

Abstract
Little is known about the immune performance and interactions of CNS microglia/macrophages in glioma patients. We found that microglia/macrophages were the predominant immune cell infiltrating gliomas (~1% of total cells); others identified were myeloid dendritic cells (DCs), plasmacytoid DCs, and T cells. We isolated and analyzed the immune functions of CD11b/c+CD45+ glioma-infiltrating microglia/macrophages (GIMs) from postoperative tissue specimens of glioma patients. Although GIMs expressed substantial levels of Toll-like receptors (TLRs), they did not appear stimulated to produce pro-inflammatory cytokines (tumor necrosis factor α, interleukin 1, or interleukin 6), and in vitro, lipopolysaccharides could bind TLR-4 but could not induce GIM-mediated T-cell proliferation. Despite surface major histocompatibility complex class II expression, they lacked expression of the costimulatory molecules CD86, CD80, and CD40 critical for T-cell activation. Ex vivo, we demonstrate a corresponding lack of effector/activated T cells, as glioma-infiltrating CD8+ T cells were phenotypically CD8+CD25−. By contrast, there was a prominent population of regulatory CD4 T cells (CD4+CD25+FOXP3+) infiltrating the tumor. We conclude that while GIMs may have a few intact innate immune functions, their capacity to be stimulated via TLRs, secrete cytokines, upregulate costimulatory molecules, and in turn activate antitumor effector T cells is not sufficient to initiate immune responses. Furthermore, the presence of regulatory T cells may also contribute to the lack of effective immune activation against malignant human gliomas.
Keywords: costimulation, human glioma, macrophages, microglia, regulatory T cells, tumor immunotherapy
Malignant gliomas are the most common type of primary brain tumors, and glioblastoma multiforme (GBM)3 accounts for more than 50% of all intracranial gliomas (Davis et al., 2001). Although GBMs are immunogenic tumors (Fathallah-Shaykh et al., 1998; Holladay et al., 1994; Ridley, 1977), they are not eradicated by the immune system. Previously characterized impairments in glioma patient immunity have included low peripheral lymphocyte counts, reduced delayed-type hypersensitivity reactions to recall antigens, and impaired mitogen-induced blastogenic responses by peripheral blood mononuclear cells (PBMCs) (Heimberger et al., 2000). The modulation and down-regulation of T-cell function by immunosuppressive cytokines such as transforming growth factor β (TGF-β) and interleukin 10 (IL-10) elaborated by malignant gliomas has been previously characterized (Roussel et al., 1996). TGF-β and IL-10 are known potent inhibitors of macrophage function (Bogdan and Nathan, 1993; Tsunawaki et al., 1988) and of microglia production of nitric oxide synthase, NFκ-B activation, RANTES, and pro-inflammatory cytokines (Aloisi, 2001; Hu et al., 1999; Vincent et al., 1997). However, even when these factors are neutralized, efficacious immune responses still cannot be generated (Luscher et al., 1994). Furthermore, many subtypes of gliomas and cancers metastasizing to the brain do not secrete these immunosuppressive cytokines, yet still cannot generate efficacious immune responses, which indicates that other factors or cell types are involved in the lack of immunological responses.
Innate immunity is the initial antigen-nonspecific response that results in the rapid production of effector cytokines that in turn mature and activate antigen-presenting cells (APCs). One of the most important prerequisites for the immune activation of effector T-cell responses is the presence of functional APCs that process and present tumor antigens, secrete specific cytokines and chemokines, and abundantly express costimulatory molecules essential for initiating and/or propagating tumor-directed T cells (Banchereau and Steinman, 1998). Immature APCs use Toll-like receptors (TLRs) to recognize microbial or nonself tissue factors. TLR expression is a critical component of mediating innate immune responses to any infectious or traumatic challenge and activating adaptive immune responses. More specifically, the bacterial wall component lipopolysaccharide (LPS) activates through TLR-4 and the cluster of differentiation (CD) molecule CD14, peptidoglycan through TLR-2, dsRNA through TLR-3, and CpG through TLR-9 (Iwasaki and Medzhitov, 2004). Activation through the TLR results in maturation of the APC, increasing the expression of surface costimulatory molecules and cytokine production (Hemmi et al., 2000).
The activation of T cells requires two signals: The first signal is provided by the interaction of the T-cell receptor and major histocompatibility complex (MHC)/antigen complex, and the second signal is provided by the interaction of CD28 on the T cell and the costimulatory molecules CD80 (B7-1) or CD86 (B7-2) on the APC. In addition, CD40 is constitutively expressed on APCs while its ligand CD154 (CD40L) is expressed on activated T cells. The CD40-CD154 interaction provides a costimulatory signal to the APC (reverse costimulation), thereby augmenting the ability of APCs to present antigen and to deliver positive costimulatory signals, which in turn promotes T-cell activation (Tan et al., 1999). While naive T cells have been shown to be capable of entry into the CNS (Brabb et al., 2000; Krakowski and Owens, 2000), where they would require mature APC-mediated priming for activation, studies have also demonstrated that primed T cells can infiltrate and be maintained in murine brain tumors (Calzascia et al., 2003, 2005). In such situations, repeat stimulation and rerecognition of antigen by the T cells have proved necessary for tumor responses to be generated and to trigger migrant T cells to carry out their effector function (Gabrilovich et al., 1996; Oxenius and Bachmann, 1997). In either of these scenarios, if the APCs failed to provide appropriate stimulation, they could induce T-cell anergy and thereby contribute to the failure of the immune response.
The role and subtype of APCs involved in priming effector T cells within a malignant human glioma are unknown, but candidates have included microglia, monocytes/macrophages, B lymphocytes, and dendritic cells (DCs), in the latter of which there are two main subpopulations: myeloid (M-DCs) and plasmacytoid (P-DCs). Although microglia express MHC class I, controversy exists as to whether they also express MHC class II (Heimberger et al., 2000); thus it is unclear whether microglia can prime T-cell responses. Murine microglia have been shown to express the mRNA for all TLRs (TLR 1–9) (Olson and Miller, 2004) and can be strongly activated after in vitro stimulation with their specific agonists, especially LPS, which includes mounting a strong inflammatory cytokine response (Ebert et al., 2005). Studies have suggested that murine microglia can activate CD4+ helper T cells (McMahon et al., 2005) and have antigen-presenting capabilities (Juedes and Ruddle, 2001; Ulvestad et al., 1994). However, little is known about the immune performance and interactions of human microglia in the immunosuppressive glioma microenvironment, in part because of the scarcity of fresh human glioma tissue and the technical difficulty in obtaining pure microglia from CNS tissue. Previous analyses have relied on postautopsy cell isolations, primary microglial cultures, or rodent studies, which may not truly characterize these cells within human brain tumors.
Rodent microglia have been defined as having a CD11b/c+CD45Low phenotype, macrophages as having a CD11b/c+CD45High phenotype, and lymphocytes as having a CD11b/c−CD45High phenotype (Badie and Schartner, 2000; Ford et al., 1995). Although macrophages are believed to be activated microglia within the CNS, no distinct and universally accepted histological marker has been described in human brain tissue that can definitively distinguish macrophages from microglia. Previously, isolation of microglia from rodents has been performed using a series of Percoll gradients (Amersham Biosciences, Uppsala, Sweden) where the microglia were isolated from the interphase between the 1.065- and 1.072-g/ml gradients (Badie and Schartner, 2000; Ford et al., 1995). The microglia were found to express MHC I and II as well as costimulatory molecules. Most evidence for microglia cytokine production has come from immortalized human microglia cell lines that express the genes encoding IL-1β, IL-6, IL-8, IL-10, IL-12, IL-15, tumor necrosis factor α (TNF-α), and TGF-β (Nagai et al., 2001) and may not accurately depict the cytokine profile in GIMs.
In our study, we identify microglia/macrophages as the largest subpopulation infiltrating human gliomas. The GIMs do not produce cytokines reflective of activation despite known elaboration of TLR ligands by the glioma (Nakagawa et al., 1996; Steck et al., 1989). Furthermore, GIMs lack the essential costimulatory molecules CD80, CD86, and CD40, thus failing to provide adequate intratumoral T-cell stimulation, which was consistent with the failure of GIMs to activate T cells in an allogeneic mixed-lymphocyte reaction (allo-MLR). In addition, the in vivo phenotypes of glioma-infiltrating T cells were predominantly CD8+CD25− T cells and CD4+CD25+FOXP3+ regulatory T cells.
Materials and Methods
Patients
Patients’ tumors (n = 50) were graded pathologically as GBM by a neuropathologist according to the WHO classification. Fresh normal brain tissue (n = 6) can only rarely be obtained, since for most patients, efforts are made by the neurosurgeon to preserve this tissue. On occasion, normal brain tissue was obtained during an approach to a benign tumor such as a sphenoid wing meningioma, a well-circumscribed metastasis, or a low-grade lesion such as an oligodendroglioma. Additionally, peritumoral GBM tissue was obtained and separately analyzed; however, because this tissue could harbor microscopic tumor, it was not characterized as normal for the purposes of this study. The resected normal tissue demonstrated no evidence of increased signal on flair MRI preoperative imaging, and the brain tissue architecture was intraoperatively inspected microscopically by an experienced neurosurgeon (A.B.H.) and found to be consistent with that of normal brain tissue prior to submission to the laboratory. Peripheral blood was drawn from the patients intraoperatively. This study was conducted under protocol no. LAB03-0687, which was approved by the institutional review board of the University of Texas M.D. Anderson Cancer Center, and informed consent was obtained.
