

Abstract
Brain metastasis, which occurs in 20% to 40% of all cancer patients, is an important cause of neoplastic morbidity and mortality. Successful invasion into the brain by tumor cells must include attachment to microvessel endothelial cells, penetration through the blood-brain barrier, and, of relevance, a response to brain survival and growth factors. Neurotrophins (NTs) are important in brain-invasive steps. Human melanoma cell lines express low-affinity NT receptor p75NTR in relation to their brain-metastatic propensity with their invasive properties being regulated by NGF, or nerve growth factor, the prototypic NT. They also express functional TrkC, the putative receptor for the invasion-promoting NT-3. In brain-metastatic melanoma cells, NTs promote invasion by enhancing the production of extracellular matrix (ECM)-degradative enzymes such as heparanase, an enzyme capable of locally destroying both ECM and the basement membrane of the blood-brain barrier. Heparanase is an endo-β-d-glucuronidase that cleaves heparan sulfate (HS) chains of ECM HS proteoglycans, and it is a unique metastatic determinant because it is the dominant mammalian HS degradative enzyme. Brain-metastatic melanoma cells also produce autocrine/paracrine factors that influence their growth, invasion, and survival in the brain. Synthesis of these factors may serve to regulate NT production by brain cells adjacent to the neoplastic invasion front, such as astrocytes. Increased NT levels have been observed in tumor-adjacent tissues at the invasion front of human brain melanoma. Additionally, astrocytes may contribute to the brain-metastatic specificity of melanoma cells by producing NT-regulated heparanase. Trophic, autocrine, and paracrine growth factors may therefore determine whether metastatic cells can successfully invade, colonize, and grow in the CNS.
Brain metastases represent a major cause of death in cancer patients and a significant area of cancer biology research: They occur in 20% to 40% of cancer cases, and their frequency is yearly increasing. Though no single clinical therapy is preferentially superior, surgical excision, radiation, and/or chemotherapy are most commonly applied in patients with a resected primary tumor and single/multiple brain metastases. Irrespective of treatment, the prognosis for patients with brain metastasis is grim (Prados and Wilson, 1993; Sawaya et al., 1996; Soffietti et al., 2002). CNS involvement is a common feature of metastatic melanoma. The high CNS involvement associated with malignant melanoma may be due to a “homing” influence since melanocytes and neuronal subpopulations share a common embryologic origin (Fig. 1; Herlyn et al., 1985). Malignant melanoma metastasizes to the brain with one of the highest frequencies of any cancer capable of colonizing the CNS. Patients with disseminated malignant melanoma frequently develop metastatic lesions in the brain and spinal cord that can result in severe and debilitating neurological complications (Sawaya et al., 1996; Soffietti et al., 2002). Once melanoma cells colonize the brain, tumor growth often results in a rapid decline in the quality of life, and death ensues: Almost 40% of melanoma patients will be treated for complications due to brain metastases. At autopsy, an additional 30% to 40% of patients show CNS lesions (Sawaya et al., 1996; Soffietti et al., 2002).
Fig. 1.
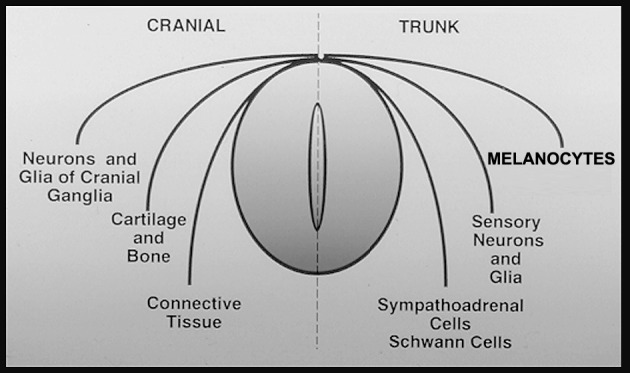
Embryologic relationship between melanocytes and the most common neuronal cell populations, both being neural-crest derived. Neuronal populations are neurotrophin-responsive and possess specific cell-surface neurotrophin receptors. Examples include neurons of peripheral nervous system sensory and sympathetic ganglia, Schwann cells, glial cells, and certain populations of CNS cholinergic neurons.
The brain, because of its anatomical and physiological properties, provides a unique target for metastasis (Steck and Nicolson, 1993). Homeostasis in the brain is highly sensitive to the slightest change in the local microenvironment because of confinement by the skull and the lack of an extensive lymphatic drainage system. Furthermore, the brain is surrounded by a formidable blood-brain barrier (BBB)3 through which brain-metastatic tumor cells must penetrate. The BBB is composed of tight junctions between brain endothelial cells, a relatively thick basement membrane (BM), and an underlying layer of astrocytes that strictly regulate the flow of ions, nutrients, and cells into the brain. Thus, to successfully colonize the brain, metastatic cells must complete a series of sequential and selective steps resulting in subpopulations of cells with different angiogenic, invasive, and metastatic properties (Fidler and Kripke, 1977). To produce brain metastases, tumor cells must reach the vasculature of the brain, attach to microvessel endothelial cells, extravasate into the parenchyma, induce angiogenesis, and proliferate by responding to growth factors (Nicolson et al., 1996; Yano et al., 2000). In addition to the above criteria, brain metastases must be able to cross the BBB and flourish in spite of inadequate lymphatic drainage.
Malignant melanomas, particularly those that advance to the brain, undergo progressive changes during their pathogenesis. Of the phenotypic changes that occur during metastatic melanoma progression, differences in the expression of receptors for paracrine growth factors and in the production of various autocrine growth factors are important (Albino et al., 1991; Herlyn et al., 1985). Although the significance of these factors in modulating the malignant properties exhibited by melanoma cells remains largely unknown, they are thought to be relevant in allowing malignant cells to survive in unusual compartments such as the brain. Neurotrophins (NTs) are growth factors that promote neuronal cell survival, differentiation, and cell death (Bradshaw et al., 1993; Lee et al., 2001; Raff, 1992; Raff et al., 1993; Snider, 1994). The involvement of NTs, neurotrophin receptors (NTRs), and NT-regulated heparanase in the development of brain metastasis is the subject of this review.
Neurotrophins and Neurotrophin Receptors
Nerve growth factor (NGF), the prototypic NT, is 1 of the 4 members that make up the mammalian NT family. The other NTs, brain-derived neurotrophic growth factor (BDNF), NT-3, and NT-4/5 (NT-5 is the mammalian homolog of Xenopus NT-4), share at least 50% amino acid homology with NGF, and they are all highly conserved in the region of the central axis of the molecule (Bradshaw et al., 1993). Neurotrophins are a family of small (≈13 kDa), highly basic (pI 9–10.5) proteins that are synthesized as pre-propeptides and subsequently N-terminally processed to contain 3 interchain disulfide bonds (Bradshaw et al., 1993). Each protein monomer contains an elongated central axis made of an antiparallel β-sheet structure with a flattened hydrophobic face that is involved in dimer formation (Bradshaw et al., 1993). It is this 26-kDa homodimer that is the circulating form of NTs. The recent discovery that NT precursor proteins and their proteolytically processed products may differentially activate pro-apoptotic and anti-apoptotic cellular responses, via preferential activation of NT receptors, promises to unveil yet another level of regulatory complexity (Lee et al., 2001).
Neurotrophin receptors have been historically divided into 2 affinity classes (Chao and Bothwell, 2002), a low-affinity receptor class (nerve growth factor receptor [NGFR] or p75NTR, KD ≈ 2 × 10−9) and a high-affinity receptor class (TrkA, TrkB, and TrkC, KD ≈ 2 × 10−11; Fig. 2; Table 1). Because of the formation of homodimers and heterodimers between the high-affinity and low-affinity receptor classes, the historical designation is an oversimplification of an increasingly complex system.
Fig. 2.
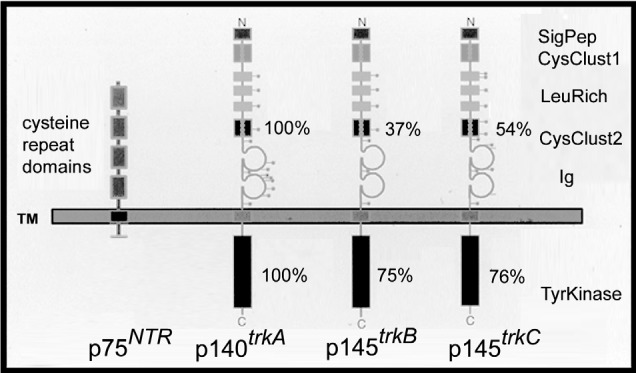
Schematic representation of the 2 different classes of neurotrophin receptors (p75NTR, TRK: TrkA, TrkB, TrkC). The p75NTR is a glycoprotein containing 4 cysteine-rich domains in its extracellular portion but no intracytoplasmic tyrosine kinase (TK) domain. However, p75NTR is capable of signaling independent of TRK presence, amplifying the TRK signal when TRK is present (Chao and Bothwell, 2002). TRK receptors contain a signal peptide sequence, cystein clusters, and leucine-rich and Ig-like regions in their extracellular domains and possess an intracytoplasmic TK domain. Percentages of similarity between the 3 main TRK receptors (TrkA, TrkB, TrkC) for their extracellular region/TK domain are respectively indicated.
