

Abstract
Angiogenesis is a critical step required for sustained tumor growth and tumor progression. The stimulation of endothelial cells by cytokines secreted by tumor cells such as vascular endothelial growth factor (VEGF) induces their proliferation and migration. This is a prominent feature of high-grade gliomas. The secretion of VEGF is greatly upregulated under conditions of hypoxia because of the transcription factor hypoxia-inducible factor (HIF)-1α, which controls the expression of many genes, allowing rapid adaptation of cells to their hypoxic microenvironment. Flavopiridol, a novel cyclin-dependent kinase inhibitor, has been attributed with antiangiogenic properties in some cancer cell lines by its ability to inhibit VEGF production. Here, we show that flavopiridol treatment of human U87MG and T98G glioma cell lines decreases hypoxia-mediated HIF-1α expression, VEGF secretion, and tumor cell migration. These in vitro results correlate with reduced vascularity of intracranial syngeneic GL261 gliomas from animals treated with flavopiridol. In addition, we show that flavopiridol downregulates HIF-1α expression in the presence of a proteasome inhibitor, an agent that normally results in the accumulation and overexpression of HIF-1α. The potential to downregulate HIF-1α expression with flavopiridol treatment in combination with a proteasome inhibitor makes this an extremely attractive anticancer treatment strategy for tumors with high angiogenic activity, such as gliomas.
Keywords: flavopiridol, proteasome inhibitor, hypoxia, HIF-1α, VEGF, glioma
Angiogenesis is a critical step that is required for sustained tumor growth and tumor progression. Gliomas are noted to be one of the most highly angiogenic solid tumors known and among the most invasive (Eberhard et al., 2000). A universal characteristic of solid tumors is hypoxia due to limited diffusion of O2 (Brown and Giaccia, 1998). Hypoxia is associated with resistance to cancer therapy (chemotherapy and radiation) (Unruh et al., 2003). Hypoxia-inducible factor-1 (HIF-1)3 is a heterodimeric transcription factor that regulates O2 homeostasis in response to changes in levels of O2 (Guillemin and Krasnow, 1997; Semenza, 1999; Wang et al., 1995). It consists of two subunits, HIF-1α and HIF-1α. The molecular pathway leading from the sensing of hypoxia to the activation of HIF-1 is critically dependent on the relative abundance and stabilization of the HIF-1α subunit (Huang et al., 1996; Jiang et al., 1996). The half-life of the HIF-1α protein is extremely short in normoxic conditions but is markedly prolonged during hypoxic stimulation because its normal degradation by the proteasome no longer occurs in hypoxic conditions (Huang et al., 1998; Kallio et al., 1999; Salceda and Caro, 1997). HIF-1 upregulates the expression of many genes that contain hypoxic responsive promoter elements, such as vascular endothelial growth factor (VEGF), a highly potent cytokine known to promote angiogenesis (Carmeliet et al., 1998; Forsythe et al., 1996).
Previously, we studied the expression of the HIF-1α subunit in gliomas in relation to the extent of neovascularization (Zagzag et al., 2000). The pattern of VEGF expression reported in glioma is remarkably similar to the pattern of HIF-1α expression we have described (Shweiki et al., 1992). The most striking similarity involves pseudopalisading cells around areas of necrosis that are known to express VEGF (Damert et al., 1997). The co-localization of HIF-1α with VEGF expression within glioma suggested a distinct pathway for angiogenesis mediated by VEGF. HIF-1α was highly expressed in areas of tumor adjacent to necrosis, the pseudopalisading cells of glioma, suggesting that the pattern of expression in glioma is modulated by tumor oxygenation. HIF-1 has been shown to regulate VEGF expression both in cultured cells and in tumors (Iyer et al., 1998; Jiang et al., 1997; Maxwell et al., 1997; Ravi et al., 2000). Thus, it is likely that hypoxia induces HIF-1α expression in glioma, which activates VEGF gene transcription, leading to increased VEGF production and angiogenic activity.
A cardinal feature of gliomas and a major reason for the failure of neurosurgical and adjunctive therapies is their invasive properties. We have observed strong HIF-1α expression in invading glioma cells at the edge of gliomas that implied a role in invasion (Zagzag et al., 2000). Recently, we showed that geldanamycin, an inhibitor of heat shock protein 90 function, interfered with the hypoxia-mediated induction of HIF-1α concomitant with inhibition of glioma cell migration (Zagzag et al., 2003). Indeed, others have reported an association between hypoxia and HIF-1α overexpression and cell migration (Brat et al., 2004; Krishnamachary et al., 2003). For example, hypoxia or overexpression of HIF-1α induced invasion of HCT116 colon carcinoma cells, which could be inhibited by small interfering RNA targeting HIF-1α (Krishnamachary et al., 2003). Importantly, exposure to hypoxia stimulated migration of human glioma cell lines as compared with normoxia (Brat et al., 2004). The proteolytic activity of membrane type 1 matrix metalloproteinase (MT1-MMP) and that of MMP-2 have been shown to play critical roles in the degradation of extra-cellular matrix (ECM) proteins that would promote cell invasion (Ellerbroek et al., 2001; Hotary et al., 2000; Koshikawa et al., 2000). Overexpression of VEGF has been correlated with upregulation of MT1-MMP and the MMP-2 and MMP-9 in human gliomas (Munaut et al., 2003). Inducing overexpression of MT1-MMP in transfected glioma U251 cells produced aggressive tumor growth in vivo concomitant with elevated levels of angiogenesis associated with increased production of VEGF (Deryugina et al., 2002). Thus, controlling HIF-1α expression could not only limit the angiogenic response of endothelial cells to increased levels of VEGF in the tumor microenvironment and decrease tumor vascularization but also limit tumor cell invasion.
Flavopiridol, a synthetic flavone with potent cyclin-dependent kinase activity, has shown promise in several clinical trials of human cancers (Newcomb, 2004; Senderowicz, 2003). Previously, we studied the effect of flavopiridol treatment on human glioma U87MG and T98G cell lines and showed that it induced apoptosis and reduced expression of key cell cycle regulators, independently of retinoblastoma and p53 tumor suppressor alterations (Alonso et al., 2003). More recently, induction of VEGF production in human monocytes or in human neuroblastoma cell lines by inducers of HIF-1α expression such as hypoxia (1% O2) or an iron-chelating agent (desferrioxamine) was inhibited by flavopiridol treatment (Melillo et al., 1999; Rapella et al., 2002). The mechanism of action of flavopiridol inhibition of VEGF production was the decreased expression of HIF-1α. Thus, flavopiridol can inhibit the hypoxia-mediated induction of HIF-1α, which in turn prevents the production of VEGF under a variety of conditions and in different cell types.
Here we demonstrate that flavopiridol treatment of human glioma U87MG and T98G cell lines decreases hypoxia-mediated HIF-1α expression, VEGF secretion, and tumor cell migration. These in vitro results correlate with reduced vascularity of intracranial syngeneic GL261 gliomas from animals treated with flavopiridol. In addition, we show that flavopiridol downregulates HIF-1α expression in the presence of a proteasome inhibitor, an agent that normally results in the accumulation and overexpression of HIF-1α. The potential to down-regulate HIF-1α expression with flavopiridol treatment in combination with a proteasome inhibitor makes this an extremely attractive anticancer treatment strategy for tumors with high angiogenic activity, such as gliomas.