Antibodies
The cell surface was stained with phycoerythrin (PE)-labeled, fluorescein isothiocyanate (FITC)–labeled, or allophycocyanin-labeled antibodies against the following proteins: CD3, CD4, CD8, CD11b, CD11c, CD14, CD16, CD19, CD25, CD32, CD40, CD45, CD56, CD80, and CD86 (Pharmingen, San Diego, Calif.); CD14 and HLA DP, DQ, and DR (Dako Cytomation, Carpinteria, Calif.); TLR-1, TLR-2, TLR-3, and TLR-4 (eBiosciences, San Diego, Calif.); and CD1c and BDCA-2 (Miltenyi Biotec, Auburn, Calif.). For intracellular cytokine staining, we used PE-labeled or FITC-labeled antibodies against interferon γ (IFN-γ), TNF-α, IL-1β, IL-6, IL-10, and IL-12 (R&D Systems, Minneapolis, Minn.); and FOXP3 (eBiosciences). Appropriate isotype controls were used for every antibody.
Staining Fresh Tumor Specimens
Freshly isolated tumor tissue was mechanically dissociated, passed through a 70-μm-pore-size mesh, and washed. Nonspecific binding was blocked by incubation with purified anti-CD16 antibody (Pharmingen) and rabbit serum (Invitrogen, Carlsbad, Calif.) for 20 min and then stained with the fluorescence-labeled primary antibody or isotype control for 30 min at 4°C.
GBM or normal CNS tissue was stained with a three-color isotype control panel, which included FITC mouse immunoglobulin G1 (mIgG1), PE mIgG1, and APC mIgG1, as described in Results. Since the isotype controls were identical for both, we have displayed under Results only the isotype controls performed on GBMs. Approximately 1 × 106 live, gated events based on forward scatter and side scatter were assessed during fluorescence-activated cell sorting (FACS) using the FACs-Calibur instrument (BD Biosciences, San Jose, Calif.) and analyzed with CellQuest software (BD Immunocytometry Systems, San Jose, Calif.). The double-negative populations were not present because of the gating strategy that was employed as described in Fig. 1.
Fig. 1.

Gating strategy for immune cell analysis from CNS tissue. Cells were stained with IgG1-FITC, IgG1-PE, and IgG1-APC (isotype control) or CD1c-FITC, CD11c-PE, and BDCA-2-APC (stained) to identify myeloid DCs. In this example, GBM cells were analyzed by using a side-scatter versus forward-scatter dot plot on both the isotype (A) and stained cells (B) to select live cells (G1). The G1 gate was then applied to a CD11c versus CD1c dot plot (D) and its corresponding isotype (C). A statistical gate (G2) was drawn to indicate the CD11c+CD1c+ double positive (C and D). In the alternative gating strategy, isotype cells (E) and stained cells (F) were plotted on side scatter versus FITC, and FITC+ cells were gated (gate G3). By using the G3 gate, isotype and stained cells were analyzed on a CD11c versus CD1c dot plot (G and H, respectively). A statistical gate (G4) was drawn to indicate the CD11c+CD1c+ population. Percentages in C and D and in G and H indicate the percentage of cells within G2 or G4, respectively.
Isolation of Microglia/Macrophages from Human Brain Tumor Tissue
To purify microglia/macrophages from human brain tissue, we adapted a modified Percoll-gradient isolation technique previously described in rodent CNS tissue (Ford et al., 1995) after evaluation of the phenotype of each interphase. This technique minimized artificial activation of the microglia/macrophages, and these cells were isolated usually within 3 h of surgical resection. Briefly, after resection, freshly isolated tumor or normal brain tissue was mechanically dissociated through a stainless steel sieve. The dissociated material was centrifuged, washed, and layered onto an isotonic Percoll gradient diluted with PBS to 1.095 g/ml and overlaid with 1.03 g/ml Percoll. After centrifugation, the cell layer between the 1.095-g/ml and 1.03-g/ml layers was removed, washed, layered on top of a second gradient (2-ml increments of isotonic Percoll diluted to 1.12, 1.088, 1.072, 1.065, and 1.03 g/ml), and centrifuged. Microglia/macrophages were collected from the interface between the 1.065-g/ml and 1.03-g/ml layers and washed, and their viability was determined by the Trypan blue dye–exclusion method.
Prioritization of Characterization
Microglia/macrophages could never be isolated in sufficient numbers from a single GBM specimen to perform all of the assays concurrently. Furthermore, there is significant variability in the numbers of microglia/macrophages obtained from each clinical specimen that could not be predicted preoperatively. A minimum of 5 g of tissue was necessary in order to isolate sufficient microglia/macrophages for the basic phenotypic characterizations. Purity was established on all specimens regardless of the assay priority. Fresh surgical tissue was analyzed as follows: (1) If large numbers (3–4 million) of microglia/macrophages could be generated from a GBM specimen (>10 g), the priority went to performing the allogeneic mixed-lymphocyte responses, and routinely drawn intraoperative peripheral blood was accessed for the controls. (2) For specimens >5 g, the microglia/macrophages were analyzed for MHC and costimulator molecules, then intracellular cytokine analysis, then TLR expression, and then TLR responses to incubation with LPS-FITC. (3) For specimens <5 g, priority went to performing the characterization of the immune cells infiltrating the unprocessed glioma or normal brain tissue.
Surface Marker and Intracellular Cytokine Staining
Microglia/macrophages were blocked for nonspecific binding by using purified anti-CD16 antibody (Pharmingen) for 20 min, and after washing, the cells were incubated with the fluorescence-labeled primary antibody or isotype control for 30 min at 4°C. For the intracellular cytokine analysis, microglia/macrophages were fixed with Cytofix/Cytoperm (BD Biosciences), washed in PermWash (BD Biosciences), and then stained with fluorescence-labeled monoclonal antibodies or isotype controls for 30 min at 4°C. Positive controls for all of the monoclonal antibodies consisted of either stimulated human lymphocytes (Pharmingen) or A375 cells (E.G.). Approximately 1 × 104 live, gated events were assessed during FACS by using an Epics XL-MCL cytometer (Beckman-Coulter, Fullerton, Calif.) and analyzed with IsoContour software (Verity Software House, Turramurra, Australia).
For the FOXP3 intracellular staining, the cells were first surface stained as described above with CD4 and CD25, followed by fixation and permeabilization with the respective buffers (eBiosciences), and then stained with FOXP3 antibody or the respective isotype control for 30 min at 4°C. Cells were analyzed with a FACSCalibur flow cytometer with CellQuest software.
Allogeneic Mixed Lymphocyte Responses
Stimulator cells were either microglia/macrophages isolated from patients’ tumors as described above or APCs isolated from the peripheral blood of the respective glioma patients as follows. The buffy layer was isolated from patient peripheral blood after Ficoll density centrifugation. Cells were washed and labeled with CD3 microbeads (Miltenyi Biotec) at 20 μl/107 cells for 15 min at 4°C. The cells were washed and passed through a magnetic separation column (LD MACS Column; Miltenyi Biotec) placed in the magnetic field of a MACS separator (Miltenyi Biotec). Magnetically labeled CD3+ cells were retained on the column while the unlabeled cell fraction that was CD3− ran through. Those CD3− cells were washed and determined by flow cytometry staining to be 97% pure for CD14+, CD19+, CD11c+, CD11b+, MHC II+, and CD3− cells.
Responder T cells from peripheral blood obtained from a normal donor were isolated, stained with CD3 micro-beads, and separated as described above, except that the unlabeled CD3− cells were allowed to run through, the column was washed, and the magnetically labeled CD3+ fraction was flushed from the column. The CD3+ cells were washed and determined to be 97% T cells by flow cytometry. Responder T cells were incubated with carboxyfluorescein diacetate succinimidyl ester (CFSE) for 15 min and washed thoroughly. Responder T cells were then labeled with CFSE as described earlier under the Antibodies section. For the mixed-lymphocyte reaction, in a 96-well plate, CFSE-labeled responder allo-T cells (0.75 × 105 cells/50 μl/well) were added to the patient stimulators (2.25 × 105 cells/50 μl/well) with or without LPS (5 μg/ml) (purified LPS [smooth] from Escherichia coli 0111:B4; Sigma-Aldrich Corp., St. Louis, Mo.) and incubated for four days at 37°C, 5% CO2. Controls included T cells incubated in media alone, each stimulator in media alone, and T cells stimulated with 5 μg/ml of purified anti-CD3 antibody (Pharmingen). Following incubation, cells were labeled with PE-anti-CD3 antibody to further gate out the CFSE-labeled responder T cells for analysis by FACS.
LPS-FITC Binding to TLR
Purified microglia/macrophages (5 × 105/well) isolated from patients’ glioma tissue were incubated with 1 μg/ml of LPS-FITC (purified LPS [smooth] from E. coli 0111: B4 conjugated with FITC; Sigma-Aldrich Corp.) for 5-min and 30-min incubation periods at 37°C, 5% CO2. Following incubation, cells were washed thoroughly in staining buffer and stained with TLR-4. Appropriate controls included cells stained with the isotype control for TLR-4 and cells incubated in media alone with no LPS-FITC stimulation.