Table 1.
Receptors and ligands
| Receptor | Ligand* | Characteristics** |
|---|---|---|
| P75NTR | NGF, BDNF, NT-3, NT-4/5 | Lacks tyrosine kinase domain
Binds all TRK receptors Possesses a signaling function independent of TRK receptors Provides differentiation and survival cues to neuronal tissues Triggers apoptosis in certain virally transformed neuronal cells Triggers survival when expressed in neutrophils |
| TrkA | NGF | Tyrosine kinase intracytoplasmic domain |
| NT-3 | Omission produces defects in the cervical ganglia of mice | |
| TrkB | BDNF | Tyrosine kinase intracytoplasmic domain |
| NT-3, NT-4/5 | Influences vestibular ganglia | |
| TrkC | NT-3 | Tyrosine kinase intracytoplasmic domain
Regulates proliferation and survival of neuronal precursors Regulates the collateral branching of axons into target fields |
Abbreviation: TRK, tropomyosin receptor kinase.
The primary ligands are indicated in plain text. Secondary neurotrophin cross-reactivities are listed in italics.
Characteristics for TRK receptor functions derived from either gene targeting or knockout mouse studies.
The low-affinity receptor p75NTR is capable of binding all members of the NT family (Fig. 3; Table 1), and it mediates cellular responses to NT dimers in neuronal tissues (Chao and Bothwell, 2002). Cloned by Chao and coworkers (Johnson et al., 1986), the human gene encodes a 75-kDa cell-surface glycoprotein that, as sequence analysis revealed, lacks a tyrosine kinase (TK) consensus sequence (Johnson et al., 1986). However, despite the absence of a TK domain, transfection of p75NTR into non-neuronal cells was shown to enhance TK phosphorylation following NGF stimulation (Ohmichi et al., 1992a). The p75NTR provides differentiation and survival cues to neuronal tissues (Chao and Bothwell, 2002). Furthermore, the molecule possesses a signaling function involving ceramide (Chao, 1992) that operates independently of the high-affinity tropomyosin receptor kinase (TRK) receptors (Barrett and Bartlett, 1994; Chao, 1992; Saltiel et al., 1994; Verdi et al., 1994). In addition, p75NTR provides retrograde transport to neuronal cell types, which triggers apoptosis in certain virally transformed neuronal cells (Rabizadeh et al., 1993) or survival when expressed in neutrophils (Kannan et al., 1992).
Fig. 3.
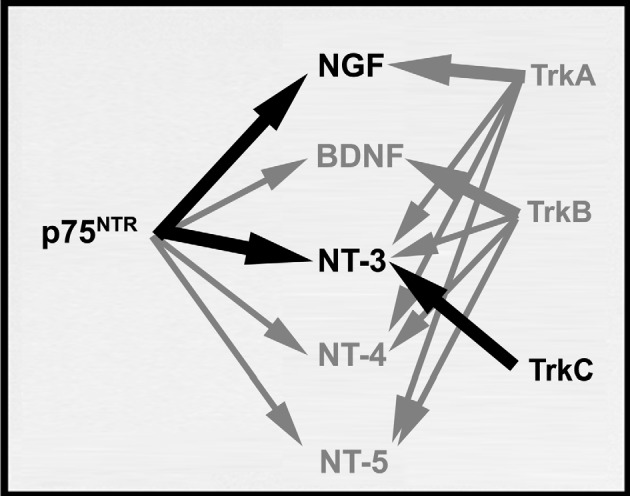
Functional cross-reactivities among neurotrophins and neurotrophin receptors (p75NTR and TRK). P75NTR binds all NT members equally well. The primary ligand for each TRK receptor is indicated by heavy arrows. Black heavy arrows denote interactions between neurotrophin receptors and neurotrophins in brain-metastatic melanoma.
Original studies established that the biological effects of NGF involve TK activity (Maher, 1988; Miyasaka et al., 1990). The search for a high-affinity NGF receptor with TK activity led to the discovery of the TRK family of NTRs (Barbacid, 1993; Chao, 1992; Meakin and Shooter, 1992; Saltiel and Decker, 1994). This family of tyrosine receptor protein kinases consists of several receptor molecules with varying degrees of specificity for the different members of the NT family. The TRK family members are widely distributed in neuronal tissues and hematopoietic cells (Barbacid, 1993; Chao, 1992; Meakin and Shooter, 1992; Saltiel and Decker, 1994). Each receptor selectively binds only a subset of the NTs (Table 1), all functions of which have been derived from either gene targeting or knockout mouse studies. TrkA mainly binds NGF, TrkB interacts with BDNF, while TrkC is the putative high-affinity receptor for NT-3 (Chao, 1992; Chao and Bothwell, 2002; Fig. 3).
Developmental changes in NT dependence parallel the increase in p75NTR production coincident with the progression of melanocytes to malignant melanoma. There are a number of reasons why NTRs are upregulated in melanoma. Melanoma cells frequently exhibit genetic instability (Nicolson, 1987). Second, because melanoma has the same developmental origin as neuroectodermal cells (Yaar and Gilchrest, 1991), a predisposition to switching expression of NTR genes is not unlikely. Those that upregulate NTRs are more apt to survive in the brain. Finally, 12-O-tetradecanoylphorbol-13-acetate has also been shown to induce p75NTR and other TRK receptors in melanocytes (Peacocke et al., 1988; Yaar and Gilchrest, 1991; Yaar et al., 1994). Interestingly, primary melanocyte cultures express low levels of TrkC that are enhanced by 12-O-tetradecanoylphorbol-13-acetate stimulation (Peacocke et al., 1988; Yaar and Gilchrest, 1991; Yaar et al., 1994). Our laboratory has observed high levels of both p75NTR and TrkC expression in melanoma cells (Herrmann et al., 1993; Marchetti et al., 2003a). Importantly, the presence of these NTRs (p75NTR, TrkC) in brain-metastatic melanoma resulted in enhancement of melanoma cell invasion (Marchetti and Nicolson, 1997a, b; Marchetti et al., 1996; Nicolson et al., 1994). Furthermore, we have recently reported that TrkC receptor functionality in these cells occurs via the association of TrkC with a purine-analog sensitive kinase (Marchetti et al., 2003a).
Based upon the observed NTR overexpression in brain-metastatic melanoma cells and NT regulation of their invasive properties (see below), we have formulated the hypothesis that brain metastases essentially represent a traumatic event related to brain-injury processes. Following mechanical or chemical brain insults, NT synthesis is increased and NT/NTR presence is imperative in neuronal regeneration (Chao and Bothwell, 2002). These changes are paralleled by brain-invasive melanoma cells whose invasion and colonization within the brain microenvironment triggers NT production and secretion by surrounding brain cells. Similarly, melanoma cells overexpressing NTRs (p75NTR, TrkC) can benefit from such a synergistic microenvironment in terms of survival, growth, and further invasion into the brain parenchyma (Nicolson et al., 1996). We suggest that NTRs may play important roles in melanoma progression to the brain while NT-regulated heparanase can be critical to this process. Although we have validated this hypothesis with in vitro models of brain-metastatic melanoma, further supporting evidence must be obtained by in vivo experiments.
Neurotrophin Receptor Signaling Mechanisms
The complexity of functional interactions between p75NTR and TRK receptors rivals that of other complex receptor systems (Chao and Bothwell, 2002; Hempstead et al., 1991; Lee et al., 1992, 1994; Verdi et al., 1994). It is generally agreed that TK receptors are involved in sequences of events that include ligand binding, leading to receptor dimer formation and transactivation, resulting in tyrosine phosphorylation, with the eventual activation of serine/threonine (ser/thr) phosphorylation cascades (Barbacid, 1993; Chao, 1992; Meakin and Shooter, 1992; Ohmichi et al., 1991, 1992b; Saltiel and Decker, 1994). Active signaling complexes are frequently formed by interactions between receptor phosphotyrosines and proteins containing SH2, or Src homology-2, tyrosine-binding domains (Avruch et al., 1994; Batistatou et al., 1992; Borrello et al., 1994; Lange-Carter and Johnson, 1994; Obermeier et al., 1993a, b, 1994; Ohmichi et al., 1994; Rozakis-Adcock et al., 1992; Satoh et al., 1992; Stephens et al., 1994; Taylor et al., 1994). Formation of this complex leads to tyrosine phosphorylation on Shc and the association of Shc with Grb2, another SH2-containing protein (Borrello et al., 1994; Obermeier et al., 1993a, b, 1994; Ohmichi et al., 1994; Stephens et al., 1994). The association of Shc with Grb2 can lead to further complex formation with the p21ras nucleotide exchange factor Son of sevenless-1 (Sos-1). This may result in increased GTP binding and activation of p21ras, a GTP-binding oncoprotein originally identified in a rat sarcoma virus (Satoh et al., 1992).