Materials and Methods
Cells and Reagents
Human glioma U87MG and T98G cell lines were obtained from the American Type Culture Collection (Manassas, Va.). Because p53 and PTEN mutations can affect HIF-1α expression in response to hypoxia (Ravi et al., 2000; Zundel et al., 2000), we selected glioma cell lines on the basis of their differences in p53 (U87MG, wild type; T98G, mutant) and PTEN (U87MG, deletion exon 3; T98G, point mutation codon 42 status) (Ishii et al., 1999). Cell lines were cultured in 5% CO2 and 95% humidified air atmosphere at 37°C in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco BRL, Grand Island, N.Y.). The medium was supplemented with 10% fetal bovine serum (FBS) (Atlanta Biologicals, Norcross, Ga.), 1% penicillin and streptomycin, and 2 mM glutamine (Gibco BRL). Cells were split every three days to ensure logarithmic growth. Flavopiridol (Aventis Pharmaceuticals, Bridgewater, N.J.) was dissolved in water, and stock solution (4 mg/ml) was stored at −20°C. HIF-1α expression can be stabilized in cells under normoxic conditions by using hypoxia mimetics such as CoCl2 (Chan et al., 2002; Zagzag et al., 2000). CoCl2 (Sigma-Aldrich, St. Louis, Mo.) was prepared fresh in water for use at a final concentration of 125 μM. The 26S proteasome inhibitor MG-262 (Biomol, Plymouth Meeting, Pa.) was dissolved in dimethyl sulfoxide, and stock solution (500 μM) was stored at −20°C.
Experimental Culture Conditions with CoCl2 and Proteasome Inhibitor
Cells (2 × 106) were seeded in 10-cm dishes in 10 ml of complete growth medium for 24 h. To investigate HIF-1α expression, normoxic cells were cultured with 125 μM of CoCl2 for 5 h, alone or together with 300 nM flavopiridol or 500 nM MG-262. After incubation for 5 h, cells were harvested from each culture condition by washing three times with cold phosphate-buffered saline (PBS), and the cells were scraped into radioimmunoprecipitation buffer (400 μl/plate). Lysates were incubated on ice for 1 h, clarified by centrifugation for 1 h at 4ºC at 16,000 rpm, and stored at −80ºC until quantification for protein.
Western Blot Analysis
Cells were lysed in radioimmunoprecipitation buffer (150 mM of NaCl, 1% Nonidet P-40 [Roche Applied Sciences, Indianapolis, Ind.], 1% deoxycholate, 0.1% sodium dodecyl sulfate [SDS], 10 mM Tris-HCl, pH 8.0, 1 mM EDTA, pH 8.0) supplemented with protease inhibitors (1 mM phenyl methyl sulfonyl fluoride, 0.2 mM sodium orthovanadate, and 1 μg/ml aprotinin). Quantitation of protein was carried out with the bicinchoninic acid reagent (Pierce Biotechnology Inc., Rockford, Ill.). Equal amounts of protein (30 μg) in the presence of 5% β-mercaptoethanol (Sigma-Aldrich) were electrophoresed on 7.5% SDS-PAGE gels and subsequently transferred to Immobilon-P membranes (Millipore, Bedford, Mass.) by electroblotting at 4ºC at 33 V overnight. Western blot analysis was performed as described (Alonso et al., 2003) with the following antibodies: mouse anti-HIF-1α monoclonal antibody used at 1:1000 (clone 54, BD Transduction Laboratories, San Jose, Calif.), mouse anti-actin monoclonal antibody used at 1:50,000 (clone C4, Chemicon International, Inc., Temecula, Calif). Monoclonal sheep antimouse IgG (Amersham Life Pharmacia Biotech, Piscataway, N.J.) horseradish peroxidase-conjugated secondary antibodies were used at 1:2000. Immunodetection was carried out with either the Supersignal West Femto (HIF-1α) or Pico (actin) Maximum Sensitivity Substrate ECL detection system (Pierce Biotechnology Inc.). Visualization and quantitation of protein bands were performed with the BioRad Fluor-S apparatus (BioRad, Hercules, Calif.) with Quantity One (version 4.2.1) software or with the National Institutes of Health (NIH) Image software (version 1.62).
Isolation and Analysis of RNA
Total RNA was isolated from monolayers of U87MG cells by using the RNAeasy kit (Qiagen Inc., Valencia, Calif.) according to the manufacturer’s directions. RNA was quantitated by absorbance at 260 nm. For reverse transcription, 2 μg of total RNA was reverse-transcribed by using Super Script II RNase H reverse transcriptase (Invitrogen, Carlsbad, Calif.) and random hexamer primers (Invitrogen) at 25°C for 10 min and 42°C for 1 h for cDNA synthesis; 1 μl of the reverse transcription product was used as a template for polymerase chain reaction (PCR) amplifications. PCR was performed under standard conditions in a 50-μl reaction mix containing 1× PCR buffer, 1 unit of Platinum Taq polymerase (Invitrogen), 200 μM of deoxynucleoside triphosphate mix, 1.5 mM of MgCl2, 200 nM of HIF-1α primers (5-GTCGGACAGCCTCAC-CAAACAGAGC-3‚-sense, 5-GTTAACTTGATC-CAAAGCTCTGAG-3‚-antisense) or 30 nM of β-actin primers (5-GTACCACTGGCATCGTGATGGACT-3‚-sense, 5-ATCCACACGGAGTACTTGCGCTCA-3‚-antisense). The PCR conditions consisted of 3 min of an initial denaturation step (95°C) followed by 23 cycles of denaturation (95°C, 30 s), annealing (55°C, 30 s), and extension (72°C, 50 s), followed by a final elongation step of 7 min at 72°C. Next, 20 μl of PCR product was analyzed on 3% agarose gels stained with ethidium bromide. Four independent experiments were performed. Samples in each PCR assay were run in duplicate, and each assay was repeated at least once, with similar results. Quantitation of bands was performed with the BioRad Fluor-S apparatus (BioRad) with Quantity One (version 4.2.1) software. Data from four independent experiments were pooled for statistical analysis.
VEGF Quantification
VEGF protein released into the conditioned medium was measured by using a commercial enzyme-linked immunosorbent assay (ELISA) kit (R & D Systems, Minneapolis, Minn.) according to the manufacturer’s instructions. Cells (5 × 105) were seeded in 6-well plates in 2 ml of complete growth medium. After 24 h, the cells were washed twice with PBS and preconditioned for 1 h at 37°C in 1 ml of DMEM containing 2% FBS. The preconditioned medium was replaced with 1 ml DMEM containing 2% FBS alone or with 125 μM CoCl2 in the absence or presence of 300 nM of flavopiridol. After a 24-h incubation to allow VEGF protein secretion under the various culture conditions, medium was collected and 1 mM of phenyl methyl sulfonyl fluoride was added. The supernatant was clarified by centrifugation for 5 min at 16,000 rpm, aliquoted, and stored at −80°C until quantification for VEGF. The assay was run in triplicate and repeated three times with similar results. Data from three independent experiments were pooled for statistical analysis.
Migration Assay
BD Biocoat chambers (catalog #354578, BD Bioscience Discovery Labware, Bedford, Mass.) with 8-μm pore size polycarbonate filter inserts for 24-well plates were used according to the manufacturer’s instructions and as described (Zagzag et al., 2002). Briefly, cells (2 × 104) in 500 μl of DMEM medium with 10% FBS were seeded onto the upper compartment of each chamber and placed into wells containing 750 μl of complete medium. Cells were allowed to adhere for 1.5 h, when the medium in the upper chamber was replaced with complete medium alone or with 125 μM CoCl2 in the absence or presence of 300 nM flavopiridol. The migration chambers were incubated for 24 h in 5% CO2 and 95% humidified air atmosphere at 37°C to allow cells to migrate through the membrane. Following incubation, the inserts were fixed in absolute methanol for 2 min at room temperature and stained with 1% toluidine blue in 1% borax for 3 min. The cells on the upper surface of the insert were mechanically removed with a cotton-tipped swab. Quantitation of migrating cells on the lower surface of each filter was done by counting six random fields under a light microscope (final magnification ×100). Images were quantified for number of cells/10× field by using NIH Image software (version 1.62). Each assay was performed in duplicate and repeated three times with similar results. The data from independent experiments were pooled for statistical analysis.