Results
Flow Cytometry Gating Strategy to Identify Rare Cell Populations in Glioma Tissue
Tumor suspensions are markedly heterogeneous. Secondary to the rarity of the immune cells within these specimens, we used a specific flow cytometry approach to identify these cells in glioma tissue. In Fig. 1, as a representative sample, we demonstrate our flow cytometry strategy for analysis of CD11c+CD1c+ M-DC from glioma tissue. On a side-scatter versus forward-scatter plot, dead cells were excluded when using gate G1 on both the isotype (Fig. 1A) and surface marker–stained cells (Fig. 1B). We then applied this G1 gate on a CD11c versus CD1c dot plot and its corresponding isotype (Fig. 1D and C, respectively). A quadrant gate was drawn on the CD11c versus CD1c dot plot based on the isotype control to exclude the double-negative-stained cells. A statistical gate (G2) was then drawn to identify the CD11c+CD1c+ double-positive population in the upper right quadrant. The G2 gate excludes the autofluorescent population that is seen in human CNS tissue suspensions (Watters et al., 2005). Some of the double-positive cells may be lost in the autofluorescent population; however, the number of autofluorescent cells in the stained specimens and that in the corresponding isotypes remain almost identical. Therefore, we excluded this population in our analysis of double-positive cells. In addition, with the same compensation settings, single-stained (with only one fluorochrome marker) tumor samples that are similarly analyzed show distinct positive cell populations in the respective upper left or lower right quadrant (data not shown).
Since the double-positive cells are rare in CNS tissues (0.2% [Fig. 1D]) and can appear masked by the abundant background cells, to identify these immune cells successfully and gain a better perspective on their distribution in glioma tissue, we adopted a different gating strategy that enabled us to focus solely on these infrequent events. As shown in Fig. 1E–H, we analyzed the data on a side-scatter versus FITC plot. The gate for the FITC+ cells (G3) included only the CD1c−FITC+ cells (Fig. 1F) when compared to the isotype control (Fig. 1E). This G3 gate (CD1c+ cells) was then applied to a CD11c versus CD1c dot plot and its corresponding isotype (Fig. 1H and G, respectively). Finally, a statistical gate (G4) was drawn to identify the CD11c+CD1c+ double-positive population in the upper right quadrant of these two plots but to exclude the autofluorescent population (Fig. 1G and H). The same gating strategy was used in Fig. 1E–H to identify the different immune cells (microglia, plasmacytoid DCs, B cells, and T cells), as demonstrated in Fig. 2 and described under the section Intratumoral T Cells.
Fig. 2.

Immune cells infiltrating GBMs and normal brain tissue. Dissociated GBM or normal tissue was triple stained with fluorescent conjugated antibodies and analyzed by flow cytometry. The percentage indicated in each respective quadrant/gate denotes the number of gated cells that are positive for the expression of the respective surface markers and subtracts the isotype background. The panel is representative of one glioblastoma tissue specimen and one normal brain specimen. A. All CD11c+ cells were gated, and these cells were then observed for simultaneous CD11b and CD45 surface marker expression. Microglia/macrophages are the CD11c+CD11b+CD45+ cells as indicated by the gate in the upper right quadrant for both GBM tissue and normal tissue. B. CD1c+ gated cells that were BDCA-2− and CD11c+ representing myeloid DCs are indicated by the gate in the upper right quadrant for GBM tissue and normal brain tissue. C. BDCA-2+ gated cells that were CD11c− and CD1c− representing plasmacytoid DCs are in the upper left quadrant for GBM tissue and normal brain tissue. D. The upper right quadrant represents the B cells that are MHC II+ and CD19+ in both gliomas and normal brain. (E) Numbers in each quadrant are CD3+ that are either CD4+ (upper left quadrant) or CD8+ (lower right quadrant) T cells within the normal brain parenchyma or GBM tissue. An autofluorescent population was identified in both normal and GBM tissue samples during flow cytometry analysis and is also consistently present in the respective isotype controls. This population was excluded when calculating the percentages of positive fluorescing cells.
Characterization of the Immune Cells Infiltrating Human Gliomas
To determine the different subsets of immune cells within human gliomas, we stained freshly isolated patient glioma tissue with fluorescence-labeled antibodies to specific surface markers. In the glioma, we found a predominant population of microglia/macrophages (Fig. 2A and Table 1). In contrast, microglia/macrophages were present in strikingly lower numbers in normal brain tissue, emphasizing the infiltration of these cells into glioma tissue. In GBM specimens, we observed far fewer infiltrating DCs relative to microglia/macrophages; in comparison to normal tissue, DCs were more common relative to microglia/macrophages. However, 20% of the CD11c+ gated cells were M-DCs (CD1c+) in GBM specimens, which was significantly greater than that seen in normal brain tissue (1% CD1c+CD11c+ gated cells) (Fig. 2B) and represented an absolute increase the in total number of M-DCs in GBM as compared to that in normal brain tissue (Table 1). In contrast, 2% of BDCA-2+ gated cells were P-DCs (CD11c−) in GBM specimens, which was less than that seen in normal brain tissue (15%) (Fig. 2C), but this does not represent an absolute increase in the total number of P-DCs (Table 1). B cells were infrequent in both GBM and normal tissue (MHC class II+CD19+) (Fig. 2D). As expected, the GBM tissue contained both infiltrating CD4+ and CD8+ T cells (Fig. 2E). Since the majority of intratumoral immune cells were microglia/macrophages, we proceeded to a more detailed analysis of these cells, including an assessment of phenotype and functional capabilities to participate in innate and adaptive immune responses.
Table 1.
Frequency of immune cells within total GBM tissue versus normal brain tissue*
| Microglia | M-DCs | P-DCs | B cells | T cells | T cells | ||
|---|---|---|---|---|---|---|---|
| Tissue | CD45+ CD11b+ CD11c+ | CD1c+ CD11c+ BDCA-2− | BDCA-2+ CD11c− CD1c− | MHC II+ CD19+ | CD3+ CD4+ | CD3+ CD8+ | |
| GBM | N | 6 | 6 | 6 | 5 | 6 | 6 |
| Range (%) | 0.11–2.88 | 0.025–0.38 | 0.0009–0.03 | 0.0009–0.14 | 0.0045–0.2 | 0.0023–0.074 | |
| SD | 1.06 | 0.14 | 0.01 | 0.06 | 0.08 | 0.03 | |
| Mean (%) | 0.825 | 0.132 | 0.015 | 0.034 | 0.099 | 0.039 | |
| Median (%) | 0.365 | 0.063 | 0.014 | 0.01 | 0.088 | 0.043 | |
| Normal | N | 2 | 2 | 2 | 2 | 2 | 2 |
| Range (%) | 0.0035–0.0099 | 0.0008–0.0013 | 0.009–0.015 | 0.0001–0.0003 | 0.0004–0.00013 | 0.00076–0.0014 | |
| SD | 0.005 | 0.0003 | 0.004 | 0.0001 | 0.0002 | 0.0005 | |
| Mean (%) | 0.007 | 0.001 | 0.012 | 0.0002 | 0.0003 | 0.001 | |
| Median (%) | 0.0067 | 0.001 | 0.012 | 0.0002 | 0.0003 | 0.001 |
Abbreviations: CD, cluster of differentiation; GBM, glioblastoma multiforme; M-DCs, myeloid dendritic cells; MHC, major histocompatibility complex; N, total number of specimens analyzed; P-DCs, plasmacytoid dendritic cells.
The percentage of positive cells (numerator) was calculated by flow cytometry analysis from ungated tissue specimens. The denominator is the total of all cells prior to any type of purification, cell sorting, or gating.
Purity of Microglia/Macrophages Isolated from Human Gliomas
To obtain human microglia/macrophages, we performed a series of Percoll-gradient purifications from normal human brain tissue and from freshly resected human glioma tissue. To determine the purity of our microglia/macrophage isolation following Percoll-gradient purification, cells were stained with fluorescence-labeled antibodies to CD11b, CD11c, and CD45. We observed the largest population of CD11b/c+CD45+ cells at the inter-phase between the 1.03-g/ml and 1.065-g/ml gradients (Fig. 3A), unlike in the rodent studies, where the inter-phase between the 1.065-g/ml and 1.072-g/ml gradients was determined to contain the largest fraction of CD11b/c+CD45+ cells (Ford et al., 1995). The expression levels of CD45 and CD11b/c were not significantly different in the cells isolated from each interphase (Fig. 3A). Thus, in contrast to the rodent purification of microglia/macrophages in which distinct CD11b/c+CD45High and CD11b/c+CD45Low cells were identified (Ford et al., 1995), our purifications of human microglia did not reveal this heterogeneity in any GBM or normal brain tumor specimens. The human GIMs expressed Fc receptor II (FcR II) and FcR III, as expected (Fig. 3B). All the GIMs were positive for CD14 expression (Fig. 3C), an activation marker that plays a critical role in LPS-mediated TLR-4 activation. To characterize the cell populations from each interphase further, the cell populations were also stained with antibodies to CD3, CD4, CD8, CD56, and BDCA-2+ and found to be negative for B cells, T cells, natural killer (NK) cells, and DCs (Fig. 3D and data not shown).
Fig. 3.

GIM can be isolated as a pure population. A. Each interphase from the second Percoll gradient was analyzed for expression of the surface markers CD11b and CD45. The gated cells in the upper right quadrant indicate the percentage of CD45+ gated cells that were positive for CD11b and represent GIM. An autofluorescent population was identified in both normal and GBM tissue samples during flow cytometry analysis and is also consistently present in the respective isotype controls. This population was excluded when calculating the percentages of positive fluorescing cells. B. GIMs express the Fc receptors CD16 and CD32. C. The GIM cells express CD14. D. The GIM population does not contain T cells (CD3+) or NK cells (CD56+). In each histogram of panels B–D, the data plot on the left represents the isotype control, and the plot on the right represents cells that express the respective surface marker. The data are from one tumor specimen but are representative of at least 20 tumor tissue samples from patients with GBM.
The average yield of microglia/macrophages isolated from human GBM was 3.2 × 106 cells per gram of tumor (range, 1.8 × 105 to 3.7 × 107 cells/g; n = 15) but was only 1.4 × 105 cells per gram of normal brain (range, 4.6 × 104 to 3 × 105; n = 5). This represented an average, although highly variable, 30% recovery and a 60-fold enrichment of the microglia/macrophages. The purity of GIM isolated from human glioma specimens can vary because of disparities in each excised individual tissue sample.