The downstream effectors of p21ras include proteins involved in ser/thr phosphorylation cascades (Avruch et al., 1994). Studies have demonstrated that p21ras can coordinate the NGF-mediated, phosphorylation-dependent activation of several key growth and differentiation molecules (Kaplan and Miller, 2000), including (1) c-Raf-1, a cytoplasmic ser/thr kinase discovered as the oncoprotein v-raf in a mouse sarcoma virus, (2) the mitogen-activated protein kinase/extracellular-signal-regulated kinase (MAPK/ERK [MEK]), and (3) MAPK. The activation of MAPK can transiently induce the expression of a number of primary response genes that encode transcription factors, such as c-fos, c-jun, NGFI-B, and kirx24 (Batistatou et al., 1992). The MAPK activity can also affect other ser/thr kinases and cytoskeletal elements (Kaplan and Miller, 2000; Taylor et al., 1994). MEKK, a ser/thr kinase that can activate MEKs independently of c-Raf-1, has been observed to phosphorylate MEK in PC12 cells as they respond to NGF (Kaplan and Miller, 2000; Lange-Carter and Johnson, 1994).
Coexpression of p75NTR and TrkA resulted in increases in downstream signaling and NT responses, including mitotic arrest following neurite extension and neuronal maturation, relative to cells expressing only TrkA (Barbacid, 1993; Berg et al., 1991; Chao, 1992; Hempstead et al., 1989). According to another model, p75NTR procures and presents bound NT molecules to members of the TRK receptor tyrosine kinase family to initiate signal transduction (Chao, 1992; Chao and Bothwell, 2002). Little is known about p75NTR cooperative interactions with other NTRs, but recent evidence based on anti-p75NTR antibody injections into chick embryos suggests that BDNF and NT-3 may cooperate with p75NTR to form functional signaling pathways (von Bartheld et al., 1994). Collectively, these data emphasize the importance of cooperativity between the TRK family of receptors and p75NTR for enhancing NT capabilities of cells. Biochemical and functional interactions between TrkA and p75NTR have also been detected after co-immunoprecipitation and Western blotting (Bibel et al., 1999).
Dependent upon the cellular context in which it is expressed, p75NTR shows alternative functions independent of TRK presence. In addition to receiving differentiation or survival signals in neuronal cells, p75NTR provides retrograde transport in certain neuronal cell types (Verdi et al., 1994), triggering apoptosis in certain virally transformed neuronal cells (Rabizadeh et al., 1993) or survival when expressed in neutrophils (Kannan et al., 1992). The p75NTR cytoplasmic tail contains a 14-amino acid mastoparan-like domain (Feinstein and Larhammar, 1990; Johnson et al., 1986). Activation of a G-stimulatory protein complex in the presence (or absence) of NGF may lead to the production of cyclic AMP (cAMP) by adenylate cyclase and activation of protein kinase A, followed by transcription factor activation (Knipper et al., 1993). Transfection studies involving sequence deletions in p75NTR of small segments in the cytoplasmic tail proved that these deletions are essential for high-affinity NGF binding involving TrkA in PC12 and NIH3T3 cells (Hantzopoulos et al., 1994; Hempstead et al., 1990). Certain properties of p75NTR also allow it to function in regulating survival and death of melanoma cells. In this regard, p75NTR is analogous to members of the tumor necrosis factor (TNF) receptor superfamily, such as Fas (Apo I), TNFRI, and TNFRII, and the B cell antigen CD40, all of which regulate programmed cell death (Beutler and van Huffel, 1994; Smith et al., 1994). Therefore, it is apparent that p75NTR plays a bifunctional role as a molecular switch that signals either cell survival or cell death (Barrett and Bartlett, 1994).
Of relevance, a ser/thr protein kinase that is sensitive to purine analogs and known as protein kinase N (PKN) has been isolated with p75NTR following NGF stimulation of PC12 cells (Volonté et al., 1993; Volonté and Greene, 1992). The activation of this PKN in association with stimulation of ornithine decarboxylase activity plays a potentially important role in the signaling pathways associated with p75NTR (Ohmichi et al., 1991). We have recently demonstrated that there is an association between TrkC receptors and a purine-analog-sensitive kinase in human brain-metastatic melanoma cells (Marchetti et al., 2003a). We have also determined that this kinase is similar to the one known to associate with p75NTR and possesses an activity under the specific regulation by TrkC putative ligand, NT-3 (Marchetti et al., 2003a). Therefore, purine-analog-sensitive kinases like PKN can represent a signaling component(s) common to NTR, playing roles in melanoma cell pathogenesis by generating constitutive downstream signaling. In relation to p75NTR, the cooperative interaction of downstream signals from p75NTR/PKN in amplifying signals pre-established by TRK may be important in brain-metastatic melanoma. In this case, when NT concentrations are high, the low-affinity activation of p75NTR/PKN signals amplify the TRK response pathway. In contrast, when NT levels are low, p75NTR signals are driven along an alternate pathway, allowing p75NTR to act as a sensitive molecular switch because of its low affinity for NTs.
Activation of the sphyngomyelin cycle serves as an alternate signaling pathway for p75NTR (Dobrowsky et al., 1994). The sphyngomyelin pathway also seems to be important during signaling by TNFα receptors (p75NTR is a member of the TNF receptor superfamily), and this pathway appears to involve a ceramide-activated protein phosphatase (Wolff et al., 1994). This alternate form of signal transduction by p75NTR may be important to cells invading the brain. Brain tissue injured by tumor cell invasion can provide a ready source of ceramide that might also influence invading cells.
Neurotrophin Receptors and Progression of Malignant Melanoma Cells
During malignant progression, melanoma cells show progression-associated increases in the expression of p75NTR (Herlyn et al., 1985; Nicolson et al., 1996), as witnessed by in situ examination of p75NTR levels in advanced stages of malignant melanoma (Ross et al., 1984; Marchetti et al., 1995). Human melanoma cells established in short-term tissue culture from brain metastases exhibit characteristic chromosomal alterations (Morse et al., 1992). Importantly, although p75NTR expression was not examined in these cells (Morse et al., 1992), the gene is located at 17q21–22 and may be amplified in tumor cells containing the isochromosome.
We have examined the role of NTs and NTRs in brain invasion and colonization of malignant melanomas. Using the human melanoma variant cell subline 70W, which has the capacity to form brain colonies in nude mice, we have studied the effects of NTs and growth factors on their malignant properties. The 70W subline was derived as one of the series of human MeWo melanoma cell variants selected by treatment with wheat germ agglutinin (Ishikawa et al., 1988). Parental MeWo cells exhibit intermediate metastatic potential as compared with other cell lines such as the nonmetastatic 3S5 and the brain-metastatic 70W cell variants. Of note, 3S5 and 70W cells possess opposite metastatic capabilities when injected in vivo in nude mice: While 3S5 cells are nonmetastatic, 70W cells are highly aggressive and brain-metastatic (Marchetti et al., 1993), being the first reported example of human melanoma cells capable of metastasizing to the brain when injected intravenously in nude mice (Ishikawa et al., 1988). Furthermore, target organ site colonization by the 70W line is similar to the clinical presentation of melanoma metastasis. Using the MeWo melanoma cellular system (MeWo parental, 3S5, and 70W variants), we have shown that overexpression of p75NTR is associated with brain colonization, enhancement of extracellular matrix (ECM) invasion (Herrmann et al., 1993; Marchetti et al., 1993), and heparanase activity (Marchetti et al., 1993).
