain a very simple composition of only a few amino acids. In future studies, sapeptides with different composition and sequences could be designed and tailored to systematically dissect and analyze neuronal attachment, neurite extension, and synapse formation to understand further these complex events as well as to define specific neuronal properties. In this study, the neuronal synapses were monitored by using FM1–43, a fluorescence dye that is widely used to follow the process of endocytosis. The protocol of FM staining used in this study was designed specifically to label the location of active synaptic terminals, because FM1–43 dye perfusion was coupled with strong presynaptic stimulation. Under these conditions, most FM1–43 spots represent individual synapses.
Sapeptide Scaffolds Are Tolerated in Vivo.
EAK and RAD sapeptides were injected into the leg muscles of Fisher 344 rats to test for their induction of inflammation. In parallel control experiments, saline was injected into leg muscles of animals. The rats were killed 9 days to 5 wk after the injections. The area of injection was examined by light microscopy. No apparent inflammation was observed in the muscles that were injected with either saline or the sapeptides. Additionally, the sapeptides elicited no detectable immune responses after injection of sapeptide or sapeptide conjugated with carrier BSA protein in rabbits and goats (data not shown). These results suggest that in vivo tissue repair applications may be developed by using these sapeptide scaffolds.
A Biological Scaffold for Tissue Engineering.
The self-assembling sapeptide scaffold described here belongs to a class of biologically inspired materials. The self-assembly event creating the sapeptide scaffold takes place under physiological conditions, and these materials are readily transportable to different environments. These new biological materials will become increasingly important in developing approaches for a wide range of innovative medical technologies. These technologies include controlled drug release, new scaffolds for cell-based therapies, tissue engineering, and biomineralization for hard tissue repair.
In combination with stem cell technology, we can anticipate the encapsulation of stem cells in the sapeptide scaffold, allowing them to differentiate into desired cell types with specific growth factors and cytokines, and then the application of cell-scaffold systems into needed tissues. These biocompatible and biodegradable sapeptide scaffolds developed through molecular self assembly will likely have a broad range of applications for tissue repair and tissue engineering.
Acknowledgments
We thank Roger Kamm and Neeta Verma for the construction of the sapeptide-tape processing device, John Kisiday for producing the sapeptide-sheet processing device, Michel Didier, Meredith Fisher, and Rachel Kindt (McLean Hospital/Harvard Medical School, Belmont, MA) for providing primary neurons, and da Vinci for inspiration. This work was supported in part by grants from Hercules, Inc., the U.S. Army Research Office, the National Institutes of Health (Grant R41), and the Whitaker Foundation.
Abbreviations
- sapeptides
self-assembling peptides
- ECM
extracellular matrix
- NGF
nerve growth factor
References
- 1.Lanza R P, Langer R S, Chick W L. Principles of Tissue Engineering. Austin, TX: Academic; 1997. [Google Scholar]
- 2.Langer R L, Vacanti J P. Science. 1993;260:920–926. doi: 10.1126/science.8493529. [DOI] [PubMed] [Google Scholar]
- 3.Patrick C W, Jr, Mikos A G, McIntire L V. Frontiers on Tissue Engineering. New York: Pergamon; 1998. [Google Scholar]
- 4.Vacanti J P, Langer R S. Lancet. 1999;354 Suppl 1:S32–S34. doi: 10.1016/s0140-6736(99)90247-7. [DOI] [PubMed] [Google Scholar]
- 5.Shakesheff K, Cannizzaro S, Langer R S. J Biomater Sci Polymer Ed. 1998;9:507–518. doi: 10.1163/156856298x00596. [DOI] [PubMed] [Google Scholar]
- 6.Langer R S, Vacanti J P. Sci Am. 1999;280:86–89. doi: 10.1038/scientificamerican0499-86. [DOI] [PubMed] [Google Scholar]
- 7.Ellis D L, Yannas I V. Biomaterials. 1996;17:291–299. doi: 10.1016/0142-9612(96)85567-0. [DOI] [PubMed] [Google Scholar]
- 8.Mueller S M, Shortkroff S, Schneider T O, Breinan H A, Yannas I V, Spector M. Biomaterials. 1999;20:701–709. doi: 10.1016/s0142-9612(98)00189-6. [DOI] [PubMed] [Google Scholar]