TLR Expression on GIM
To determine whether GIM expressed the TLRs, we stained the purified GIM with fluorescence-labeled antibodies to TLR-1, -2, -3, and -4. Although TLR-1 was not expressed at significant levels, TLR-2, -3, and -4 were highly expressed on GIM as determined by fluorescence intensity. Similar expression levels of fluorescent intensity of TLRs were found on microglia/macrophages isolated from normal brain tissue (Fig. 4 and Table 2) or from microglia/macrophages obtained from peritumoral GBM (data not shown).
Fig. 4.

Expression of TLR on GIM and microglia/macrophages isolated from normal brain. Purified microglia/macrophages from GBM tissue and normal brain tissue were double-stained for CD45 and TLR expression. An autofluorescent population was identified in both normal and GBM tissue samples during flow cytometry analysis and is also consistently present in the respective isotype controls. This population was excluded when calculating the percentages of positive fluorescing cells.
Table 2.
Percentages of purified glioma-infiltrating microglia or microglia isolated from normal brain tissue expressing the listed cell surface marker or cytokine*
|
Tissue |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
|
GBM |
Normal Brain |
||||||||||
| Cell Surface Marker or Cytokine | Gate | Number of Specimens Analyzed | Median (%) | Mean (%) | Range (%) | SD | Number of Specimens Analyzed | Median (%) | Mean (%) | Range (%) | SD |
| HLA+ | CD11b+ | 9 | 8 | 15.7 | 5–36 | 12.7 | 4 | 14.6 | 14.6 | 8–21 | 7.2 |
| CD80+ | CD11b+ | 12 | 0 | 0.4 | 0–3 | 0.9 | 5 | 0.8 | 2.0 | 0–7 | 2.9 |
| CD86+ | CD11b+ | 11 | 2.5 | 4.1 | 0–13 | 4.4 | 5 | 2.17 | 2.8 | 0.7–7 | 2.4 |
| TNF-α+ | CD45+ | 5 | 0 | 0 | 0 | — | 2 | 0 | 0 | 0 | — |
| IFN-γ | CD45+ | 5 | 0 | 0 | 0 | — | 2 | 0 | 0 | 0 | — |
| IL-1β+ | CD45+ | 5 | 0 | 0 | 0 | — | 2 | 0 | 0 | 0 | — |
| IL-6+ | CD45+ | 5 | 0.3 | 1.2 | 0–5 | 2.1 | 2 | 0.65 | 0.65 | 0.3–1 | 0.5 |
| IL-10+ | CD45+ | 5 | 0 | 0 | 0 | — | 2 | 0 | 0 | 0 | — |
| IL-12+ | CD11b+ | 5 | 20 | 21.6 | 14–33 | 7.3 | 3 | 23 | 29.1 | 21–41 | 11.1 |
| TLR-1+ | CD45+ | 3 | 3.3 | 4.0 | 0.9–8 | 7.5 | 4 | 10 | 15.3 | 0–40 | 17.4 |
| TLR-2+ | CD45+ | 4 | 21.6 | 21.6 | 15–28 | 9 | 4 | 12 | 16.9 | 5–39 | 15.1 |
| TLR-3+ | CD45+ | 3 | 16 | 17.6 | 15–21 | 2.9 | 4 | 10.3 | 9.8 | 3–15 | 6.5 |
| TLR-4+ | CD45+ | 4 | 20 | 20.3 | 14–26 | 8 | 5 | 7.7 | 13.1 | 2.4–38 | 14.6 |
Abbreviations: CD, cluster of differentiation; GBM, glioblastoma multiforme; IFN, interferon; IL, interleukin; TLR, Toll-like receptor; TNF, tumor necrosis factor.
The percentage of positive cells was calculated after subtracting for isotype staining as previously described. All cells were gated on either CD11b+ or CD45+ (both markers are expressed on microglia) out of a total of 30,000 analyzed cells. There was no statistically significant difference in the levels of surface marker expression between glioma-infiltrating microglia and microglia from normal brain tissue.
Intracellular Cytokine Analysis of GIMs
To determine if the microglia/macrophages are activated and elaborating cytokines that may modulate immune responses, intracellular cytokine analysis was performed for IL-1β, IL-6, IL-10, IL-12, IFN-γ, and TNF-α. GIMs demonstrated significant production of only IL-12 (Fig. 5 and Table 2) when compared to the production other cytokines. However, this production of IL-12 was no different from the intracellular cytokine production in microglia/macrophages from normal brain (n = 3). All other cytokines (i.e., IL-1, IL-10, and TNF-α) were either found at minimal levels or were absent.
Fig. 5.

Cytokine production by GIM and microglia/macrophages isolated from normal brain. Purified microglia/macrophages were freshly isolated from GBM tissue and stained for the intracellular cytokines IL-6, IL-10, IL-12, TNF-α, and IFN-γ without in vitro stimulation. Cytokines were detected by using fluorescence-labeled antibodies and analyzed by flow cytometry. Positive intracellular cytokine expression was confirmed for all cytokines in either a reporter cell line (A375) or stimulated lymphocytes (data not shown). Microglia/macrophages isolated from normal brain were also analyzed for the same cytokine expression profile.
GIM Expression of MHC but Not Costimulatory Molecules
To determine whether GIM cells can participate in adaptive immunity as functional CNS APCs, these cells were analyzed for surface MHC and costimulatory molecule expression. Microglia/macrophages from GBM and those from normal tissue both express MHC class II (Fig. 6A and Table 2).
Fig. 6.

MHC class II and costimulatory marker expression on GIM and microglia/macrophages from normal brain. In each histogram, the data plot on the left represents the isotype control, and the second plot represents CD45+ or CD11b+ gated cells that express the respective surface marker. A. GIMs were gated on CD11b+ expression and were analyzed for the expression of MHC class II antigens (HLA DR, DP, or DQ). GIMs were gated on CD45+ expression and analyzed for the expression of the costimulatory molecules CD80 (B), CD86 (C), and CD40 (D). Microglia/macrophages isolated from normal brain were also assessed for surface expression of the above markers except CD40 (A–C).
To establish whether microglia/macrophages express the costimulatory molecules, these cells were stained with fluorescence-labeled anti-CD80 and anti-CD86 antibodies. GIMs did not express CD80, whereas CD86 was largely absent but was expressed at more variable levels (Fig. 6B and C). These findings were comparable to those seen in microglia/macrophages isolated from normal brain tissue and from peritumoral GBM microglia/macrophages. In addition, CD40 was not expressed on the GIM cell surface (n = 12) (Fig. 6D). Collectively, these findings indicate that GIMs do not express the costimulatory molecules necessary for them to participate in adaptive immunity.
Functional Capabilities of GIMs to Stimulate T-Cell Responses
To determine if the lack of surface expression of costimulatory molecules on GIMs affected their ability to stimulate T cells, allo-MLRs were performed with either patient GIMs or APCs isolated from the patient’s peripheral blood as stimulators and allogeneic T cells from a normal donor as responders. Negligible proliferation (2%) was seen in T cells without an activated APC population (Fig. 7A). The addition of the peripheral blood APCs enhanced T-cell proliferation slightly (9%) and was likely nonspecific activation (Fig. 7B). However, there was marked T-cell proliferation when the APCs were activated with LPS (36%) or the T cells activated with anti-CD3 antibody (48%) (Fig. 7B and A, respectively). In marked contrast, GIMs were incapable of inducing significant T-cell stimulation, regardless of whether they were stimulated by LPS; T-cell proliferation remained similar to baseline levels (8% vs. 9%) (Fig. 7C).
Fig. 7.

Functional activity of GIMs: Allo-MLR. Stimulator cells were either GIMs isolated from patients’ tumors or APCs isolated from the peripheral blood of the respective glioma patients as described in Materials and Methods. Responder T cells were from an allogeneic normal donor, CFSE labeled, and incubated with the different stimulators. Following incubation, cells were labeled with PE-conjugated anti-CD3 antibody to further gate out the CFSE-labeled responder T cells for analysis by flow cytometry. The percentage of allo-T cells that underwent proliferation, as determined by dilution of the fluorescence intensity of the CFSE dye on gated CD3+ T cells, is indicated in each data plot. A. Controls included T cells incubated in media alone and T cells stimulated with anti-CD3 antibody. B. CFSE-labeled responder allo-T cells were incubated with the patient’s peripheral blood APCs either with or without LPS. C. CFSE-labeled allo-T cells were incubated with GIMs either with or without LPS.
Binding of LPS to TLR-4 on GIMs
GIMs express significant levels of the LPS receptors TLR-4 and CD14 (Fig. 4 and 3C, respectively). To determine whether GIMs are unresponsive to LPS-mediated stimulation because of the inability of LPS to actually bind the TLR-4 receptor, we incubated freshly isolated GIMs with FITC-conjugated LPS. LPS binding is detected on the surface of GIMs when compared to cells not stimulated with LPS (Fig. 8A) and appears to be present only on the cells expressing TLR-4 (Fig. 8B). Similar data were obtained for LPS binding via CD14 (data not shown).
Fig. 8.