Neurotrophins Enhance Invasion and Heparanase Production in Brain-Metastatic Melanoma Cells
During metastasis formation, migrating tumor cells are confronted by natural tissue barriers that surround the blood vessels, such as BM (Gladson et al., 1995; Liotta et al., 1991), or ECM, which is an integral part of the BBB. The ability of malignant cells to penetrate these barriers depends upon the production and activation of enzymes capable of ECM degradation (Liotta et al., 1982; Powell and Matrisian, 1996; Sloane and Honn, 1984). ECM and BM are rigid structures formed from macromolecules such as type IV collagen, laminin, entactin, nidogen, fibronectin, and proteoglycans (Iozzo and Murdoch, 1996), one type being heparan sulfate (HS) proteoglycans, or HSPGs. It is known that HSPGs play a central role in embryonic morphogenesis, angiogenesis, neurite outgrowth, and tissue repair (Bernfield et al., 1999; Iozzo, 1988; McKeehan and Kan, 1994; Yanagishita and Hascall, 1992). ECM and BM HSPGs also provide a readily available storage depot for growth factors and cytokines (Vlodavsky and Friedmann, 2001). Since HSPGs are now recognized as active biological modulators, their degradation at the level of HS chains is expected to have significant regulatory consequences in cancer metastasis (Bernfield et al., 1999). Indeed, HSPG catabolism is observed in inflammation, wound repair, diabetes, and neoplasia, including melanoma (Marchetti, 1997; Nakajima et al., 1988). Melanoma heparanase responsible for HS degradation cleaves HS at specific intrachain sites, which results in the formation of fragments of discrete molecular weight size (Marchetti, 1997; Nakajima et al., 1986a). Therefore, heparanase was identified as an endo-β-d-glucuronidase (Nakajima et al., 1986b, 1988). Importantly, heparanase differs from heparinases or other HS-specific elimination enzymes (bacterial heparitinases), which are eliminases indiscriminately cleaving HS to disaccharide and/or tetrasaccharide units. Heparanase activities have also been described in the immune system and in cancer cells other than melanoma (Nakajima et al., 1988; Vlodavsky and Friedmann, 2001; Vlodavsky et al., 1999). Increased levels of heparanase activity are associated with metastatic melanoma and other invasive tumors types, and copious evidence has demonstrated its role in tumor cell invasion into distant organs (Nakajima et al., 1988; Vlodavsky and Friedmann, 2001).
By examining the heparanase/HSPG axis in brain-metastatic melanoma, we have demonstrated the following: (1) highly brain-invasive human melanoma cells degrade purified ECM HS and HS cell-surface subpopulations faster than sublines of lower metastatic potential (Marchetti et al., 1993, 1996; Walch et al., 1999); (2) heparanase is responsible for this HS degradation (Marchetti, 1997; Marchetti et al., 1996); (3) in direct correlation with both increased invasiveness and presence of their specific NTR, select NT members augment heparanase production in brain-metastatic melanoma, which makes it a major candidate enzyme responsible for ECM degradation (Marchetti and Nicolson, 1997b; Marchetti et al., 1993, 1995, 1996); and (4) heparanase recognizes specific motifs within HS chains associated with both the binding domains to angiogenic factors and to an HS-interacting protein, HIP, which was recently cloned and characterized (Marchetti et al., 1997b).
Of note, human heparanase had not been purified nor well characterized or cloned until 1999 (Hulett et al., 1999; Kussie et al., 1999; Toyoshima and Nakajima, 1999; Vlodavsky et al., 1999). Therefore, molecular tools to explore the potentially important roles of heparanase in disease have been lacking for almost 25 years following the first reports describing its enzymatic activity. Interestingly, the newly discovered cDNA sequences of human heparanase that is derived from normal and tumor cells represent the same gene (Hulett et al., 1999; Kussie et al., 1999; Toyoshima and Nakajima, 1999; Vlodavsky et al., 1999).
We have postulated that heparanase plays 2 critical roles in the biology of brain metastasis, which are (1) in local invasive processes by degrading the HS chains of HSPGs and (2) in the release of HS-bound angiogenic factors at the metastatic site, with the brain considered the ideal environment because of its high levels of NT production. Heparanase can therefore be dually relevant in brain-metastatic melanoma in consideration of the strong angiogenic properties exhibited by melanoma cells in the brain. In fact, although metastasizing cancer cells may produce as many as 28 different matrix-digesting proteases, the new findings show that there is only 1 heparanase. Heparanase inhibition not only inhibits the ability of cancer cells to invade but also hinders the formation of new blood vessels that feed tumors, or angiogenesis (Folkman, 2001).
To determine if poorly metastatic melanoma cells acquire an increased metastatic potential following heparanase gene upregulation, we constructed eukaryotic expression vectors that contained the full-length human heparanase cDNA and used them to transfect melanoma cells. Transfection of the human heparanase gene into cells of low invasive potential and heparanase content resulted in functional enzymatic activity and in significantly increased (7- to 14-fold) invasion of transfected cells, as demonstrated by using in vitro chemo-invasion assays with purified HSPG as an invasive barrier (Marchetti and Nicolson, 2001).
Equally relevant to the area of brain invasion and metastasis is assessing heparanase contributions to angiogenic events. Extracellular matrix HSPGs serve as a reservoir for angiogenic factors, such as basic fibroblast growth factor (bFGF), that can be extracted from subendothelial ECM produced in vitro (Aviezer et al., 1997; McKeehan and Kan, 1994). Displacement of bFGF from the ECM by heparanase can provide a novel mechanism for induction of neovascularization in normal and pathological conditions (Rodeck et al., 1991). Several studies have indicated that heparin and HS inhibit the mitogenic activity of angiogenic bFGF and at the same time stabilize and protect the molecule from inactivation (Gospodarowicz and Cheng, 1986). Basic fibroblast growth factor is stored in the ECM in a highly stable, inactive form. Its release from ECM as a complex with HS fragments can result in a form of bFGF that is more stable than free bFGF and capable of binding the high-affinity plasma-membrane receptors.
Since the abilities of bFGF to bind ECM HSPG and to act as a potent angiogenic factor have long been recognized (Folkman, 2001), we have evaluated the ability of purified human heparanase to modulate bFGF activity and its release from ECM HSPG by using the rabbit ear chamber method. This in vivo angiogenic assay has been used for the past 50 years as a well-controlled in vivo model to study angiogenesis, wound healing, and tissue responses. Implantation of HSPG-bFGF beads previously incubated with purified heparanase into rabbit ear chambers produced a significantly increased vascularization (Marchetti et al., 2003b). Furthermore, inhibition of heparanase-induced angiogenesis was achieved in the presence of a selected group of suramin analogues as newly developed potent angiogenic inhibitors, which demonstrates that these reagents inhibited heparanase-mediated degradation of subendothelial ECM (Marchetti et al., 2003b). These results further emphasize the importance of heparanase in brain invasive and angiogenic mechanisms (Vlodavsky and Friedmann, 2001) and the potential clinical application of heparanase inhibitors, such as suramin analogs or others, in angiogenic-dependent cancers like brain-metastatic melanoma.
Brain Tissue Neurotrophin Production at the Melanoma Tumor Invasion Front
Since growth factor production could influence the invasion and growth of melanoma cells in the brain, we have examined human brain-metastatic melanoma (70W) cells for synthesis of NT-regulating factors and for the presence of growth factor transcripts. Reverse transcriptase-polymerase chain reaction analysis revealed the production of NT-regulating transforming growth factor (TGF)-β 1 and bFGF, as well as other factors such as TGF-α and interleukin-1β (Menter et al., 1994). Some of these factors can stimulate brain astrocytes or oligodendrocytes to produce NT. We reasoned that, because melanoma cells could produce NT-regulating factors, these factors might influence NT production in the brain. Therefore, we have investigated whether brain-invading melanoma cells induced changes in NT concentration or NT distribution at the brain invasion front of melanoma tumors in vivo. Brain-tissue samples from human melanoma metastases and uninvolved brain tissues in adjacent sections were examined immunohistochemically for NT presence. Staining of serial sections with anti-NT monoclonal antibodies revealed increased concentrations of NT (in particular, NGF and NT3) in tumor-adjacent tissues. Staining was highest at the interface between the melanoma tumor and adjacent normal brain tissue and gradually decreased in concentration until NTs were undetectable at more distant sites (Marchetti et al., 1995). Using these methodologies, we observed that controls without primary antibody or uninvolved brain tissue progressively distant from the melanoma lesion possessed very low or undetectable levels of NT (Marchetti et al., 1995).