Gelatin Zymography
MMP-2 released into the conditioned medium was measured by gelatin zymography. Cells (5 × 105) were seeded in 6-well plates in 2 ml complete growth medium for 24 h. Then cells were washed twice with PBS and incubated in 1 ml of serum-free DMEM for an additional 24 h in the absence or presence of 300 nM flavopiridol. Medium was collected, clarified of cellular debris by centrifugation for 5 min at 16,000 rpm, and stored at −80°C until analyzed. Samples (30 μl) were mixed with 4× SDS sample buffer without β-mercaptoethanol and electrophoresed on 7.5% SDS-PAGE gels containing 0.2% gelatin type B (Sigma-Aldrich). After electrophoresis, the gels were washed four times (15 min/wash) in 2.5% Triton-X-100 at room temperature to remove SDS and then incubated for 48 h at 37°C in development buffer (50 mM Tris-HCl, pH 7.6; 200 mM NaCl, 5 mM CaCl2, 0.02% Brij-35 [Universal Preserva-Chem, Inc., Edison, N.J.]). The gels were stained with 0.25% Coomassie brilliant blue R-250 (BioRad) in 40% methanol and 10% acetic acid for 6 h at room temperature and then destained overnight in 40% methanol and 10% acetic acid. Proteolytic activity for MMP-2 in the gel was visualized as clear white bands at 72 kDa against a blue background. Gels were photographed and quantified by NIH Image software (version 1.62). The gelatinase standard (Chemicon International, Inc.) was used as a positive control for gelatin zymography. The data from three independent experiments were pooled for statistical analysis.
Analysis of Tumor Vascularity
The GL261 glioma cells were implanted into brains of C57BL/6 female mice as described previously (Newcomb et al., 2004). Animals received one cycle of treatment either with flavopiridol or with vehicle on days 7 through 11 after tumor implantation and then were sacrificed on days 14, 21, or 28. At sacrifice the brains were removed and fixed as described (Newcomb et al., 2004). Fixed brains were cut into coronal slabs at 0.2- to 0.3-cm intervals; the slabs were processed into paraffin blocks, sectioned at 6 μm, and stained with hematoxylin and eosin. To assess vascular density, new sections were stained by using standard avidin-biotin immunoperoxidase methods with anti-CD31 antibody (clone MEC13.3, BD Pharmingen, San Diego, Calif.) diluted 1:20. Briefly, tissue sections were first digested with alkaline endopeptidase (8 min at 37ºC) (Ventana Medical System, Tucson, Ariz.), and endogenous peroxidase activity was blocked with hydrogen peroxide (4 min). After washing, slides were incubated overnight with primary antibody at room temperature. Bound anti-CD31 was detected with biotinylated goat antimouse (BD Pharmingen) diluted 1:20 (32 min at 37ºC), followed by application of streptavidin–horseradish peroxidase conjugate.
Vascular density was scored with use of a multihead microscope by two neuropathologists (D.C.M., M.F.) who were blinded as to whether the slides were from animals treated with flavopiridol or with vehicle alone. Tumor vascularity was scored semiquantitatively by consensus on an ascending scale (1+, 2+, 3+, 4+) in multiple 100× and 400× fields selected from the most vascularized portions of the tumors.
Statistical Analysis
Experiments were performed at least twice, and determinations were performed in replicates. Results are expressed as mean ± standard error (SE). All analyses for the conditions being compared were performed by using a two-sided Student’s t test for significance (P < 0.05). All analyses used Stat View software (SAS Institute, Cary, N.C.).
Results
Flavopiridol Downregulates Hypoxia-Mediated HIF-1α Expression by a Proteasome-Independent Pathway
U87MG and T98G cells, grown under normoxic conditions, were or were not exposed to 125 μM CoCl2 for 5 h in the absence or presence of 300 nM flavopiridol. Cells were harvested after 5 h, and lysates were immunoblotted to detect levels of HIF-1α expression. Results of a representative experiment are shown (Fig. 1). Under normoxic culture conditions, U87MG or T98G cells showed baseline levels of HIF-1α expression. However, treatment with CoCl2 upregulated HIF-1α expression approximately sixfold in U87MG cells or twofold in T98G cells as compared with the respective untreated control cultures (Fig. 1, lane 2 vs. 1). Hypoxia mimetics, such as CoCl2, stabilize HIF-1 protein expression under normoxic conditions by inhibiting prolyl hydroxylation of the HIF-1α subunit, thereby preventing its ubiquitination and subsequent degradation by the proteasome (Chan et al., 2002). Simultaneous addition of 300 nM flavopiridol together with CoCl2 to the cultures interfered with the ability of CoCl2 to upregulate HIF-1α expression during the 5-h time interval (Fig. 1, lane 4 vs. 2). The level of expression of HIF-1α in the presence of flavopiridol was decreased approximately 80% in U87MG cells or 25% in T98G cells.
Fig. 1.

HIF-1α expression as a function of the presence of flavopiridol. Results show that flavopiridol downregulates hypoxia-mediated HIF-1α expression independently of the proteasome degradation pathway. U87MG and T98G cells that were or were not exposed to CoCl2 or MG-262 in the absence or presence of flavopiridol were harvested, and lysates were immunoblotted to detect levels of HIF-1α expression. The expression of β-actin was used as the loading control. Representative Western blot results are shown from one of three independent experiments. Protein bands were quantitated by densitometry.
To investigate whether flavopiridol treatment induced degradation of HIF-1α via the proteasome pathway, we tested the effect of the 26S proteasome inhibitor MG-262 on levels of HIF-1α expression in cells grown under normoxic conditions. The level of expression of HIF-1α in the presence of the proteasome inhibitor MG-262 was upregulated in both the U87MG and in T98G cells as compared with untreated control cultures (Fig. 1, lane 5 vs. 1). For T98G cells, accumulation of HIF-1α expression was markedly enhanced by treatment with the MG-262 proteasome inhibitor approximately elevenfold compared with CoCl2 treatment alone (Fig. 1, lane 2 vs. 5). This may be explained by the fact that inhibitors of the prolyl hydroxylase enzyme, such as CoCl2, act upstream of the proteasome degradation step, whereas inhibitors of the proteasome, such as MG-262, act directly on the proteasome to prevent degradation of ubiquitinated proteins (Chan et al., 2002). Regardless of the mechanism for HIF-1α protein accumulation, simultaneous addition of flavopiridol with MG-262 decreased accumulation of HIF-1α in both U87MG and T98G cells, which resulted in >60% inhibition of HIF-1α protein expression (Fig. 1, lane 6 vs. 5). Thus, under two different conditions producing accumulation of HIF-1α protein due to post-transcriptional stabilization of the protein, flavopiridol induced downregulation of HIF-α protein expression. Because flavopiridol did not appear to target HIF-1α protein for proteasomal degradation, we considered whether flavopiridol might be inducing downregulation of gene expression by preventing transcription of the HIF-1α gene.