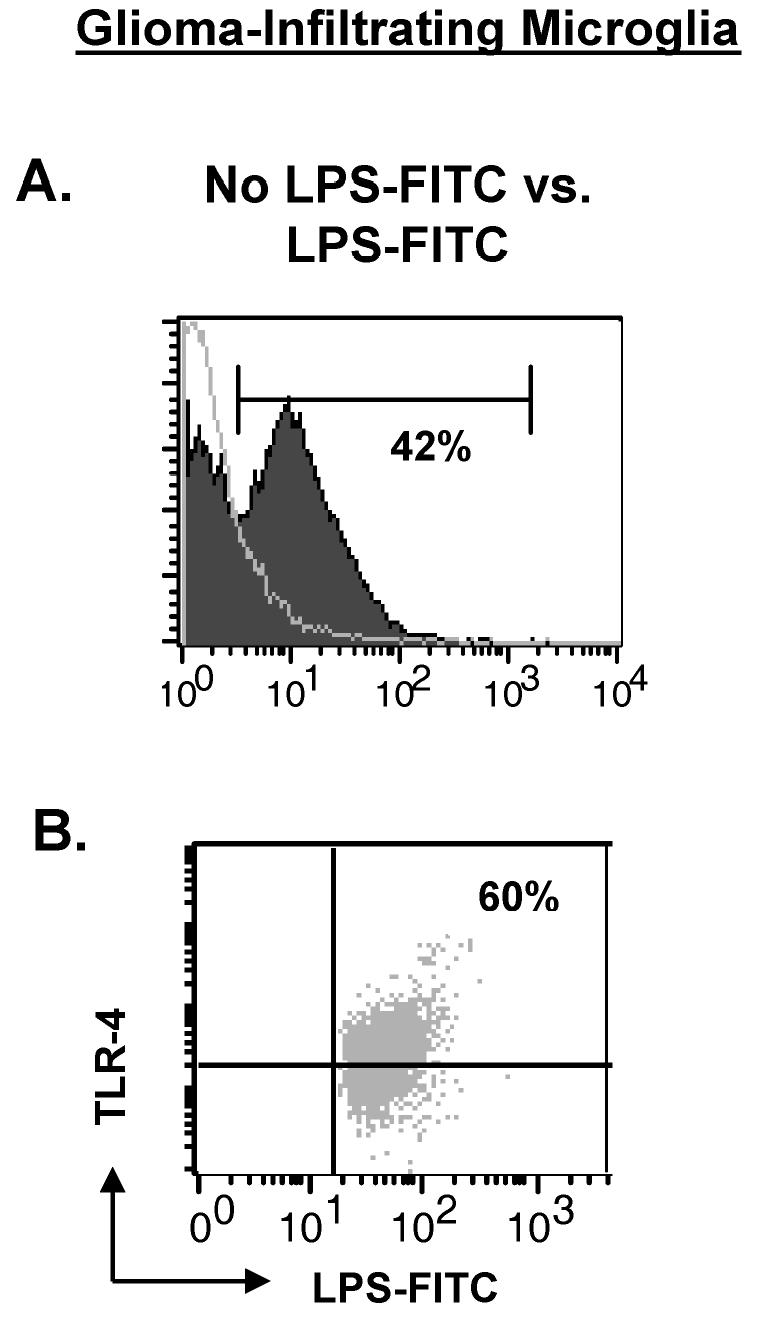
LPS can bind the surface of GIMs via TLR-4. Purified microglia/macrophages from GBMs tissue (n = 2) were incubated with LPS-FITC for 30 min and stained with TLR-4. Controls included GIMs that were stained with TLR-4 but not stimulated with LPS-FITC and GIMs that were incubated with LPS-FITC and stained with isotype to TLR-4 (data not shown). A. Histogram overlay demonstrating LPS binding to the surface of GIM stimulated with LPS (filled graph) compared to GIM without LPS stimulation (clear graph). B. GIMs were gated on LPS+ expression and examined for an LPS+TLR-4+ double-positive population (upper right quadrant).
Intratumoral T Cells
Since GIM produced IL-12, a CD8+ T-cell-stimulating cytokine, we wanted to determine if there was a population of activated tumor-infiltrating T cells in the gliomas. Gliomas were dissociated and stained for the CD8+ and CD4+ subsets (Fig. 9A). The tumor-infiltrating CD8+ T cells were phenotypically CD8+CD25− (Fig. 9B), which indicated that these cells were not activated. CD4+ T cells were more numerous than CD8+ T cells within glioma tissue (20% vs. 11% gated lymphocytes) (Fig. 9C), and a significant number were also CD25+, which indicated possible regulatory T-cell phenotype. This was confirmed by demonstrating that glioma-infiltrating CD4+ T cells, as well as peripheral blood CD4+ T cells from patients, were not only CD25+ but also positive for the intracellular marker FOXP3 (Fig. 9D), a transcription factor that is elevated only in regulatory T cells (Treg cells) and not in activated effector CD4+ T cells (Fontenot and Rudensky, 2005).
Fig. 9.

T-cell phenotype in GBM. Patients’ glioma cells and PBMCs were stained with fluorescent conjugated antibodies and analyzed by flow cytometry. An autofluorescent population was identified in GBM tissue samples during flow cytometry analysis and is consistently present in the respective isotype controls. This population was excluded when calculating the percentages of positive fluorescing cells. The panel is representative of one glioblastoma tissue specimen and one PBMC specimen. A. GBM cells were gated on CD3+ cells and examined for CD4+ (upper left quadrant) and CD8+ (lower right quadrant) expression. B. GBM cells were gated on CD8+ expression (by using the R1 gate on Fig. 8A) and examined for CD8+CD25+ expression (upper right quadrant). C. GBM CD4+ T cells were gated (by using the R2 gate on Fig. 8A) and examined for CD4+CD25+ expression in the upper right quadrant. D. GBM CD4+ T cells were gated (by using the R2 gate on Fig. 8A) and examined for FOXP3 expression in the upper right quadrant. E. Patients’ PBMCs were gated on lymphocytes by size and density and examined for CD4+ (upper left quadrant) and CD8+ (lower right quadrant) expression. F. PBMCs were gated on CD8+ expression (by using the R3 gate on Fig. 8A) and examined for CD8+CD25+ expression (upper right quadrant). G. PBMC CD4+ T cells were gated (by using the R4 gate on Fig. 8A) and examined for CD4+CD25+ expression in the upper right quadrant. H. PBMC CD4+ T cells were gated (by using the R4 gate on Fig. 8A) and examined for FOXP3 expression in the upper right quadrant.
Discussion
For the first time, we have isolated human microglia/macrophages from human brain and malignant gliomas and examined their phenotype and function. In stark contrast to the identification of distinct CD45Low and CD45High subpopulations in rodent studies, in our study, these subpopulations were not identified in microglia/macrophages isolated from human glioma tissue as has been previously suggested (Watters et al., 2005). Macrophages are considered to be activated microglia, and thus our isolated “microglia” may all be activated within the malignant glioma. However, we would have anticipated that these subpopulations would be identified in the “microglia” isolated from normal brain without any type of initiator for inflammation. Alternatively, this distinction may be artifactual to the monoclonal antibodies or to the model system. Our data indicate that the cells we have isolated are not contaminated with other immune cells such as B cells, T cells, NK cells, or DCs. Previous investigators (Ford et al., 1995) determined in their studies on normal rodent brains that the CD45HighCD11b/c+ cells classified as macrophages had significant APC activity, whereas the CD45Low CD11b/c+ microglia clearly did not. Although we cannot definitely determine whether our isolated population from human brain tissue represents microglia or macrophages or a cell population within the continuum, it is clear from our data that none of the cells isolated have functional APC activity or APC phenotype, which reinforces the possibility that the cells are mostly microglia. Since these cells are clearly very prominent and common within malignant gliomas, their role in controlling immunological responses within the CNS is nonetheless important, especially in the setting of immunotherapeutic approaches.
Within human gliomas, microglia/macrophages are a predominant immune subset. However, they do not appear to be fully able to modulate innate and adaptive immune responses. Activation of innate immunity triggers cytokine production and the subsequent activation of adaptive immune responses (Akira et al., 2001; Ito et al., 2002) that are essential for tumor eradication/suppression. Although it has been argued that tumors fail to provide sufficient pro-inflammatory responses, others have suggested otherwise (Matzinger, 1994; Pardoll, 2003). Factors that induce innate immune responses similar to microbial pathogens include heat shock proteins (Asea et al., 2002; Ohashi et al., 2000; Srivastava, 2002), hyaluronic acid (Termeer et al., 2002), and heparin sulfates (Johnson et al., 2002), which are all prodigiously produced by gliomas (Glimelius et al., 1978; Nakagawa et al., 1996; Neuhaus-Steinmetz and Rensing, 1997; Steck et al., 1989). These factors are TLR-4 agonists (Beg, 2002; Habich et al., 2002; Kakimura et al., 2002), and although TLR-4 is expressed on GIM cells, we were surprised to find that, unlike the GIMs of the murine system (Ebert et al., 2005), these human GIMs did not express cytokines that would be reflective of the innate immune arm being activated. Instead, the cytokine profile of GIMs is similar to that of resting microglia isolated from normal brain tissue, which suggests that TLR agonists within malignant gliomas do not activate GIMs. Furthermore, LPS, a known TLR-4 agonist and stimulator of murine microglia activity (Ebert et al., 2005), did not improve the ability of GIMs to stimulate allogeneic T cells in vitro. We cannot exclude the possibilities that activation of the innate immune system could occur with alternative TLR agonists, cytokine-mediated stimulation, or even alternative CNS cells. Finally, despite the presence of TLR-4 and its ability to bind its agonist LPS, the GIMs are apparently unresponsive to LPS-induced stimulation, which suggests a breakdown of key signaling pathways following the triggering of the TLR-4-CD14 complex. Studies are currently under way to ascertain if the failure of LPS response despite adequate expression of TLR is secondary to a breakdown in signaling pathways following triggering of the TLR-4-CD14 complex. The use of TLR agonists for immunotherapy in glioma patients needs to be carefully considered because this may not necessarily result in activation, especially in the presence of immunosuppressive cytokines. Although GIM and microglia/macrophages isolated from normal brain tissue express MHC class II, neither population expressed significant levels of costimulatory molecules. The expression of these costimulatory molecules and that of MHC appear to vary between species, model systems, and disease states. Previously, human microglia have been shown to express CD86 but not CD80 during autoimmunity (multiple sclerosis) and infection (HIV) (Dangond et al., 1997; Satoh et al., 1995). On the other hand, parenchymal microglia from normal adult mice express high levels of CD80, CD86, and MHC class II (Zhang et al., 2002), while parenchymal microglia from normal rat brains express high levels of CD86 but not CD80 or MHC class II (Badie et al., 2002). Since T-cell activation requires signals through both MHC and costimulatory molecules, the expression of MHC alone on microglia/macrophages would not activate a T-cell response but could result in T-cell anergy (Hu et al., 2002; Yi-qun et al., 1997), which has previously been proposed as the means by which gliomas elude immune defense despite the presence of antigenic stimuli. Murine microglia expressing low levels of costimulatory molecules have been unable to activate either naive or primed T cells and can induce T-cell anergy (Matyszak et al., 1999). Indeed, our in vitro data from the allo-MLR indicate that human GIMs are unable to activate T cells effectively, and ex vivo analyses of the glioma infiltrate show a marked absence of activated CD8 T cells (CD8+CD25+). This phenomenon appears to occur despite the presumed secretion of IL-12 by the isolated GIMs. IL-12 is a potent stimulator of effector CD8 T cells. However, studies have previously shown that once T-cell anergy has been established, IL-12 alone is unable to reverse this state and that it takes additional factors such as CTLA4 blockade to reverse this anergy (Andrews et al., 1997; Quill et al., 1994; Van Parijs et al., 1997). We can not exclude the possibility that the rare DCs identified within the gliomas could act as APCs mediating immune responses within the immunosuppressive environment, especially since recent studies in murine experimental allergic encephalomyelitis models have described a minority population of CNS DCs that were adequate to restimulate adoptively transferred antigen-experienced effector cells (Greter et al., 2005; McMahon et al., 2005).