Enhancement by Astrocytes, Brain Environment, and Cellular Responses
Astrocytes Contribute to the Brain-Metastatic Specificity of Melanoma Cells by Producing Heparanase
Astrocytes, which are among the first brain cells encountered by extravasating melanoma cells, produce NT (Yoshida and Gage, 1992). In addition, they are capable of binding NT because they express members of the TRK receptor family and p75NTR (Yoshida and Gage, 1992). Futhermore, they are relevant because astrogliosis can be at times a pathologic trauma in response to brain-invasive events: Injury-reacting astrocytes are frequently found in areas surrounding melanotic lesions. Astrocytes can therefore play important roles in the development of brain metastases. To test this hypothesis, we have employed purified in vitro astrocytic cultures and investigated the presence of heparanase. Primary glial cells were obtained from newborn rat and mouse cerebra by established purification methods (McCarthy and de Vellis, 1980). Their identification as astrocytes was confirmed by positive immunoreactivity with an antibody against the astrocyte-specific, intermediate-filament glial fibrillary acidic protein GFAP (McCarthy and de Vellis, 1980). We next examined the astrocyte cultures for expression of heparanase. A specific heparanase transcript was detected by semiquantitative reverse transcriptase-polymerase chain reaction. This transcript was upregulated 3-fold in astrocytes previously incubated with purified and biologically active NGF (Marchetti et al., 2000). Similar results were obtained with human brain-metastatic 70W cells. Heparanase activity was also detectable and NGF regulated in cellular extracts from both purified astrocytes and brain-metastatic cells. This was shown by the appearance of distinct HS degradation products detected in agarose gel shift assays or by high-speed gel permeation column chromatography (high-performance liquid chromatography analysis; Marchetti et al., 2000).
Second, we analyzed heparanase activity in brain-metastatic melanoma cells and astrocytic cell populations in logarithmic growth by obtaining high-performance liquid chromatography–derived elution profiles of HS-digested products at various incubation times (Marchetti et al., 1993). Cultures of highly brain-metastatic 70W cells showed a gradual and time-dependent increase in heparanase activity for up to 72 h. After this time interval, the heparanase levels in cultures plateaued. Interestingly, cultures of astrocytes also produced heparanase in a time-dependent manner. Moreover, coincubation of brain-metastatic melanoma cells and astrocytes in equicellular numbers resulted in a superadditive increase of enzymatic activity, greater than that expected for both cell types.
Finally, we incubated brain-metastatic melanoma cells with astrocyte-conditioned medium (ACM) and examined its effects on their invasive characteristics. We found consistent increases in in vitro invasion following exposure of these cells to ACM. Invasion was most pronounced when ACM from NGF-treated astrocytes was used, and the invasion effects of ACM were completely abrogated in the presence of heparanase antibodies (Marchetti and Nicolson, 2001). The invasion enhancement caused by this NGF treatment was also abolished in the presence of a neutralizing NGF monoclonal antibody, which confirms the relevance of melanoma/astrocyte heparanase and its NT-regulation to brain invasion events.
The Brain as a Unique Compartment for the Invasion and Growth of Malignant Melanoma Cells
Homeostasis and the control of material flow into the brain is strictly regulated by the BBB. Brain-metastatic cells must breach this formidable barrier to invade and colonize the brain parenchyma. As discussed above, invasion of the brain requires that metastatic cells increase their expression of certain cell surface receptors (NTRs), degradative enzymes (heparanase), and growth factors and cytokines (TGFα/β, bFGF, interleukin-1β, and others). They must also respond to invasion-stimulating cytokines such as NTs and paracrine growth factors.
Brain-metastasizing melanoma cells express relatively high levels of BM hydrolytic enzymes such as type IV collagenases, cathepsins, plasminogen activators, and of relevance, heparanase. Although highly metastatic cells generally express higher amounts of degradative enzymes than nonmetastatic cells, some of these enzymes are induced to even higher levels by the microenvironment (paracrine invasion factors) or are provided by certain normal cells (microvessel endothelial cells and astrocytes, among others; Nicolson et al., 1994, 1996). If the appropriate paracrine signals are received by malignant cells, they can be stimulated to increase the synthesis and release of BBB-degrading enzymes.
Cellular Responses to Brain Tissue Injury as a Paradigm for Brain Metastasis
Astroglial cells constitute the primary cellular response following brain injury (Norenberg, 1994). Astrocytes are the predominant cell type in the brain, outnumbering neurons by a factor of 10 to 1. By numbers, astrocytes make up one third of the cerebral cortex; however, as a population of cells they are widely heterogenous (Kettenmann et al., 1983; Wilkin et al., 1990). One of the earliest pathological responses to brain trauma involves astrocyte swelling occurring predominantly in the perivascular astrocytic endings (Hirano et al., 1994; Kimelberg and Ransom, 1986). In experimental brain tumors, cerebral edema has been associated with significant alterations in vascular permeability (Lantos et al., 1984). If the BBB is compromised, astrocyte swelling may involve vasogenic edema. In this case, the astrocytes swell as they take up proteins and water that may become cytotoxic because of increased potassium and glutamate (Hirano et al., 1994; Klatzo et al., 1980; Norenberg, 1994). It is generally believed that astrocyte swelling is caused by increases in intracellular osmolarity followed by water influx. This can occur without loss of BBB integrity and perhaps simply represents a redistribution of water from the neuronal to the astrocytic cell compartment. This form of astrocyte swelling is generally not as severe as the astrocyte swelling that can result from vasogenic edema associated with the trauma caused by tumor cell invasion. If astrocyte swelling becomes too severe, it can cause depolarization of astroglial cells, leading to the loss of homeostatic ion gradients and membrane rupture, resulting in cell death. These dynamic astrocyte changes in response to tumor cell invasion can lead to increased intracranial pressure and further complications. The tumor-induced response by astrocytes is being investigated as a cause, or one of the causes, of brain metastases causing severe symptoms such as paralysis, headache, seizures, and impaired cognition.
Vasogenic edema leads to the influx of thrombin, platelet-derived growth factor, steroids, insulin, and various cytokines from the blood and lymphocytes, as well as endothelin, ATP, and bFGF from endothelial cells (Fig. 4). The induction of reactive astrocytes, when associated with tumor cell invasion, is likely triggered by endogenous factors in the brain in addition to those provided by the invading tumor cells (Fig. 4). We have observed reactive gliosis in brain tissue associated with the melanoma invasion front, illustrating the cellular response of the adjacent brain tissue (Nicolson et al., 1996). In addition to morphological changes, the adjoining brain cells produce high levels of NTs in comparison with uninvolved brain tissue (Marchetti et al., 1995). Thus, brain-metastatic melanoma cells may induce the production of brain cytokines such as NTs that aid in the invasion and survival of melanoma cells in the CNS.
Fig. 4.

Reciprocal interactions between brain-invading melanoma cells and normal cells in the brain microenvironment. Tumor cells release cytokines that can affect host cells such as parenchymal cells, endothelial and glial cells, astrocytes, and brain tissue extracellular matrix (ECM). Reactive astrocytes can arise from stimulation by factors released by invading melanoma cells. In turn, brain cells can release factors that stimulate tumor cell motility and invasion. Astrocytes, oligodendrocytes, and neurons can release NT and ECM degradative enzymes, for example, heparanase produced by astrocytes (Marchetti et al., 2000) in response to brain-invading melanoma. Conversely, these cells secrete growth factors and cytokines which can synergistically regulate NT synthesis and activity in normal brain cells.
Concluding Remarks and Scientific Challenges
The points raised in this review raise questions more than provide answers in dealing with brain-metastatic melanoma. Clearly, we have much more to learn about the mechanisms used by melanoma cells in colonizing the brain. Some of the scientific challenges are the following.
There is no simple mechanism that can explain the organ preference of brain metastasis. Although we know much more about the growth and invasive properties of brain-metastatic melanoma cells, we are now beginning to identify melanoma and brain properties involved in brain metastasis. Nonetheless, some of these properties, such as the expression of cell-surface NTRs, response to NTs, and production/secretion of NT-regulated heparinase, may be general properties of tumor cells that metastasize to the brain.
Various brain cells other than astrocytes may contribute enzymes, motility factors, growth factors, growth inhibitors, and many other cytokines that have not yet been fully characterized. These may provide an appropriate microenvironment for invasion and brain colonization by brain-metastatic melanoma cells. This needs further research emphasis and will require further examination since we must obtain a greater understanding of the brain microenvironment and the reciprocal cytokine regulation of growth. Because many of the cytokines involved in brain metastasis are effective at stimulating both the brain tissue and invading melanoma cells, this will be one of the most difficult problems to decipher.
The frequency of distribution of certain cancers to sites that follow their developmental potentials suggests that inappropriately expressed developmental genes could contribute to the organ preference of metastasis. Unfortunately, we know little about the developmental processes that control embryonic cell motility and organ colonization by normal cells during development. It is interesting, however, to note that NTs are important molecules, signaling growth and differentiation of specific neuronal cell subpopulations at specific times during development. It may not be a coincidence that brain-metastatic melanoma cells may use paracrine NTs as source of growth in the brain. This will require further research efforts by performing in vivo experiments to validate causative roles of the NT/NTR axis and heparanase in melanoma brain invasion in vivo.