Flavopiridol Downregulates HIF-1α Expression by Interfering with Gene Transcription
To investigate whether flavopiridol downregulated HIF-1α expression through its known ability to inhibit gene transcription (Blagosklonny, 2004; Chao and Price, 2001; de Azevedo et al., 2002), we next determined the effect of flavopiridol treatment on levels of HIF-1α mRNA expression. U87MG cells were grown under normoxia in the absence or presence of CoCl2 and in the absence or presence of flavopiridol, and total RNA was extracted from harvested cells for gene expression studies. Figure 2 shows representative results of one of four independent PCR assays together with relative expression levels of HIF-1α in each treatment group. Treatment with flavopiridol decreased normoxic levels of HIF-1α mRNA by 24% (Fig. 2, lane 1 vs. 2), a significant reduction in comparison with the untreated culture (P < 0.001). CoCl2 treatment increased HIF-1α mRNA levels by 32% relative to the uninduced culture (Fig. 2, lane 1 vs. 3). However, simultaneous addition of 300 nM flavopiridol together with CoCl2 to the cultures significantly decreased HIF-1α mRNA by >60% as compared with the culture treated with only CoCl2 (Fig. 2, lane 3 vs. 4) (P < 0.001).
Fig. 2.

Levels of RNA (top) and HIF-1α (bottom) in four treatment groups. Results show that flavopiridol downregulates HIF-1α expression by interfering with gene transcription. U87MG cells were grown under normoxia in the absence (lanes 1 and 2) or in the presence (lanes 3 and 4) of 125 μM CoCl2 and in the absence (lanes 1 and 3) or in the presence (lanes 2 and 4) of 300 nM of flavopiridol. Total RNA was extracted from harvested cells for gene expression studies. Levels of HIF-1α and β-actin gene expression were measured by using a semiquantitative RT-PCR assay. Samples in each PCR assay were run in duplicate and repeated at least once, with similar results. PCR products were analyzed on 3% agarose gels stained with ethidium bromide. DNA bands were quantitated by densitometry. Results of one representative PCR assay are shown from one of four independent experiments. Bar graphs represent data pooled from four independent experiments for statistical analysis comparing relative levels of HIF-1α expression in the different treatment groups. Error bars indicate the range of the determinations (mean ± SE). *P < 0.001, significantly different from the respective control.
Previously, we reported that downregulation of MDM2 expression in U87MG cells by flavopiridol was due to decreased levels of MDM2 mRNA by Northern analysis rather than changes in the half-life of the MDM2 protein (Alonso et al., 2003). Similarly, we tested the effect of flavopiridol treatment on the half-life of HIF-1α protein. U87MG cells were first stimulated with CoCl2 to induce upregulation of HIF-1α expression and subsequently incubated with the protein synthesis inhibitor cycloheximide. Inhibition of protein synthesis for 1 h greatly decreased HIF-1α expression, which was barely detectable after 2 h of treatment. The half-life of HIF-1α protein did not change in the presence of flavopiridol (data not shown). Taken together, these results indicate that downregulation of HIF-1α expression by flavopiridol is most likely due to inhibition of HIF-1α mRNA gene transcription. This is consistent with our previous results and those of others and is supported by the fact that flavopiridol inhibits the activity of the transcriptional elongation factor P-TEFb, which is essential for gene transcription (Blagosklonny, 2004; Chao and Price, 2001; de Azevedo et al., 2002; Gojo et al., 2002).
Flavopiridol Downregulates Hypoxia-Mediated Secretion of VEGF
Because increased HIF-1α expression promotes transcriptional activation of many target genes, one of which is VEGF (Forsythe et al., 1996), we next wanted to determine the levels of VEGF expression that are associated with levels of HIF-1α expression under various treatment conditions. Thus, U87MG and T98G cells were or were not exposed to CoCl2 in the absence or presence of flavopiridol, and levels of VEGF protein secreted into the conditioned medium were measured by ELISA. The pooled results from three independent experiments are shown in Fig. 3. Treatment of the U87MG and T98G cells with CoCl2 induced a twofold increase in the level of VEGF secretion compared with untreated controls. Flavopiridol treatment significantly inhibited CoCl2-stimulated levels of VEGF secretion (P < 0.001). Decreased secretion of VEGF in flavopiridol-treated cultures was not due to loss of cell viability. Previously, we reported >90% viability of U87MG or T98G cells after 24 h of flavopiridol treatment (Alonso et al., 2003). Thus, flavopiridol treatment downregulates HIF-1α protein expression (Fig. 1), which is correlated with inhibition of VEGF secretion.
Fig. 3.
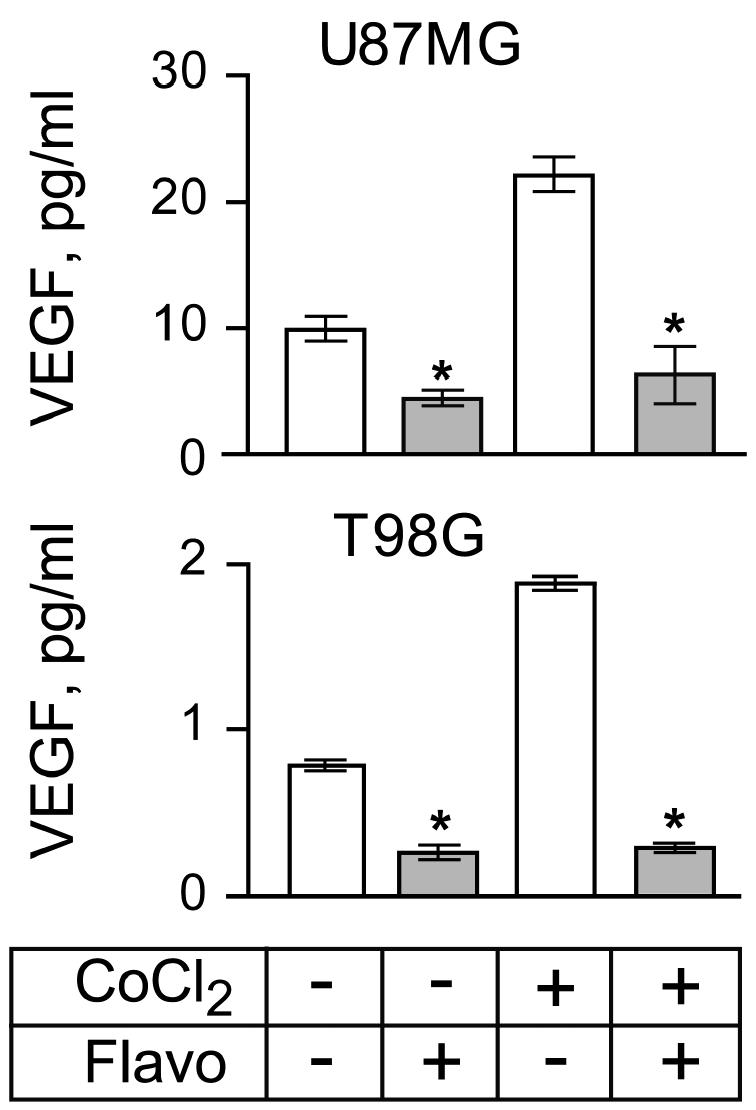
VEGF protein secreted by U87MG and T98G cells as a function of treatment with CoCl2 or flavopiridol. ELISA results show that flavopiridol downregulates hypoxia-mediated secretion of VEGF. Cells (5 × 105) were seeded in 6-well plates in 2 ml of complete growth medium. The assay was run in triplicate and repeated three times with similar results. Bar graphs represent data pooled from three independent experiments for statistical analysis comparing VEGF secretion in cultures with and without CoCl2 or flavopiridol treatment. Error bars indicate the range of the determinations (mean ± SE). *P < 0.001, significantly different from the respective control.
Flavopiridol Inhibits Hypoxia-Mediated Glioma Cell Migration
Next we determined the effect of flavopiridol treatment on cellular migration using BD Biocoat migration chambers (BD Biosciences, San Jose, Calif.). The U87MG and T98G cells were plated on the chamber inserts and allowed to adhere and spread on the insert surface prior to addition of CoCl2 in the absence or presence of flavopiridol. After an incubation for an additional 24 h, the number of cells that migrated through the membrane was quantitated. The pooled results from three independent experiments performed in triplicate are shown in Fig. 4. CoCl2 treatment stimulated migration of U87MG and T98G cells by 33% and 18%, respectively, compared with untreated control cultures. Flavopiridol treatment significantly inhibited CoCl2-stimulated migration of U87MG and T98G cells (P < 0.001).