Immune recognition of gliomas appears to have occurred, as evidenced by the presence CD4+ and CD8+ T cells within the glioma. If T cells were passively present, secondary to the breakdown of the blood-brain barrier, we would have expected a similar ratio of CD4+ to CD8+ T cells seen in peripheral blood, but this was not the case. Instead, there was an elevated CD4+ T-cell population of a regulatory T-cell phenotype (CD4+CD25+FOXP3+). Functionally, Treg cells can prevent the activation or expansion of both CD4+ and CD8+ T cells through mechanisms that are still poorly understood, but could involve the secretion of suppressive cytokines (IL-10 or TGF-β), the restraining of antigen-presenting capacity, or mediation of cell-to-cell contact inhibition (Karpus and Swanborg, 1991; Shevach et al., 2001). Furthermore, Treg cells do not require costimulatory signals through the CD80/86 interaction to mediate their suppressor activity (Thornton et al., 2004). They have been identified in the peripheral blood of glioma patients (Akasaki et al., 2004) and in the tumor microenvironment of other solid tumors, such as pancreatic, colon, lung, and breast adenocarcinomas (Liyanage et al., 2002; Woo et al., 2002). However, we are the first laboratory to demonstrate the presence of Treg cells within malignant gliomas. Apart from inhibition of effector CD8+ T-cell activity, Treg cells can also suppress pro-inflammatory cytokine production (TNF-α, IFN-γ, and IL-6); inhibit expression of MHC class II, CD40, CD80, and CD86; and impair antigen presentation by monocytes and macrophages (Misra et al., 2004; Taams et al., 2005), phenomena similar to what we have demonstrated in our GIM population. Thus, in glioma patients, Treg cells appear to have a role in the lack of immune eradication or suppression of the tumor by modulating innate and adaptive immune responses (Mendez et al., 2004). The Treg cells are probably physiologic in maintaining tolerance to self-antigens and minimizing inflammatory responses, especially when significant cerebral edema could be fatal. Overcoming Treg immune suppression may be a future therapeutic approach, albeit one that will have to be exploited cautiously through immunization strategies that incorporate tumor-specific antigen approaches. The significance of Treg cells in malignant glioma patients has yet to be determined, but their presence has been found to be a negative prognostic indicator within patients with ovarian carcinoma (Curiel et al., 2004). Of note, microglia/macrophages failed to secrete the immunosuppressive cytokine IL-10 as has been previously suggested (Huettner et al., 1997). Microglia/macrophages from human glioblastomas, identified solely by CD68+ expression, were shown by in situ hybridization and IL-10 mRNA expression to secrete IL-10 (Wagner et al., 1999). Although CD68 antigen is expressed by tissue macrophages and cells of myeloid/mononuclear lineage, it is also constitutively expressed in NK cells, gamma/delta T cells, and activated CD4+ and CD8+ T cells (Hameed et al., 1994), which suggests that the aforementioned IL-10 secretion could be from nonmicroglial cells such as Th2 CD4+ T cells or T reg cells.
Together, these data demonstrate that while microglia/macrophages are the largest immune cell population infiltrating human glioma tissue, they are likely acting in an inherent regulatory mechanism by which rapid intra-cerebral T-cell expansion is prevented in the CNS microenvironment. This suppressive activity may be bolstered or even induced by the action of the CD4+CD25+ Treg cells also present in the glioma tissue, affecting subsequent innate and adaptive immune responses. Accumulating evidence indicates that immune recognition of tumors occurs (Dunn et al., 2004). However, our data indicate that negative immunoregulation is occurring within the tumor microenvironment. Thus, treatments directed at improving antitumor responses will need to consider these two key populations, thereby reinforcing the importance of the complexities involved in immune interactions within the glioma microenvironment.
Acknowledgments
We thank Karen Ramirez for assistance with flow cytometry data analysis, Kenneth Hess for help with the statistical analysis, and Betty L. Notzon and David Galloway for critical review of the manuscript.
Footnotes
Studies performed by the authors were initiated and supported by The University of Texas M.D. Anderson Cancer Center Department of Neurosurgery Recruitment start-up funds, the American Association of Neurological Surgeons Young Clinical Investigator Award, and the American Brain Tumor Association in memory of Shawn Hansen (A.B.H.). This data was presented at the Proceedings of the American Association of Cancer Research, 96th Annual Meeting, Anaheim, Calif., April 16–20, 2005.
Abbreviations used are as follows: allo-MLR, allogeneic mixed-lymphocyte reaction; APC, antigen-presenting cell; CD, cluster of differentiation; CFSE, carboxyfluorescein diacetate succinimidyl ester; DC, dendritic cell; FACS, fluorescence-activated cell sorting; FcR, Fc receptor; FITC, fluorescein isothiocyanate; GBM, glioblastoma multiforme; GIM, glioma-infiltrating microglia/macrophage; IFN, interferon; IL, interleukin; LPS, lipopolysaccharide; M-DC, myeloid dendritic cell; MHC, major histocompatibility complex; mIgG1, mouse immunoglobulin G1; NK, natural killer; PBMC, peripheral blood mononuclear cell; P-DC, plasmacytoid dendritic cell; PE, phycoerythrin; TGF-β, transforming growth factor β; TLR, Toll-like receptor; TNF, tumor necrosis factor; Treg cell, regulatory T cell.