Challenges in growth factor and growth inhibitor responses during melanoma progression may be important to the continued survival and growth of malignant cells in secondary compartments, and these challenges are probably driven by the selective pressures of the host. The inherent instability of malignant cells presents a daunting problem because it is likely that the most unstable, rapidly progressing melanoma cells will be the ones that metastasize to critical sites like the brain and resist therapy.
The implications of these unsolved phenomena in brain metastasis may be particularly profound for the brain tumor biologist, the neuro-oncologist, and, most importantly, the brain cancer patient affected with such devastating disease.
Acknowledgment
The authors thank Kathleen Kirvin for her secretarial assistance.
Footnotes
This work was supported by grants from the U.S. National Institutes of Health to Dario Marchetti.
Abbreviations used are as follows: ACM, astrocyte-conditioned medium; BBB, blood-brain barrier; BDNF, brain-derived neurotrophic growth factor; bFGF, basic fibroblast growth factor; BM, basement membrane; ECM, extracellular matrix; HS, heparan sulfate; HSPG, heparan sulfate proteoglycan; MAPK, mitogen-activated protein kinase; MEK, mitogen-activated protein kinase/extracellular-signal-regulated kinase; NGF, nerve growth factor; NGFR, nerve growth factor receptor; NT, neurotrophin; NTR, neurotrophin receptor; PKN, protein kinase N; ser/thr, serine/threonine; TGF, transforming growth factor; TK, tyrosine kinase; TNF, tumor necrosis factor; TRK, tropomyosin receptor kinase.
References
- Albino AP, Davis BM, Nanus DM. Induction of growth factor RNA expression in human malignant melanoma: Markers of transformation. Cancer Res. 1991;51:4815–4820. [PubMed] [Google Scholar]
- Aviezer D, Iozzo RV, Noonan DM, Yayon A. Suppression of autocrine and paracrine functions of basic fibroblast growth factor by stable expression of perlecan antisense cDNA. Mol Cell Biol. 1997;17:1938–1946. doi: 10.1128/mcb.17.4.1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avruch J, Zhang XF, Kyriakis JM. Raf meets Ras: Completing the framework of a signal transduction pathway. Trends Biochem Sci. 1994;19:279–283. doi: 10.1016/0968-0004(94)90005-1. [DOI] [PubMed] [Google Scholar]
- Barbacid M. Nerve growth factor: A tale of two receptors. Oncogene. 1993;8:2033–2042. [PubMed] [Google Scholar]
- Barrett GL, Bartlett PF. The p75 nerve growth factor receptor mediates survival or death depending on the stage of sensory neuron development. Proc Natl Acad Sci USA. 1994;91:6501–6505. doi: 10.1073/pnas.91.14.6501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batistatou A, Volonté C, Greene LA. Nerve growth factor employs multiple pathways to induce primary response genes in PC12 cells. Mol Biol Cell. 1992;3:363–371. doi: 10.1091/mbc.3.3.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg MM, Sternberg DW, Hempstead BL, Chao MV. The low-affinity p75 nerve growth factor (NGF) receptor mediates NGF-induced tyrosine phosphorylation. Proc Natl Acad Sci USA. 1991;88:7106–7110. doi: 10.1073/pnas.88.16.7106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernfield M, Gotte M, Park PW, Reizes O, Fitzgerald ML, Lincecum J, Zako M. Functions of cell surface heparan sulfate proteoglycans. Annu Rev Biochem. 1999;68:729–777. doi: 10.1146/annurev.biochem.68.1.729. [DOI] [PubMed] [Google Scholar]
- Beutler B, van Huffel C. Unraveling function in the TNF ligand and receptor families. Science. 1994;264:667–668. doi: 10.1126/science.8171316. [DOI] [PubMed] [Google Scholar]
- Bibel M, Hoppe E, Barde YA. Biochemical and functional interactions between the neurotrophin receptors trk and p75NTR. EMBO J. 1999;18:616–622. doi: 10.1093/emboj/18.3.616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borrello MG, Pelicci G, Arighi E, De Filippis L, Greco A, Bongarzone I, Rizzetti M, Pelicci PG, Pierotti MA. The oncogenic versions of the Ret and Trk tyrosine kinases bind Shc and Grb2 adaptor proteins. Oncogene. 1994;9:1661–1668. [PubMed] [Google Scholar]
- Bradshaw RA, Blundell TL, Lapatto R, McDonald NQ, Murray-Rust J. Nerve growth factor revisited. Trends Biochem Sci. 1993;18:48–52. doi: 10.1016/0968-0004(93)90052-o. [DOI] [PubMed] [Google Scholar]
- Chao MV. Neurotrophin receptors: A window into neuronal differentiation. Neuron. 1992;9:583–593. doi: 10.1016/0896-6273(92)90023-7. [DOI] [PubMed] [Google Scholar]
- Chao MV, Bothwell M. Neurotrophins: To cleave or not to cleave. Neuron. 2002;33:9–12. doi: 10.1016/s0896-6273(01)00573-6. [DOI] [PubMed] [Google Scholar]
- Dobrowsky RT, Werner MH, Castellino AM, Chao MV, Hannun YA. Activation of the sphingomyelin cycle through the low-affinity neurotrophin receptor. Science. 1994;265:1596–1599. doi: 10.1126/science.8079174. [DOI] [PubMed] [Google Scholar]
- Feinstein DL, Larhammar D. Identification of a conserved protein motif in a group of growth factor receptors. FEBS Lett. 1990;272:7–11. doi: 10.1016/0014-5793(90)80437-n. [DOI] [PubMed] [Google Scholar]
- Fidler IJ, Kripke ML. Metastasis results from preexisting variant cells within a malignant tumor. Science. 1977;197:893–895. doi: 10.1126/science.887927. [DOI] [PubMed] [Google Scholar]
- Folkman J. Angiogenesis-dependent diseases. Semin Oncol. 2001;28:536–542. doi: 10.1016/s0093-7754(01)90021-1. [DOI] [PubMed] [Google Scholar]
- Gladson CL, Wilcox JN, Sanders L, Gillespie GY, Cheresh DA. Cerebral microenvironment influences expression of the vitronectin gene in astrocytic tumors. J Cell Sci. 1995;108:947–956. doi: 10.1242/jcs.108.3.947. [DOI] [PubMed] [Google Scholar]
- Gospodarowicz D, Cheng J. Heparin protects basic and acidic FGF from inactivation. J Cell Physiol. 1986;128:475–484. doi: 10.1002/jcp.1041280317. [DOI] [PubMed] [Google Scholar]
- Hantzopoulos PA, Suri C, Glass DJ, Goldfarb MP, Yancopoulos GD. The low-affinity NGF receptor, p75, can collaborate with each of the Trks to potentiate functional responses to the neurotrophins. Neuron. 1994;13:187–201. doi: 10.1016/0896-6273(94)90469-3. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Schleifer LS, Chao MV. Expression of functional nerve growth factor receptors after gene transfer. Science. 1989;243:373–375. doi: 10.1126/science.2536190. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Martin-Zanca D, Kaplan DR, Parada LF, Chao MV. High-affinity NGF binding requires coexpression of the trk proto-oncogene and the low-affinity NGF receptor. Nature. 1991;350:678–683. doi: 10.1038/350678a0. [DOI] [PubMed] [Google Scholar]
- Hempstead BL, Patil N, Thiel B, Chao MV. Deletion of cytoplasmic sequences of the nerve growth factor receptor leads to loss of high-affinity ligand binding. J Biol Chem. 1990;265:9595–9598. [PubMed] [Google Scholar]
- Herlyn M, Thurin J, Balaban G, Bennicelli JL, Herlyn D, Elder DE, Bondi E, Guerry D, Nowell P, Clark WH. Characteristics of cultured human melanocytes isolated from different stages of tumor progression. Cancer Res. 1985;45:5670–5676. [PubMed] [Google Scholar]
- Herrmann JL, Menter DG, Hamada J, Marchetti D, Nakajima M, Nicolson GL. Mediation of NGF-stimulated extracellular matrix invasion by the human melanoma low-affinity p75 neurotrophin receptor: Melanoma p75 functions independently of trkA. Mol Biol Cell. 1993;4:1205–1216. doi: 10.1091/mbc.4.11.1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirano A, Kawanami T, Llena JF. Electron microscopy of the blood-brain barrier in disease. Microsc Res Tech. 1994;27:543–556. doi: 10.1002/jemt.1070270609. [DOI] [PubMed] [Google Scholar]
- Hulett MD, Freeman C, Hamdorf BJ, Baker RT, Harris MJ, Parish CR. Cloning of mammalian heparanase, an important enzyme in tumor invasion and metastasis. Nat Med. 1999;5:803–809. doi: 10.1038/10525. [DOI] [PubMed] [Google Scholar]
- Iozzo RV. Cell surface heparan sulfate proteoglycan and the neoplastic phenotype. J Cell Biochem. 1988;37:61–78. doi: 10.1002/jcb.240370107. [DOI] [PubMed] [Google Scholar]
- Iozzo RV, Murdoch AD. Proteoglycans of the extracellular environment: Clues from the gene and protein side offer novel perspectives in molecular diversity and function. FASEB J. 1996;10:598–614. [PubMed] [Google Scholar]
- Ishikawa M, Dennis JW, Man S, Kerbel RS. Isolation and characterization of spontaneous wheat germ agglutinin-resistant human melanoma mutants displaying remarkably different metastatic profiles in nude mice. Cancer Res. 1988;48:665–670. [PubMed] [Google Scholar]
- Johnson D, Lanahan A, Buck CR, Sehgal A, Morgan C, Mercer E, Bothwell M, Chao M. Expression and structure of the human NGF receptor. Cell. 1986;47:545–554. doi: 10.1016/0092-8674(86)90619-7. [DOI] [PubMed] [Google Scholar]
- Kannan Y, Usami K, Okada M, Shimizu S, Matsuda H. Nerve growth factor suppresses apoptosis of murine neutrophils. Biochem Biophys Res Commun. 1992;186:1050–1056. doi: 10.1016/0006-291x(92)90853-d. [DOI] [PubMed] [Google Scholar]
- Kaplan DR, Miller FD. Neurotrophin signal transduction in the nervous system. Curr Opin Neurobiol. 2000;10:381–391. doi: 10.1016/s0959-4388(00)00092-1. [DOI] [PubMed] [Google Scholar]
- Kettenmann H, Orkand RK, Schachner M. Coupling among identified cells in mammalian nervous system cultures. J Neurosci. 1983;3:506–516. doi: 10.1523/JNEUROSCI.03-03-00506.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimelberg, H.K., and Ransom, B.R. (1986) Physiological and pathological aspects of astrocytic swelling. In: Fedoroff, S., and Vernadakis, A. (Eds.), Astrocytes: Cell Biology and Pathology of Astrocytes, Volume 3 Orlando: Academic Press, pp. 129–166.