Fig. 4.
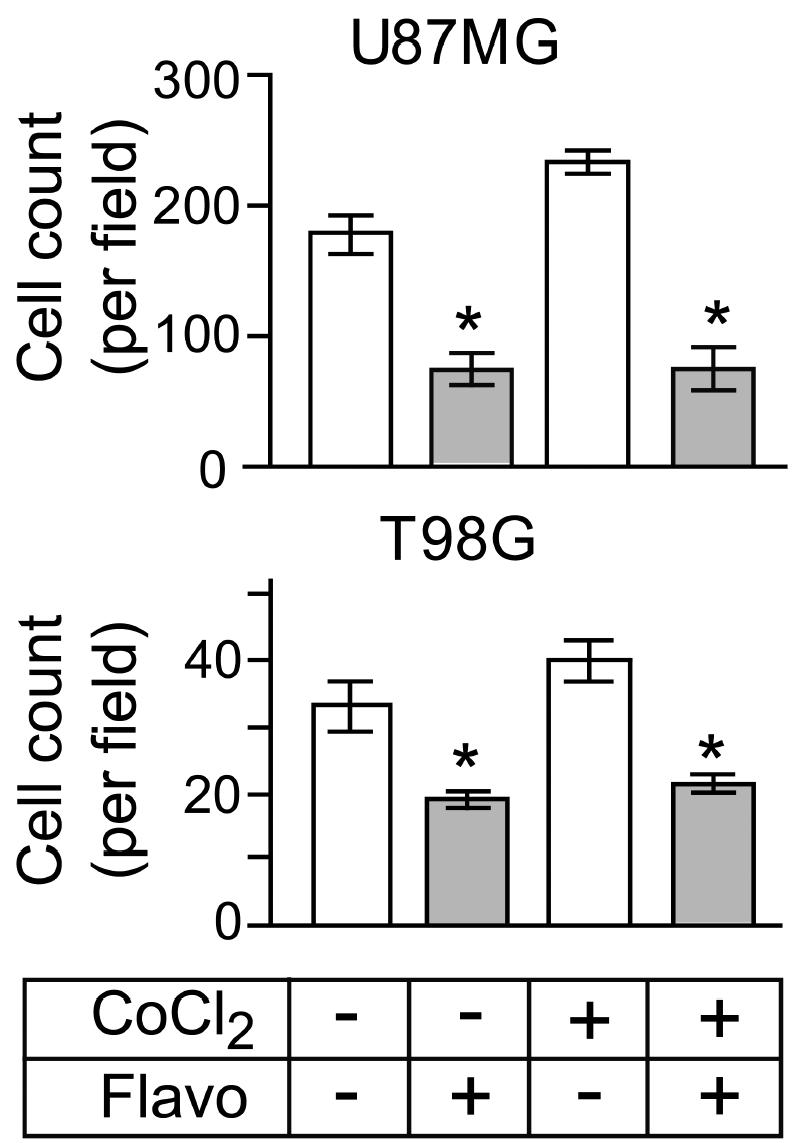
Number of U87MG and T98G cells that migrated through the membrane insert as a function of treatment with CoCl2 with and without flavopiridol. Results show that flavopiridol inhibits hypoxia-mediated glioma cell migration. After 24 h of incubation, the number of cells on the upper and lower surfaces of the inserts was determined. Bar graphs represent data pooled from three independent experiments for statistical analysis comparing migration in cultures with and without CoCl2 or flavopiridol treatment. Error bars indicate the range of the determinations (mean ± SE). *P < 0.001, significantly different from the respective control.
Flavopiridol Downregulates Secretion of MMP-2
Glioma tumors characterized by MMP-2 expression induce proteolysis of the surrounding ECM, favoring invasion (Deryugina et al., 2002; Munaut et al., 2003). We therefore determined the effect of flavopiridol treatment on the secretion of MMP-2. U87MG and T98G cells were grown in the absence or presence of flavopiridol for 24 h, and the level of MMP-2 protein secreted into the conditioned medium was measured by gelatin zymography. Results are shown in Fig. 5. The results of one representative gelatin zymography experiment are shown (Fig. 5A) together with the quantitative data pooled from three independent experiments (Fig. 5B). Flavopiridol treatment decreased the secretion of MMP-2 significantly compared with that of the untreated controls by approximately 40% to 50% (P < 0.01).
Fig. 5.

MMP-2 protein secreted from U87MG and T98G cells. Results show that flavopiridol downregulates secretion of MMP-2. Cells were incubated in the absence (C) or presence (F) of 300 nM of flavopiridol, and medium was analyzed for MMP-2 activity. (A) Representative zymography results are shown from one of three independent experiments. (B) Bar graphs represent data pooled from three independent experiments for statistical analysis comparing MMP-2 secretion in control cultures (C) or flavopiridol-treated cultures (F). The gelatinase standard (Std) was used as a positive control for gelatin zymography. Error bars indicate the range of the determinations (mean ± SE). *P < 0.01, significantly different from the respective control.
Flavopiridol Decreases Tumor Vascularity
Because the in vitro studies conducted here showed flavopiridol downregulated both HIF-1α and VEGF expression that might correlate with inhibition of angiogenic activity in vivo, we next determined whether flavopiridol treatment influenced tumor vascularity in vivo. We recently tested flavopiridol in an experimental intracranial animal model of GL261 murine glioma (Newcomb et al., 2004). We demonstrated that tumor volume at day 14 after one course of flavopiridol showed a significant decrease compared with the control treatment group (Newcomb et al., 2004). This effect disappeared by day 21. We immunostained the same tumors for CD31 to semiquantitate tumor vascularity. Animals received one cycle of treatment either with flavopiridol or with vehicle on days 7 through 11 after tumor implantation, and the brains obtained on days 14 and 21 were stained with anti-CD31 to determine tumor vascularity. Figure 6 shows representative CD31 staining results of brains from a control and a drug-treated animal (Fig. 6A) at day 14, together with semiquantitative CD31 scoring data (Fig. 6B). Animals treated with flavopiridol (n = 4) had significantly reduced CD31 scores in comparison with the controls (n = 4) at day 14. The average CD31 score at day 14 for drug-treated animals was 1.75 ± 0.5 compared with 2.75 ± 0.5 in the control group (P < 0.03). Consistent with our published findings on tumor volumes at day 21, the CD31 scores of tumors at day 21 in control and drug treatment groups (n = 5/group) showed no significant difference.
Fig. 6.

Flavopiridol decreases tumor vascularity. Animals received one cycle of treatment either with flavopiridol or with vehicle on days 7 through 11 after tumor implantation, and brains were harvested on days 14, 21, or 28. Tissue sections were stained with anti-CD31 antibody to reveal the tumor vasculature. Tumor vascularity was scored semiquantitatively, as described in Materials and Methods. A. Representative CD31 staining results of brains from a control (3+, moderately high) and a drug-treated animal (1+, low). Arrows denote some of the CD31-positive microvessels. Magnification ×400. B. Bar graphs represent the semiquantitative CD31 scoring data obtained from the analysis of brain tumor sections of control or flavopiridol-treated animals from day 14 and day 21. Error bars indicate the range of the determinations (mean ± SE). *P < 0.03, significantly different from the respective control.