References
- Akasaki Y, Liu G, Chung NH, Ehtesham M, Black KL, Yu JS. Induction of a CD4+ T regulatory type 1 response by cyclooxygenase-2-overexpressing glioma. J Immunol. 2004;173:4352–4359. doi: 10.4049/jimmunol.173.7.4352. [DOI] [PubMed] [Google Scholar]
- Akira S, Takeda K, Kaisho T. Toll-like receptors: Critical proteins linking innate and acquired immunity. Nat Immunol. 2001;2:675–680. doi: 10.1038/90609. [DOI] [PubMed] [Google Scholar]
- Aloisi F. Immune function of microglia. Glia. 2001;36:165–179. doi: 10.1002/glia.1106. [DOI] [PubMed] [Google Scholar]
- Andrews C, Swain SL, Muralidhar G. CD4 T cell anergy in murine AIDS: Costimulation via CD28 and the addition of IL-12 are not sufficient to rescue anergic CD4 T cells. J Immunol. 1997;159:2132–2138. [PubMed] [Google Scholar]
- Asea A, Rehli M, Kabingu E, Boch JA, Bare O, Auron PE, Stevenson MA, Calderwood SK. Novel signal transduction pathway utilized by extracellular HSP70: Role of toll-like receptor (TLR) 2 and TLR4. J Biol Chem. 2002;277:15028–15034. doi: 10.1074/jbc.M200497200. [DOI] [PubMed] [Google Scholar]
- Badie B, Schartner JM. Flow cytometric characterization of tumor-associated macrophages in experimental gliomas. Neurosurgery. 2000;46:957–961. doi: 10.1097/00006123-200004000-00035. [DOI] [PubMed] [Google Scholar]
- Badie B, Bartley B, Schartner J. Differential expression of MHC class II and B7 costimulatory molecules by microglia in rodent gliomas. J Neuroimmunol. 2002;133:39–45. doi: 10.1016/s0165-5728(02)00350-8. [DOI] [PubMed] [Google Scholar]
- Banchereau J, Steinman RM. Dendritic cells and the control of immunity. Nature. 1998;392:245–252. doi: 10.1038/32588. [DOI] [PubMed] [Google Scholar]
- Beg AA. Endogenous ligands of Toll-like receptors: Implications for regulating inflammatory and immune responses. Trends Immunol. 2002;23:509–512. doi: 10.1016/s1471-4906(02)02317-7. [DOI] [PubMed] [Google Scholar]
- Bogdan C, Nathan C. Modulation of macrophage function by transforming growth factor beta, interleukin-4, and interleukin-10. Ann NY Acad Sci. 1993;685:713–739. doi: 10.1111/j.1749-6632.1993.tb35934.x. [DOI] [PubMed] [Google Scholar]
- Brabb T, von Dassow P, Ordonez N, Schnabel B, Duke B, Goverman J. In situ tolerance within the central nervous system as a mechanism for preventing autoimmunity. J Exp Med. 2000;192:871–880. doi: 10.1084/jem.192.6.871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calzascia T, Di Berardino-Besson W, Wilmotte R, Masson F, de Tribolet N, Dietrich PY, Walker PR. Cutting edge: Cross-presentation as a mechanism for efficient recruitment of tumor-specific CTL to the brain. J Immunol. 2003;171:2187–2191. doi: 10.4049/jimmunol.171.5.2187. [DOI] [PubMed] [Google Scholar]
- Calzascia T, Masson F, Di Berardino-Besson W, Contassot E, Wilmotte R, Aurrand-Lions M, Ruegg C, Dietrich PY, Walker PR. Homing phenotypes of tumor-specific CD8 T cells are predetermined at the tumor site by crosspresenting APCs. Immunity. 2005;22:175–184. doi: 10.1016/j.immuni.2004.12.008. [DOI] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P, Evdemon- Hogan M, Conejo-Garcia JR, Zhang L, Burow M, Zhu Y, Wei S, Kryczek I, Daniel B, Gordon A, Myers L, Lackner A, Disis ML, Knutson KL, Chen L, Zou W. Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med. 2004;10:942–949. doi: 10.1038/nm1093. [DOI] [PubMed] [Google Scholar]
- Dangond F, Windhagen A, Groves CJ, Hafler DA. Constitutive expression of costimulatory molecules by human microglia and its relevance to CNS autoimmunity. J Neuroimmunol. 1997;76:132–138. doi: 10.1016/s0165-5728(97)00043-x. [DOI] [PubMed] [Google Scholar]
- Davis FG, Kupelian V, Freels S, McCarthy B, Surawicz T. Prevalence estimates for primary brain tumors in the United States by behavior and major histology groups. Neuro-Oncology. 2001;3:152–158. doi: 10.1093/neuonc/3.3.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn GP, Old LJ, Schreiber RD. The immunobiology of cancer immunosurveillance and immunoediting. Immunity. 2004;21:137–148. doi: 10.1016/j.immuni.2004.07.017. [DOI] [PubMed] [Google Scholar]
- Ebert S, Gerber J, Bader S, Muhlhauser F, Brechtel K, Mitchell TJ, Nau R. Dose-dependent activation of microglial cells by Toll-like receptor agonists alone and in combination. J Neuroimmunol. 2005;159:87–96. doi: 10.1016/j.jneuroim.2004.10.005. [DOI] [PubMed] [Google Scholar]
- Fathallah-Shaykh HM, Gao W, Cho M, Herrera MA. Priming in the brain, an immunologically privileged organ, elicits anti-tumor immunity. Int J Cancer. 1998;75:266–276. doi: 10.1002/(sici)1097-0215(19980119)75:2<266::aid-ijc16>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- Fontenot JD, Rudensky AY. A well adapted regulatory contrivance: Regulatory T cell development and the forkhead family transcription factor Foxp3. Nat Immunol. 2005;6:331–337. doi: 10.1038/ni1179. [DOI] [PubMed] [Google Scholar]
- Ford AL, Goodsall AL, Hickey WF, Sedgwick JD. Normal adult ramified microglia separated from other central nervous system macrophages by flow cytometric sorting. Phenotypic differences defined and direct ex vivo antigen presentation to myelin basic protein-reactive CD4+ T cells compared. J Immunol. 1995;154:4309–4321. [PubMed] [Google Scholar]
- Gabrilovich DI, Patterson S, Timofeev AV, Harvey JJ, Knight SC. Mechanism for dendritic cell dysfunction in retroviral infection of mice. Clin Immunol Immunopathol. 1996;80:139–146. doi: 10.1006/clin.1996.0107. [DOI] [PubMed] [Google Scholar]
- Glimelius B, Norling B, Westermark B, Wasteson A. Composition and distribution of glycosaminoglycans in cultures of human normal and malignant glial cells. Biochem J. 1978;172:443–456. doi: 10.1042/bj1720443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greter M, Heppner FL, Lemos MP, Odermatt BM, Goebels N, Laufer T, Noelle RJ, Becher B. Dendritic cells permit immune invasion of the CNS in an animal model of multiple sclerosis. Nat Med. 2005;11:328–334. doi: 10.1038/nm1197. [DOI] [PubMed] [Google Scholar]
- Habich C, Baumgart K, Kolb H, Burkart V. The receptor for heat shock protein 60 on macrophages is saturable, specific, and distinct from receptors for other heat shock proteins. J Immunol. 2002;168:569–576. doi: 10.4049/jimmunol.168.2.569. [DOI] [PubMed] [Google Scholar]
- Hameed A, Hruban RH, Gage W, Pettis G, Fox WM., 3rd Immunohistochemical expression of CD68 antigen in human peripheral blood T cells. Hum Pathol. 1994;25:872–876. doi: 10.1016/0046-8177(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Heimberger, A.B., Bigner, D.D., and Sampson J.J. (2000) Biological principles of brain tumor immunotherapy. In: Liau, L.M., Becker, D.P., Cloughesy, T.F., and Bigner D.D. (Eds.), Brain Tumor Immunotherapy Totowa, N.J.: Humana Press Inc., pp. 101–130.
- Hemmi H, Takeuchi O, Kawai T, Kaisho T, Sato S, Sanjo H, Matsumoto M, Hoshino K, Wagner H, Takeda K, Akira S. A Toll-like receptor recognizes bacterial DNA. Nature. 2000;408:740–745. doi: 10.1038/35047123. [DOI] [PubMed] [Google Scholar]
- Holladay FP, Choudhuri R, Heitz T, Wood GW. Generation of cytotoxic immune responses during the progression of a rat glioma. J Neurosurg. 1994;80:90–96. doi: 10.3171/jns.1994.80.1.0090. [DOI] [PubMed] [Google Scholar]
- Hu HM, Winter H, Ma J, Croft M, Urba WJ, Fox BA. CD28, TNF receptor, and IL-12 are critical for CD4-independent cross-priming of therapeutic antitumor CD8+ T cells. J Immunol. 2002;169:4897–4904. doi: 10.4049/jimmunol.169.9.4897. [DOI] [PubMed] [Google Scholar]
- Hu S, Chao CC, Ehrlich LC, Sheng WS, Sutton RL, Rockswold GL, Peterson PK. Inhibition of microglial cell RANTES production by IL-10 and TGF-beta. J Leukoc Biol. 1999;65:815–821. doi: 10.1002/jlb.65.6.815. [DOI] [PubMed] [Google Scholar]
- Huettner C, Czub S, Kerkau S, Roggendorf W, Tonn JC. Interleukin 10 is expressed in human gliomas in vivo and increases glioma cell proliferation and motility in vitro. Anticancer Res. 1997;17:3217–3224. [PubMed] [Google Scholar]
- Ito T, Amakawa R, Kaisho T, Hemmi H, Tajima K, Uehira K, Ozaki Y, Tomizawa H, Akira S, Fukuhara S. Interferon-alpha and interleukin-12 are induced differentially by Toll-like receptor 7 ligands in human blood dendritic cell subsets. J Exp Med. 2002;195:1507–1512. doi: 10.1084/jem.20020207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki A, Medzhitov R. Toll-like receptor control of the adaptive immune responses. Nat Immunol. 2004;5:987–995. doi: 10.1038/ni1112. [DOI] [PubMed] [Google Scholar]
- Johnson GB, Brunn GJ, Kodaira Y, Platt JL. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 2002;168:5233–5239. doi: 10.4049/jimmunol.168.10.5233. [DOI] [PubMed] [Google Scholar]
- Juedes AE, Ruddle NH. Resident and infiltrating central nervous system APCs regulate the emergence and resolution of experimental autoimmune encephalomyelitis. J Immunol. 2001;166:5168–5175. doi: 10.4049/jimmunol.166.8.5168. [DOI] [PubMed] [Google Scholar]
- Kakimura J, Kitamura Y, Takata K, Umeki M, Suzuki S, Shibagaki K, Taniguchi T, Nomura Y, Gebicke-Haerter PJ, Smith MA, Perry G, Shimohama S. Microglial activation and amyloid-beta clearance induced by exogenous heat-shock proteins. FASEB J. 2002;16:601–603. doi: 10.1096/fj.01-0530fje. [DOI] [PubMed] [Google Scholar]