- Klatzo I, Chui E, Fujiwara K, Spatz M. Resolution of vasogenic brain edema. Adv Neurol. 1980;28:359–373. [PubMed] [Google Scholar]
- Knipper M, Beck A, Rylett J, Breer H. Neurotrophin induced cAMP and IP3 responses in PC12 cells: Different pathways. FEBS Lett. 1993;324:147–152. doi: 10.1016/0014-5793(93)81382-a. [DOI] [PubMed] [Google Scholar]
- Kussie PH, Hulmes JD, Ludwig DL, Patel S, Navarro EC, Seddon AP, Giorgio NA, Bohlen P. Cloning and functional expression of a human heparanase gene. Biochem Biophys Res Commun. 1999;261:183–187. doi: 10.1006/bbrc.1999.0962. [DOI] [PubMed] [Google Scholar]
- Lange-Carter CA, Johnson GL. Ras-dependent growth factor regulation of MEK kinase in PC12 cells. Science. 1994;265:1458–1461. doi: 10.1126/science.8073291. [DOI] [PubMed] [Google Scholar]
- Lantos, P.L., Luthert, P.J., and Deane, B.R. (1984) Vascular permeability and cerebral oedema in experimental brain tumors. In: Inaba, Y., Klatzo, I., and Spatz, I. (Eds.), Brain Edema New York: Springer, pp. 40–47.
- Lee KF, Li E, Huber LJ, Landis SC, Sharpe AH, Chao MV, Jaenisch R. Targeted mutation of the gene encoding the low affinity NGF receptor p75 leads to deficits in the peripheral sensory nervous system. Cell. 1992;69:737–749. doi: 10.1016/0092-8674(92)90286-l. [DOI] [PubMed] [Google Scholar]
- Lee KF, Bachman K, Landis S, Jaenisch R. Dependence on p75 for innervation of some sympathetic targets. Science. 1994;263:1447–1449. doi: 10.1126/science.8128229. [DOI] [PubMed] [Google Scholar]
- Lee R, Kermani P, Teng KK, Hempstead BL. Regulation of cell survival by secreted proneurotrophins. Science. 2001;294:1945–1948. doi: 10.1126/science.1065057. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Thorgeirsson UP, Garbisa S. Role of collagenases in tumor cell invasion. Cancer Metastasis Rev. 1982;1:277–288. doi: 10.1007/BF00124213. [DOI] [PubMed] [Google Scholar]
- Liotta LA, Steeg PS, Stetler-Stevenson WG. Cancer metastasis and angiogenesis: An imbalance of positive and negative regulation. Cell. 1991;64:327–336. doi: 10.1016/0092-8674(91)90642-c. [DOI] [PubMed] [Google Scholar]
- Maher PA. Nerve growth factor induces protein-tyrosine phosphorylation. Proc Natl Acad Sci USA. 1988;85:6788–6791. doi: 10.1073/pnas.85.18.6788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchetti D. Specific degradation of subendothelial matrix proteoglycans by brain-metastatic melanoma and brain endothelial cell heparanases. J Cell Physiol. 1997;172:334–342. doi: 10.1002/(SICI)1097-4652(199709)172:3<334::AID-JCP7>3.0.CO;2-P. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Nicolson GL. Human melanoma cell invasion: Selected neurotrophin enhancement of invasion and heparanase activity. J Invest Dermatol Symp Proc. 1997a;2:99–105. doi: 10.1038/jidsymp.1997.19. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Nicolson GL. Neurotrophin stimulation of human melanoma cell invasion: Selected enhancement of heparanase activity and heparanase degradation of specific heparan sulfate subpopulations. Adv Enzyme Regul. 1997b;37:111–134. doi: 10.1016/s0065-2571(96)00019-2. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Nicolson GL. Human heparanase: A molecular determinant of brain metastasis. Adv Enzyme Regul. 2001;41:343–359. doi: 10.1016/s0065-2571(00)00016-9. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Menter D, Jin L, Nakajima M, Nicolson GL. Nerve growth factor effects on human and mouse melanoma cell invasion and heparanase production. Int J Cancer. 1993;55:692–699. doi: 10.1002/ijc.2910550430. [DOI] [PubMed] [Google Scholar]
- Marchetti D, McCutcheon IE, Ross MJ, Nicolson GL. Inverse expression of neurotrophins and neurotrophin receptors at the invasion front of human-melanoma brain metastases. Int J Oncol. 1995;7:87–94. doi: 10.3892/ijo.7.1.87. [DOI] [PubMed] [Google Scholar]
- Marchetti D, McQuillan DJ, Spohn WC, Carson DD, Nicolson GL. Neurotrophin stimulation of human melanoma cell invasion: Selected enhancement of heparanase activity and heparanase degradation of specific heparan sulfate subpopulations. Cancer Res. 1996;56:2856–2863. [PubMed] [Google Scholar]
- Marchetti D, Liu S, Spohn WC, Carson DD. Heparanase and a synthetic peptide of heparan sulfate-interacting protein recognize common sites on cell surface and extracellular matrix heparan sulfate. J Biol Chem. 1997;272:15891–15897. doi: 10.1074/jbc.272.25.15891. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Li J, Shen R. Astrocytes contribute to the brain-metastatic specificity of melanoma cells by producing heparanase. Cancer Res. 2000;60:4767–4770. [PubMed] [Google Scholar]
- Marchetti D, Murry B, Galjour J, Wilke-Greiter A. Human melanoma TrkC: Its association with a purine-analog-sensitive kinase activity. J Cell Biochem. 2003a;88:865–872. doi: 10.1002/jcb.10473. [DOI] [PubMed] [Google Scholar]
- Marchetti D, Reiland J, Erwin B, Roy M. Inhibition of heparanase activity and heparanase-induced angiogenesis by suramin analogues. Int J Cancer. 2003b;104:167–174. doi: 10.1002/ijc.10930. [DOI] [PubMed] [Google Scholar]
- McCarthy KD, de Vellis J. Preparation of separate astroglial and oligodendroglial cell cultures from rat cerebral tissue. J Cell Biol. 1980;85:890–902. doi: 10.1083/jcb.85.3.890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKeehan WL, Kan M. Heparan sulfate fibroblast growth factor receptor complex: Structure-function relationships. Mol Reprod Dev. 1994;39:69–81. doi: 10.1002/mrd.1080390112. [DOI] [PubMed] [Google Scholar]
- Meakin SO, Shooter EM. The nerve growth factor family of receptors. Trends Neurosci. 1992;15:323–331. doi: 10.1016/0166-2236(92)90047-c. [DOI] [PubMed] [Google Scholar]
- Menter DG, Herrmann JL, Marchetti D, Nicolson GL. Involvement of neurotrophins and growth factors in brain metastasis formation. Invasion Metastasis. 1994;14:372–384. [PubMed] [Google Scholar]
- Miyasaka T, Chao MV, Sherline P, Saltiel AR. Nerve growth factor stimulates a protein kinase in PC-12 cells that phosphorylates microtubule-associated protein-2. J Biol Chem. 1990;265:4730–4735. [PubMed] [Google Scholar]