Discussion
Hypoxia is associated with resistance to cancer therapies (radiation and chemotherapy) (Brown and Giaccia, 1998; Shannon et al., 2003; Unruh et al., 2003). In the absence of an adequate supply of O2, tumor cells upregulate the transcription factor HIF-1, together with a large number of hypoxia-inducible genes that allow the cells to rapidly adapt to their microenvironment facilitating their survival. One gene involved in this adaptive response is VEGF, a potent angiogenic factor associated with the aggressive growth of hypoxic tumors, particularly gliomas. To facilitate treatment of resistant tumors, an effective anticancer agent should target features associated with the unrestrained growth of tumor cells, such as their ability to sustain angiogenesis.
Here we demonstrate that flavopiridol, a novel cyclin-dependent kinase inhibitor, rapidly downregulates hypoxia-mediated HIF-1α expression in U87MG and T98G cells in vitro, with an associated decrease in secretion of the angiogenic factor VEGF. These results provide a plausible mechanism for flavopiridol’s reported antiangiogenic activity in vivo (Kerr et al., 1999). Surprisingly, in a recent screen of 2000 compounds in the National Cancer Institute chemical repository for drugs able to inhibit hypoxia-mediated induction of HIF-1α, flavopiridol was not among the compounds identified (Rapisarda et al., 2002). The potential to downregulate HIF-1α expression together with VEGF makes flavopiridol an extremely attractive agent for gliomas that are highly angiogenic tumors and refractory to current therapies. We recently tested flavopiridol in an experimental intracranial animal model of GL261 murine glioma (Newcomb et al., 2004). As a monotherapy, flavopiridol showed a significant inhibition of GL261 glioma tumor growth at day 14. Here we show that our in vitro results correlate with reduced vascularity of intracranial syngeneic GL261 gliomas from animals treated with flavopiridol. Taken together, our data provide a strong rationale for the use of flavopiridol in the treatment of human gliomas.
Gliomas are known for their typical infiltrating growth pattern into the brain adjacent to the tumor (Zagzag et al., 2000). Glioma cell invasion has been attributed in part to secretion of proteolytic enzymes, MMPs, which promote localized degradation of ECM molecules to create a permissive environment for tumor cell invasion and migration. Upregulation of VEGF expression has been correlated with MMP expression in gliomas (Munaut et al., 2003), and we and others have reported an association between HIF-1α expression and cell migration (Brat et al., 2004; Krishnamachary et al., 2003; Zagzag et al., 2000). Here we show that flavopiridol treatment of human U87MG and T98G glioma cells downregulated MMP-2 secretion and inhibited cell migration. Similarly, studies using human breast cancer cell lines treated with flavopiridol showed decreased secretion of MMP-2 and MMP-9, concomitant with inhibition of migration in Matrigel (BD Biosciences) invasion chambers (Li et al., 2000). In addition, human kidney cells treated with flavopiridol downregulated expression of MMP-9 that was associated with inactivation of nuclear factor-κB (NF-κB) (Takada and Aggarwal, 2004). Because NF-κB regulates expression of genes such as VEGF and MMPs, and flavopiridol is known to suppress NF-κB activation, one mechanism for flavopiridol inhibition of MMP secretion, together with decreased migration, may be through modulation of NF-κB activity (Takada and Aggarwal, 2004).
Proteasome inhibitors result in increased levels of many cellular proteins normally targeted for destruction via the proteasome degradation pathway (Voorhees et al., 2003). For example, treatment of U87MG and T98G cells with the proteasome inhibitor lactacystin promoted the rapid accumulation of p53, MDM2, and the cell cycle regulator p27KIP1 within 6 h (Kitagawa et al., 1999). Previously, we showed that flavopiridol treatment of the U87MG and T98G cells promoted the rapid downregulation of the same proteins and that this was due to decreased levels of mRNA and not associated with changes in the half-life of the proteins (Alonso et al., 2003). In the studies performed here, we showed that upregulation of HIF-1α expression by the proteasome inhibitor MG-262 could be prevented by flavopiridol treatment. This result indicated that flavopiridol did not target HIF-1α to the proteasome degradation pathway. Because flavopiridol plays a major role in the regulation of gene transcription (Blagosklonny, 2004; Chao and Price, 2001; de Azevedo et al., 2002; Gojo et al., 2002), we tested the effect of flavopiridol on HIF-1α mRNA. We found that hypoxia-mediated upregulation of HIF-1α mRNA levels was inhibited with flavopiridol treatment. Thus, the mechanism by which flavopiridol downregulates HIF-1α expression is associated with inhibition of HIF-1α gene transcription. This observation has implications for combination treatments of proteasome inhibitors with flavopiridol as a novel anticancer therapy. The selective and potent proteasome inhibitor bortezomib (formerly PS-341) has proven effective as an anticancer agent in a wide variety of preclinical studies (Adams et al., 1999; Nawrocki et al., 2004; Voorhees et al., 2003). Recent studies have shown that bortezomib given in combination with standard cytotoxic drugs, such as camptothecin CPT-11, doxorubicin, and melphalan, can overcome resistance of several cell lines to radiation and chemotherapy treatments (Voorhees et al., 2003). More importantly, bortezomib used in combination with flavopiridol was able to potentiate the induction of apoptosis in human leukemia cell lines, in part by disrupting the NF-κB pathway (Dai et al., 2003). An increased level of NF-κB occurs in many human cancers, and its expression becomes activated in response to chemotherapy and radiation treatments, contributing to tumor resistance (Orlowski and Baldwin, 2002). Thus, the NF-κB transcription factor has been considered as a chemotherapeutic target for inhibition in order to increase response of tumors to various cancer treatments. Most recently, proteasome inhibitors were shown to block activation of NF-κB activity, suggesting that they could be used to advantage as adjuvants in chemotherapy (Tergaonkar et al., 2003). Here we show that flavopiridol downregulates HIF-1α expression in the presence of a proteasome inhibitor, an agent that normally results in the accumulation and overexpression of HIF-1α. The potential to downregulate HIF-1α expression with flavopiridol treatment in combination with proteasome inhibitors, such as bortezomib, make this an extremely attractive anticancer treatment strategy for tumors with high angiogenic activity, such as gliomas.
Footnotes
This work was supported by grant CA90290 from the National Institutes of Health.
Abbreviations used are as follows: DMEM, Dulbecco’s Modified Eagle Medium; ECM, extracellular matrix; ELISA, enzyme-linked immunosorbent assay; FBS, fetal bovine serum; HIF-1, hypoxia-inducible factor-1; MMP, matrix metalloproteinase; MT1, membrane type 1; NF-κB, nuclear factor-κB; NIH, National Institutes of Health; PBS, phosphate-buffered saline; PCR, polymerase chain reaction; SDS, sodium dodecyl sulfate; SE, standard error; VEGF, vascular endothelial growth factor.