- Karpus WJ, Swanborg RH. CD4+ suppressor cells inhibit the function of effector cells of experimental autoimmune encephalomyelitis through a mechanism involving transforming growth factor-beta. J Immunol. 1991;146:1163–1168. [PubMed] [Google Scholar]
- Krakowski ML, Owens T. Naive T lymphocytes traffic to inflamed central nervous system, but require antigen recognition for activation. Eur J Immunol. 2000;30:1002–1009. doi: 10.1002/(SICI)1521-4141(200004)30:4<1002::AID-IMMU1002>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- Liyanage UK, Moore TT, Joo HG, Tanaka Y, Herrmann V, Doherty G, Drebin JA, Strasberg SM, Eberlein TJ, Goedegebuure PS, Linehan DC. Prevalence of regulatory T cells is increased in peripheral blood and tumor microenvironment of patients with pancreas or breast adenocarcinoma. J Immunol. 2002;169:2756–2761. doi: 10.4049/jimmunol.169.5.2756. [DOI] [PubMed] [Google Scholar]
- Luscher U, Filgueira L, Juretic A, Zuber M, Luscher NJ, Heberer M, Spagnoli GC. The pattern of cytokine gene expression in freshly excised human metastatic melanoma suggests a state of reversible anergy of tumor-infiltrating lymphocytes. Int J Cancer. 1994;57:612–619. doi: 10.1002/ijc.2910570428. [DOI] [PubMed] [Google Scholar]
- Matyszak MK, Denis-Donini S, Citterio S, Longhi R, Granucci F, Ricciardi-Castagnoli P. Microglia induce myelin basic protein–specific T cell anergy or T cell activation, according to their state of activation. Eur J Immunol. 1999;29:3063–3076. doi: 10.1002/(SICI)1521-4141(199910)29:10<3063::AID-IMMU3063>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Matzinger P. Tolerance, danger, and the extended family. Annu Rev Immunol. 1994;12:991–1045. doi: 10.1146/annurev.iy.12.040194.005015. [DOI] [PubMed] [Google Scholar]
- McMahon EJ, Bailey SL, Castenada CV, Waldner H, Miller SD. Epitope spreading initiates in the CNS in two mouse models of multiple sclerosis. Nat Med. 2005;11:335–339. doi: 10.1038/nm1202. [DOI] [PubMed] [Google Scholar]
- Mendez S, Reckling SK, Piccirillo CA, Sacks D, Belkaid Y. Role for CD4(+) CD25(+) regulatory T cells in reactivation of persistent leishmaniasis and control of concomitant immunity. J Exp Med. 2004;200:201–210. doi: 10.1084/jem.20040298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra N, Bayry J, Lacroix-Desmazes S, Kazatchkine MD, Kaveri SV. Cutting edge: Human CD4+CD25+ T cells restrain the maturation and antigen-presenting function of dendritic cells. J Immunol. 2004;172:4676–4680. doi: 10.4049/jimmunol.172.8.4676. [DOI] [PubMed] [Google Scholar]
- Nagai A, Nakagawa E, Hatori K, Choi HB, McLarnon JG, Lee MA, Kim SU. Generation and characterization of immortalized human microglial cell lines: Expression of cytokines and chemokines. Neurobiol Dis. 2001;8:1057–1068. doi: 10.1006/nbdi.2001.0437. [DOI] [PubMed] [Google Scholar]
- Nakagawa T, Kubota T, Kabuto M, Kodera T. Hyaluronic acid facilitates glioma cell invasion in vitro. Anticancer Res. 1996;16:2917–2922. [PubMed] [Google Scholar]
- Neuhaus-Steinmetz U, Rensing L. Heat shock protein induction by certain chemical stressors is correlated with their cytotoxicity, lipophilicity and protein-denaturing capacity. Toxicology. 1997;123:185–195. doi: 10.1016/s0300-483x(97)00124-8. [DOI] [PubMed] [Google Scholar]
- Ohashi K, Burkart V, Flohe S, Kolb H. Cutting edge: Heat shock protein 60 is a putative endogenous ligand of the toll-like receptor-4 complex. J Immunol. 2000;164:558–561. doi: 10.4049/jimmunol.164.2.558. [DOI] [PubMed] [Google Scholar]
- Olson JK, Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol. 2004;173:3916–3924. doi: 10.4049/jimmunol.173.6.3916. [DOI] [PubMed] [Google Scholar]
- Oxenius A, Bachmann MF. Similar ligand densities required for restimulation and effector function of cytotoxic T cells. Cell Immunol. 1997;179:16–21. doi: 10.1006/cimm.1997.1146. [DOI] [PubMed] [Google Scholar]
- Pardoll D. Does the immune system see tumors as foreign or self? Annu Rev Immunol. 2003;21:807–839. doi: 10.1146/annurev.immunol.21.120601.141135. [DOI] [PubMed] [Google Scholar]
- Quill H, Bhandoola A, Trinchieri G, Haluskey J, Peritt D. Induction of interleukin 12 responsiveness is impaired in anergic T lymphocytes. J Exp Med. 1994;179:1065–1070. doi: 10.1084/jem.179.3.1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridley A. Immunological aspects of gliomas. Proc R Soc Med. 1977;70:867–869. doi: 10.1177/003591577707001212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roussel E, Gingras MC, Grimm EA, Bruner JM, Moser RP. Predominance of a type 2 intratumoural immune response in fresh tumour-infiltrating lymphocytes from human gliomas. Clin Exp Immunol. 1996;105:344–352. doi: 10.1046/j.1365-2249.1996.d01-753.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satoh J, Lee YB, Kim SU. T-cell costimulatory molecules B7-1 (CD80) and B7-2 (CD86) are expressed in human microglia but not in astrocytes in culture. Brain Res. 1995;704:92–96. doi: 10.1016/0006-8993(95)01177-3. [DOI] [PubMed] [Google Scholar]
- Shevach EM, McHugh RS, Piccirillo CA, Thornton AM. Control of T-cell activation by CD4+ CD25+ suppressor T cells. Immunol Rev. 2001;182:58–67. doi: 10.1034/j.1600-065x.2001.1820104.x. [DOI] [PubMed] [Google Scholar]
- Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. 2002;2:185–194. doi: 10.1038/nri749. [DOI] [PubMed] [Google Scholar]
- Steck PA, Moser RP, Bruner JM, Liang L, Freidman AN, Hwang TL, Yung WK. Altered expression and distribution of heparan sulfate proteoglycans in human gliomas. Cancer Res. 1989;49:2096–2103. [PubMed] [Google Scholar]
- Taams LS, van Amelsfort JM, Tiemessen MM, Jacobs KM, de Jong EC, Akbar AN, Bijlsma JW, Lafeber FP. Modulation of monocyte/macrophage function by human CD4+CD25+ regulatory T cells. Hum Immunol. 2005;66:222–230. doi: 10.1016/j.humimm.2004.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J, Town T, Paris D, Mori T, Suo Z, Crawford F, Mattson MP, Flavell RA, Mullan M. Microglial activation resulting from CD40-CD40L interaction after beta-amyloid stimulation. Science. 1999;286:2352–2355. doi: 10.1126/science.286.5448.2352. [DOI] [PubMed] [Google Scholar]
- Termeer C, Benedix F, Sleeman J, Fieber C, Voith U, Ahrens T, Miyake K, Freudenberg M, Galanos C, Simon JC. Oligosaccharides of Hyaluronan activate dendritic cells via toll-like receptor 4. J Exp Med. 2002;195:99–111. doi: 10.1084/jem.20001858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornton AM, Piccirillo CA, Shevach EM. Activation requirements for the induction of CD4+CD25+ T cell suppressor function. Eur J Immunol. 2004;34:366–376. doi: 10.1002/eji.200324455. [DOI] [PubMed] [Google Scholar]
- Tsunawaki S, Sporn M, Ding A, Nathan C. Deactivation of macrophages by transforming growth factor-beta. Nature. 1988;334:260–262. doi: 10.1038/334260a0. [DOI] [PubMed] [Google Scholar]
- Ulvestad E, Williams K, Bjerkvig R, Tiekotter K, Antel J, Matre R. Human microglial cells have phenotypic and functional characteristics in common with both macrophages and dendritic antigen-presenting cells. J Leukoc Biol. 1994;56:732–740. doi: 10.1002/jlb.56.6.732. [DOI] [PubMed] [Google Scholar]
- Van Parijs L, Perez VL, Biuckians A, Maki RG, London CA, Abbas AK. Role of interleukin 12 and costimulators in T cell anergy in vivo. J Exp Med. 1997;186:1119–1128. doi: 10.1084/jem.186.7.1119. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Vincent VA, Tilders FJ, Van Dam AM. Inhibition of endotoxin-induced nitric oxide synthase production in microglial cells by the presence of astroglial cells: A role for transforming growth factor beta. Glia. 1997;19:190–198. doi: 10.1002/(sici)1098-1136(199703)19:3<190::aid-glia2>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Wagner S, Czub S, Greif M, Vince GH, Suss N, Kerkau S, Rieckmann P, Roggendorf W, Roosen K, Tonn JC. Microglial/macrophage expression of interleukin 10 in human glioblastomas. Int J Cancer. 1999;82:12–16. doi: 10.1002/(sici)1097-0215(19990702)82:1<12::aid-ijc3>3.0.co;2-o. [DOI] [PubMed] [Google Scholar]
- Watters JJ, Schartner JM, Badie B. Microglia function in brain tumors. J Neurosci Res. 2005;81:447–455. doi: 10.1002/jnr.20485. [DOI] [PubMed] [Google Scholar]
- Woo EY, Yeh H, Chu CS, Schlienger K, Carroll RG, Riley JL, Kaiser LR, June CH. Cutting edge: Regulatory T cells from lung cancer patients directly inhibit autologous T cell proliferation. J Immunol. 2002;168:4272–4276. doi: 10.4049/jimmunol.168.9.4272. [DOI] [PubMed] [Google Scholar]
- Yi-qun Z, Lorre K, de Boer M, Ceuppens JL. B7-blocking agents, alone or in combination with cyclosporin A, induce antigen-specific anergy of human memory T cells. J Immunol. 1997;158:4734–4740. [PubMed] [Google Scholar]
- Zhang GX, Li J, Ventura E, Rostami A. Parenchymal microglia of naive adult C57BL/6J mice express high levels of B7.1, B7.2, and MHC class II. Exp Mol Pathol. 2002;73:35–45. doi: 10.1006/exmp.2002.2441. [DOI] [PubMed] [Google Scholar]