- 9.Zhang S, Holmes T C, Lockshin C, Rich A. Proc Natl Acad Sci USA. 1993;90:3334–3338. doi: 10.1073/pnas.90.8.3334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang S, Holmes T C, DiPersio C M, Hynes R O, Su X, Rich A. Biomaterials. 1995;16:1385–1393. doi: 10.1016/0142-9612(95)96874-y. [DOI] [PubMed] [Google Scholar]
- 11.Xiong H, Buckwalter B L, Shieh H M, Hecht M H. Proc Natl Acad Sci USA. 1995;92:6349–6353. doi: 10.1073/pnas.92.14.6349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S, Lockshin C, Herbert A, Winter E, Rich A. EMBO J. 1992;11:3787–3796. doi: 10.1002/j.1460-2075.1992.tb05464.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tomaselli K J, Reichardt L F, Bixby J L. J Cell Biol. 1986;103:2659–2672. doi: 10.1083/jcb.103.6.2659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jessell T M, Hynes M A, Dodd J. Annu Rev Neurosci. 1990;13:227–255. doi: 10.1146/annurev.ne.13.030190.001303. [DOI] [PubMed] [Google Scholar]
- 15.Tawil N J, Houde M, Blacher R, Esch F, Reichardt L F, Turner D C, Carbonetto S. Biochemistry. 1990;29:6540–6544. doi: 10.1021/bi00479a028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yamada K M. J Biol Chem. 1991;266:12809–12812. [PubMed] [Google Scholar]
- 17.Hynes R O, Lander A D. Cell. 1992;68:303–322. doi: 10.1016/0092-8674(92)90472-o. [DOI] [PubMed] [Google Scholar]
- 18.Adler R, Manthorpe M, Skaper S D, Varon S. Brain Res. 1981;206:129–144. doi: 10.1016/0006-8993(81)90105-0. [DOI] [PubMed] [Google Scholar]
- 19.DiPersio C M, Jackson D A, Zaret K S. Mol Cell Biol. 1991;11:4405–4414. doi: 10.1128/mcb.11.9.4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Banker G, Goslin K. In: Primary Dissociated Cell Cultures of Neural Tissue in Culturing Nerve Cells. 2nd Ed. Banker G, Goslin K, editors. Cambridge, MA: MIT Press; 1997. pp. 41–71. [Google Scholar]
- 21.Lockhart S T, Turrigiano G G, Birren S J. J Neurosci. 1997;17:9573–9582. doi: 10.1523/JNEUROSCI.17-24-09573.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Greene L A, Tischler A S. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trenkner E, Sidman R L. J Cell Biol. 1977;75:915–940. doi: 10.1083/jcb.75.3.915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Van-Vliet B J, Sebben M, Dumuis A, Gabrion J, Bockaert J, Pin J. J Neurochem. 1989;52:1229–1239. doi: 10.1111/j.1471-4159.1989.tb01870.x. [DOI] [PubMed] [Google Scholar]
- 25.Didier M, Roux P, Piechaczyk M, Mangeat P, Verrier B, Devilliers G, Bockaert J, Pin J P. Mol Brain Res. 1992;12:249–258. doi: 10.1016/0169-328x(92)90091-o. [DOI] [PubMed] [Google Scholar]
- 26.Liu G, Tsien R W. Nature (London) 1995;375:404–408. doi: 10.1038/375404a0. [DOI] [PubMed] [Google Scholar]
- 27.Liu G, Choi S, Tsien R W. Neuron. 1999;22:395–409. doi: 10.1016/s0896-6273(00)81099-5. [DOI] [PubMed] [Google Scholar]
- 28.Reuter H. Neuron. 1995;14:773–779. doi: 10.1016/0896-6273(95)90221-x. [DOI] [PubMed] [Google Scholar]
- 29.Isaacson J S, Hille B. Neuron. 1997;18:143–152. doi: 10.1016/s0896-6273(01)80053-2. [DOI] [PubMed] [Google Scholar]
- 30.Brack A, Orgel L. Nature (London) 1975;256:383–387. doi: 10.1038/256383a0. [DOI] [PubMed] [Google Scholar]
- 31.Rippon W B, Chen H H, Walton A G. J Mol Biol. 1973;75:369–375. doi: 10.1016/0022-2836(73)90027-2. [DOI] [PubMed] [Google Scholar]
- 32.Trudelle Y. Polymer. 1975;16:9–15. [Google Scholar]
- 33.Aggeli A, Bell M, Boden N, Keen J N, Knowles P F, McLeish T C B, Pitkeathly M, Radford S E. Nature (London) 1997;386:259–262. doi: 10.1038/386259a0. [DOI] [PubMed] [Google Scholar]
- 34.Aggeli A, Bannister M L, Bell M, Boden N, Findlay J B, Hunter M, Knowles P F, Yang J C. Biochemistry. 1998;37:8121–8131. doi: 10.1021/bi972112h. [DOI] [PubMed] [Google Scholar]
- 35.McPherson D T, Morrow C, Minehan D S, Wu J, Hunter E, Urry D W. Biotechnol Progr. 1992;8:347–352. doi: 10.1021/bp00016a012. [DOI] [PubMed] [Google Scholar]
- 36.McPherson D T, Xu J, Urry D W. Protein Expression Purif. 1996;7:51–57. doi: 10.1006/prep.1996.0008. [DOI] [PubMed] [Google Scholar]
- 37.Tirrell D A. Nature (London) 1997;390:336–337. doi: 10.1038/36981. [DOI] [PubMed] [Google Scholar]
- 38.McGrath K P, Tirrell D A, Kawai M, Mason T L, Fournier M J. Biotechnol Prog. 1990;6:188–192. doi: 10.1021/bp00003a004. [DOI] [PubMed] [Google Scholar]
- 39.Valluzzi R, He S J, Gido S P, Kaplan D. Int J Biol Macromol. 1999;24:227–236. doi: 10.1016/s0141-8130(99)00005-7. [DOI] [PubMed] [Google Scholar]
- 40.Prieto A L, Edelman G M, Crossin K L. Proc Natl Acad Sci USA. 1993;90:10154–10158. doi: 10.1073/pnas.90.21.10154. [DOI] [PMC free article] [PubMed] [Google Scholar]