References
- Adams J, Palombella VJ, Sausville EA, Johnson J, Destree A, Lazarus DD, Maas J, Pien CS, Prakash S, Elliott PJ. Proteasome inhibitors: A novel class of potent and effective antitumor agents. Cancer Res. 1999;59:2615–2622. [PubMed] [Google Scholar]
- Alonso M, Tamasdan C, Miller DC, Newcomb EW. Flavopiridol induces apoptosis in glioma cell lines independent of retinoblastoma and p53 tumor suppressor pathway alterations by a caspase-independent pathway. Mol Cancer Ther. 2003;2:139–150. [PubMed] [Google Scholar]
- Blagosklonny MV. Flavopiridol, an inhibitor of transcription: Implications, problems and solutions. Cell Cycle. 2004;3:1537–1542. doi: 10.4161/cc.3.12.1278. [DOI] [PubMed] [Google Scholar]
- Brat DJ, Castellano-Sanchez AA, Hunter SB, Pecot M, Cohen C, Hammond EH, Devi SN, Kaur B, Van Meir EG. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64:920–927. doi: 10.1158/0008-5472.can-03-2073. [DOI] [PubMed] [Google Scholar]
- Brown JM, Giaccia AJ. The unique physiology of solid tumors: Opportunities (and problems) for cancer therapy. Cancer Res. 1998;58:1408–1416. [PubMed] [Google Scholar]
- Carmeliet P, Dor Y, Herbert JM, Fukumura D, Brusselmans K, Dewerchin M, Neeman M, Bono F, Abramovitch R, Maxwell P, Koch CJ, Ratcliffe P, Moons L, Jain RK, Collen D, Keshert E, Keshet E. Role of HIF-1α in hypoxia-mediated apoptosis, cell proliferation and tumour angiogenesis. Nature. 1998;394:485–490. doi: 10.1038/28867. [DOI] [PubMed] [Google Scholar]
- Chan DA, Sutphin PD, Denko NC, Giaccia AJ. Role of prolyl hydroxylation in oncogenically stabilized hypoxia-inducible factor-1α. J Biol Chem. 2002;277:40112–40117. doi: 10.1074/jbc.M206922200. [DOI] [PubMed] [Google Scholar]
- Chao SH, Price DH. Flavopiridol inactivates P-TEFb and blocks most RNA polymerase II transcription in vivo. J Biol Chem. 2001;276:31793–31799. doi: 10.1074/jbc.M102306200. [DOI] [PubMed] [Google Scholar]
- Dai Y, Rahmani M, Grant S. Proteasome inhibitors potentiate leukemic cell apoptosis induced by the cyclin-dependent kinase inhibitor flavopiridol through a SAPK/JNK- and NF-κB-dependent process. Oncogene. 2003;22:7108–7122. doi: 10.1038/sj.onc.1206863. [DOI] [PubMed] [Google Scholar]
- Damert A, Machein M, Breier G, Fujita MQ, Hanahan D, Risau W, Plate KH. Up-regulation of vascular endothelial growth factor expression in a rat glioma is conferred by two distinct hypoxia-driven mechanisms. Cancer Res. 1997;57:3860–3864. [PubMed] [Google Scholar]
- de Azevedo WF, Jr, Canduri F, da Silveira NJ. Structural basis for inhibition of cyclin-dependent kinase 9 by flavopiridol. Biochem Biophys Res Commun. 2002;293:566–571. doi: 10.1016/S0006-291X(02)00266-8. [DOI] [PubMed] [Google Scholar]
- Deryugina EI, Soroceanu L, Strongin AY. Up-regulation of vascular endothelial growth factor by membrane-type 1 matrix metalloproteinase stimulates human glioma xenograft growth and angiogenesis. Cancer Res. 2002;62:580–588. [PubMed] [Google Scholar]
- Eberhard A, Kahlert S, Goede V, Hemmerlein B, Plate KH, Augustin HG. Heterogeneity of angiogenesis and blood vessel maturation in human tumors: Implications for antiangiogenic tumor therapies. Cancer Res. 2000;60:1388–1393. [PubMed] [Google Scholar]
- Ellerbroek SM, Wu YI, Overall CM, Stack MS. Functional interplay between type I collagen and cell surface matrix metalloproteinase activity. J Biol Chem. 2001;276:24833–24842. doi: 10.1074/jbc.M005631200. [DOI] [PubMed] [Google Scholar]
- Forsythe JA, Jiang BH, Iyer NV, Agani F, Leung SW, Koos RD, Semenza GL. Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol Cell Biol. 1996;16:4604–4613. doi: 10.1128/mcb.16.9.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gojo I, Zhang B, Fenton RG. The cyclin-dependent kinase inhibitor flavopiridol induces apoptosis in multiple myeloma cells through transcriptional repression and down-regulation of Mcl-1. Clin Cancer Res. 2002;8:3527–3538. [PubMed] [Google Scholar]
- Guillemin K, Krasnow MA. The hypoxic response: Huffing and HIFing. Cell. 1997;89:9–12. doi: 10.1016/s0092-8674(00)80176-2. [DOI] [PubMed] [Google Scholar]
- Hotary K, Allen E, Punturieri A, Yana I, Weiss SJ. Regulation of cell invasion and morphogenesis in a three-dimensional type I collagen matrix by membrane-type matrix metalloproteinases 1, 2, and 3. J Cell Biol. 2000;149:1309–1323. doi: 10.1083/jcb.149.6.1309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LE, Arany Z, Livingston DM, Bunn HF. Activation of hypoxia-inducible transcription factor depends primarily upon redox-sensitive stabilization of its α subunit. J Biol Chem. 1996;271:32253–32259. doi: 10.1074/jbc.271.50.32253. [DOI] [PubMed] [Google Scholar]
- Huang LE, Gu J, Schau M, Bunn HF. Regulation of hypoxia-inducible factor 1α is mediated by an O2-dependent degradation domain via the ubiquitin-proteasome pathway. Proc Natl Acad Sci USA. 1998;95:7987–7992. doi: 10.1073/pnas.95.14.7987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii N, Maier D, Merlo A, Tada M, Sawamura Y, Diserens AC, Van Meir EG. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 1999;9:469–479. doi: 10.1111/j.1750-3639.1999.tb00536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer NV, Kotch LE, Agani F, Leung SW, Laughner E, Wenger RH, Gassmann M, Gearhart JD, Lawler AM, Yu AY, Semenza GL. Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1α. Genes Dev. 1998;12:149–162. doi: 10.1101/gad.12.2.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang BH, Semenza GL, Bauer C, Marti HH. Hypoxia-inducible factor 1 levels vary exponentially over a physiologically relevant range of O2 tension. Am J Physiol. 1996;271:C1172–C1180. doi: 10.1152/ajpcell.1996.271.4.C1172. [DOI] [PubMed] [Google Scholar]
- Jiang BH, Agani F, Passaniti A, Semenza GL. V-SRC induces expression of hypoxia-inducible factor 1 (HIF-1) and transcription of genes encoding vascular endothelial growth factor and enolase 1: Involvement of HIF-1 in tumor progression. Cancer Res. 1997;57:5328–5335. [PubMed] [Google Scholar]
- Kallio PJ, Wilson WJ, O’Brien S, Makino Y, Poellinger L. Regulation of the hypoxia-inducible transcription factor 1α by the ubiquitin-proteasome pathway. J Biol Chem. 1999;274:6519–6525. doi: 10.1074/jbc.274.10.6519. [DOI] [PubMed] [Google Scholar]
- Kerr JS, Wexler RS, Mousa SA, Robinson CS, Wexler EJ, Mohamed S, Voss ME, Devenny JJ, Czerniak PM, Gudzelak A, Jr, Slee AM. Novel small molecule alpha v integrin antagonists: Comparative anti-cancer efficacy with known angiogenesis inhibitors. Anti-cancer Res. 1999;19:959–968. [PubMed] [Google Scholar]
- Kitagawa H, Tani E, Ikemoto H, Ozaki I, Nakano A, Omura S. Proteasome inhibitors induce mitochondria-independent apoptosis in human glioma cells. FEBS Lett. 1999;443:181–186. doi: 10.1016/s0014-5793(98)01709-8. [DOI] [PubMed] [Google Scholar]
- Koshikawa N, Giannelli G, Cirulli V, Miyazaki K, Quaranta V. Role of cell surface metalloprotease MT1-MMP in epithelial cell migration over laminin-5. J Cell Biol. 2000;148:615–624. doi: 10.1083/jcb.148.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krishnamachary B, Berg-Dixon S, Kelly B, Agani F, Feldser D, Ferreira G, Iyer N, LaRusch J, Pak B, Taghavi P, Semenza GL. Regulation of colon carcinoma cell invasion by hypoxia-inducible factor 1. Cancer Res. 2003;63:1138–1143. [PubMed] [Google Scholar]
- Li Y, Bhuiyan M, Alhasan S, Senderowicz AM, Sarkar FH. Induction of apoptosis and inhibition of c-erbB-2 in breast cancer cells by flavopiridol. Clin Cancer Res. 2000;6:223–229. [PubMed] [Google Scholar]
- Maxwell PH, Dachs GU, Gleadle JM, Nicholls LG, Harris AL, Stratford IJ, Hankinson O, Pugh CW, Ratcliffe PJ. Hypoxia-inducible factor-1 modulates gene expression in solid tumors and influences both angiogenesis and tumor growth. Proc Natl Acad Sci USA. 1997;94:8104–8109. doi: 10.1073/pnas.94.15.8104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melillo G, Sausville EA, Cloud K, Lahusen T, Varesio L, Senderowicz AM. Flavopiridol, a protein kinase inhibitor, down-regulates hypoxic induction of vascular endothelial growth factor expression in human monocytes. Cancer Res. 1999;59:5433–5437. [PubMed] [Google Scholar]
- Munaut C, Noel A, Hougrand O, Foidart JM, Boniver J, Deprez M. Vascular endothelial growth factor expression correlates with matrix metalloproteinases MT1-MMP, MMP-2 and MMP-9 in human glioblastomas. Int J Cancer. 2003;106:848–855. doi: 10.1002/ijc.11313. [DOI] [PubMed] [Google Scholar]
- Nawrocki ST, Sweeney-Gotsch B, Takamori R, McConkey DJ. The proteasome inhibitor bortezomib enhances the activity of docetaxel in orthotopic human pancreatic tumor xenografts. Mol Cancer Ther. 2004;3:59–70. [PubMed] [Google Scholar]
- Newcomb EW. Flavopiridol: Pleiotropic biological effects enhance its anti-cancer activity. Anti-cancer Drugs. 2004;15:411–419. doi: 10.1097/01.cad.0000127332.06439.47. [DOI] [PubMed] [Google Scholar]
- Newcomb EW, Tamasdan C, Entzminger Y, Arena E, Schnee T, Kim M, Crisan D, Lukyanov Y, Miller DC, Zagzag D. Flavopiridol inhibits the growth of GL261 gliomas in vivo: Implications for malignant glioma therapy. Cell Cycle. 2004;3:230–234. [PubMed] [Google Scholar]
- Orlowski RZ, Baldwin AS., Jr NF-κB as a therapeutic target in cancer. Trends Mol Med. 2002;8:385–389. doi: 10.1016/s1471-4914(02)02375-4. [DOI] [PubMed] [Google Scholar]
- Rapella A, Negrioli A, Melillo G, Pastorino S, Varesio L, Bosco MC. Flavopiridol inhibits vascular endothelial growth factor production induced by hypoxia or picolinic acid in human neuroblastoma. Int J Cancer. 2002;99:658–664. doi: 10.1002/ijc.10392. [DOI] [PubMed] [Google Scholar]
- Rapisarda A, Uranchimeg B, Scudiero DA, Selby M, Sausville EA, Shoemaker RH, Melillo G. Identification of small molecule inhibitors of hypoxia-inducible factor 1 transcriptional activation pathway. Cancer Res. 2002;62:4316–4324. [PubMed] [Google Scholar]
- Ravi R, Mookerjee B, Bhujwalla ZM, Sutter CH, Artemov D, Zeng Q, Dillehay LE, Madan A, Semenza GL, Bedi A. Regulation of tumor angiogenesis by p53-induced degradation of hypoxia-inducible factor 1α. Genes Dev. 2000;14:34–44. [PMC free article] [PubMed] [Google Scholar]
- Salceda S, Caro J. Hypoxia-inducible factor 1α (HIF-1α) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem. 1997;272:22642–22647. doi: 10.1074/jbc.272.36.22642. [DOI] [PubMed] [Google Scholar]
- Semenza GL. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu Rev Cell Dev Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- Senderowicz AM. Small-molecule cyclin-dependent kinase modulators. Oncogene. 2003;22:6609–6620. doi: 10.1038/sj.onc.1206954. [DOI] [PubMed] [Google Scholar]
- Shannon AM, Bouchier-Hayes DJ, Condron CM, Toomey D. Tumour hypoxia, chemotherapeutic resistance and hypoxia-related therapies. Cancer Treat Rev. 2003;29:297–307. doi: 10.1016/s0305-7372(03)00003-3. [DOI] [PubMed] [Google Scholar]
- Shweiki D, Itin A, Soffer D, Keshet E. Vascular endothelial growth factor induced by hypoxia may mediate hypoxia-initiated angiogenesis. Nature. 1992;359:843–845. doi: 10.1038/359843a0. [DOI] [PubMed] [Google Scholar]
- Takada Y, Aggarwal BB. Flavopiridol inhibits NF-κB activation induced by various carcinogens and inflammatory agents through inhibition of IκBα kinase and p65 phosphorylation: Abrogation of cyclin D1, cyclooxygenase-2, and matrix metalloprotease-9. J Biol Chem. 2004;279:4750–4759. doi: 10.1074/jbc.M304546200. [DOI] [PubMed] [Google Scholar]
- Tergaonkar V, Bottero V, Ikawa M, Li Q, Verma IM. IκB kinase-independent IκBα degradation pathway: Functional NF-κB activity and implications for cancer therapy. Mol Cell Biol. 2003;23:8070–8083. doi: 10.1128/MCB.23.22.8070-8083.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Unruh A, Ressel A, Mohamed HG, Johnson RS, Nadrowitz R, Richter E, Katschinski DM, Wenger RH. The hypoxia-inducible factor-1 α is a negative factor for tumor therapy. Oncogene. 2003;22:3213–3220. doi: 10.1038/sj.onc.1206385. [DOI] [PubMed] [Google Scholar]
- Voorhees PM, Dees EC, O’Neil B, Orlowski RZ. The proteasome as a target for cancer therapy. Clin Cancer Res. 2003;9:6316–6325. [PubMed] [Google Scholar]
- Wang GL, Jiang BH, Rue EA, Semenza GL. Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc Natl Acad Sci USA. 1995;92:5510–5514. doi: 10.1073/pnas.92.12.5510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagzag D, Zhong H, Scalzitti JM, Laughner E, Simons JW, Semenza GL. Expression of hypoxia-inducible factor 1α in brain tumors: Association with angiogenesis, invasion, and progression. Cancer. 2000;88:2606–2618. [PubMed] [Google Scholar]
- Zagzag D, Shiff B, Jallo GI, Greco MA, Blanco C, Cohen H, Hukin J, Allen JC, Friedlander DR. Tenascin-C promotes micro-vascular cell migration and phosphorylation of focal adhesion kinase. Cancer Res. 2002;62:2660–2668. [PubMed] [Google Scholar]
- Zagzag D, Nomura M, Friedlander DR, Blanco CY, Gagner JP, Nomura N, Newcomb EW. Geldanamycin inhibits migration of glioma cells in vitro: A potential role for hypoxia-inducible factor (HIF-1α) in glioma cell invasion. J Cell Physiol. 2003;196:394–402. doi: 10.1002/jcp.10306. [DOI] [PubMed] [Google Scholar]
- Zundel W, Schindler C, Haas-Kogan D, Koong A, Kaper F, Chen E, Gottschalk AR, Ryan HE, Johnson RS, Jefferson AB, Stokoe D, Giaccia AJ. Loss of PTEN facilitates HIF-1-mediated gene expression. Genes Dev. 2000;14:391–396. [PMC free article] [PubMed] [Google Scholar]