- Morse HG, Gonzalez R, Moore GE, Robinson WA. Preferential chromosome 11q and/or 17q aberrations in short-term cultures of metastatic melanoma in resections from human brain. Cancer Genet Cytogenet. 1992;64:118–126. doi: 10.1016/0165-4608(92)90340-e. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Irimura T, Nicolson GL. A solid-phase substrate of heparanase: Its application to assay of human melanoma for heparan sulfate degradative activity. Anal Biochem. 1986a;157:162–171. doi: 10.1016/0003-2697(86)90209-5. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Irimura T, Nicolson GL. Tumor metastasis-associated heparanase (heparan sulfate endoglycosidase) activity in human melanoma cells. Cancer Lett. 1986b;31:277–283. doi: 10.1016/0304-3835(86)90148-5. [DOI] [PubMed] [Google Scholar]
- Nakajima M, Irimura T, Nicolson GL. Heparanases and tumor metastasis. J Cell Biochem. 1988;36:157–167. doi: 10.1002/jcb.240360207. [DOI] [PubMed] [Google Scholar]
- Nicolson GL. Tumor cell instability, diversification, and progression to the metastatic phenotype: From oncogene to oncofetal expression. Cancer Res. 1987;47:1473–1487. [PubMed] [Google Scholar]
- Nicolson GL, Menter DG, Herrmann J, Cavanaugh P, Jia L, Hamada J, Yun Z, Nakajima M, Marchetti D. Tumor metastasis to brain: Role of endothelial cells, neurotrophins, and paracrine growth factors. Crit Rev Oncog. 1994;5:451–471. doi: 10.1615/critrevoncog.v5.i5.20. [DOI] [PubMed] [Google Scholar]
- Nicolson GL, Menter DG, Herrmann JL, Yun Z, Cavanaugh P, Marchetti D. Brain metastasis: Role of trophic, autocrine, and paracrine factors in tumor invasion and colonization of the central nervous system. Curr Top Microbiol Immunol. 1996;213:89–115. doi: 10.1007/978-3-642-61109-4_5. [DOI] [PubMed] [Google Scholar]
- Norenberg MD. Astrocyte responses to CNS injury. J Neuropathol Exp Neurol. 1994;53:213–220. doi: 10.1097/00005072-199405000-00001. [DOI] [PubMed] [Google Scholar]
- Obermeier A, Halfter H, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A. Tyrosine 785 is a major determinant of Trk-substrate interaction. EMBO J. 1993a;12:933–941. doi: 10.1002/j.1460-2075.1993.tb05734.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obermeier A, Lammers R, Wiesmuller KH, Jung G, Schlessinger J, Ullrich A. Identification of Trk binding sites for SHC and phosphatidylinositol 3′-kinase and formation of a multimeric signaling complex. J Biol Chem. 1993b;268:22963–22966. [PubMed] [Google Scholar]
- Obermeier A, Bradshaw RA, Seedorf K, Choidas A, Schlessinger J, Ullrich A. Neuronal differentiation signals are controlled by nerve growth factor receptor/Trk binding sites for SHC and PLC gamma. EMBO J. 1994;13:1585–1590. doi: 10.1002/j.1460-2075.1994.tb06421.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohmichi M, Decker SJ, Pang L, Saltiel AR. Phospholipase C-gamma 1 directly associates with the p70 trk oncogene product through its src homology domains. J Biol Chem. 1991;266:14858–14861. [PubMed] [Google Scholar]
- Ohmichi M, Decker SJ, Saltiel AR. Nerve growth factor stimulates the tyrosine phosphorylation of a 38-kDa protein that specifically associates with the src homology domain of phospholipase C-gamma 1. J Biol Chem. 1992a;267:21601–21606. [PubMed] [Google Scholar]
- Ohmichi M, Decker SJ, Saltiel AR. Activation of phosphatidylinositol-3 kinase by nerve growth factor involves indirect coupling of the trk proto-oncogene with src homology 2 domains. Neuron. 1992b;9:769–777. doi: 10.1016/0896-6273(92)90039-g. [DOI] [PubMed] [Google Scholar]
- Ohmichi M, Matuoka K, Takenawa T, Saltiel AR. Growth factors differentially stimulate the phosphorylation of Shc proteins and their association with Grb2 in PC-12 pheochromocytoma cells. J Biol Chem. 1994;269:1143–1148. [PubMed] [Google Scholar]
- Peacocke M, Yaar M, Mansur CP, Chao MV, Gilchrest BA. Induction of nerve growth factor receptors on cultured human melanocytes. Proc Natl Acad Sci USA. 1988;85:5282–5286. doi: 10.1073/pnas.85.14.5282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell WC, Matrisian LM. Complex roles of matrix metallo-proteinases in tumor progression. Curr Top Microbiol Immunol. 1996;213:1–21. doi: 10.1007/978-3-642-61107-0_1. [DOI] [PubMed] [Google Scholar]
- Prados, M.D., and Wilson, C.B. (1993) Neoplasms of the central nervous system. In: Holland, J.F., Frei, E., III, Bast, R.C., Jr., Kufe, D.W., Morton, D.L., and Weischselbaum, R.R. (Eds.), Cancer Medicine, Third Edition Philadelphia: Lea & Febiger, pp. 1080–1119.
- Rabizadeh S, Oh J, Zhong LT, Yang J, Bitler CM, Butcher LL, Bredesen DE. Induction of apoptosis by the low-affinity NGF receptor. Science. 1993;261:345–348. doi: 10.1126/science.8332899. [DOI] [PubMed] [Google Scholar]
- Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- Raff MC, Barres BA, Burne JF, Coles HS, Ishizaki Y, Jacobson MD. Programmed cell death and the control of cell survival: Lessons from the nervous system. Science. 1993;262:695–700. doi: 10.1126/science.8235590. [DOI] [PubMed] [Google Scholar]
- Rodeck U, Becker D, Herlyn M. Basic fibroblast growth factor in human melanoma. Cancer Cells. 1991;3:308–311. [PubMed] [Google Scholar]
- Ross AH, Grob P, Bothwell M, Elder DE, Ernst CS, Marano N, Ghrist BF, Slemp CC, Herlyn M, Atkinson B, Koprowski H. Characterization of nerve growth factor receptor in neural crest tumors using monoclonal antibodies. Proc Natl Acad Sci USA. 1984;81:6681–6685. doi: 10.1073/pnas.81.21.6681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozakis-Adcock M, McGlade J, Mbamalu G, Pelicci G, Daly R, Li W, Batzer A, Thomas S, Brugge J, Pelicci PG, Schlessinger J, Pawson T. Association of the Shc and Grb2/Sem5 SH2-containing proteins is implicated in activation of the Ras pathway by tyrosine kinases. Nature. 1992;360:689–692. doi: 10.1038/360689a0. [DOI] [PubMed] [Google Scholar]
- Saltiel AR, Decker SJ. Cellular mechanisms of signal transduction for neurotrophins. Bioessays. 1994;16:405–411. doi: 10.1002/bies.950160608. [DOI] [PubMed] [Google Scholar]
- Satoh T, Nakafuku M, Kaziro Y. Function of Ras as a molecular switch in signal transduction. J Biol Chem. 1992;267:24149–24152. [PubMed] [Google Scholar]
- Sawaya R, Ligon BL, Bindal AK, Bindal RK, Hess KR. Surgical treatment of metastatic brain tumors. J Neurooncol. 1996;27:269–277. doi: 10.1007/BF00165484. [DOI] [PubMed] [Google Scholar]
- Sloane BF, Honn KV. Cysteine proteinases and metastasis. Cancer Metastasis Rev. 1984;3:249–263. doi: 10.1007/BF00048388. [DOI] [PubMed] [Google Scholar]
- Smith CA, Farrah T, Goodwin RG. The TNF receptor superfamily of cellular and viral proteins: Activation, costimulation, and death. Cell. 1994;76:959–962. doi: 10.1016/0092-8674(94)90372-7. [DOI] [PubMed] [Google Scholar]
- Snider WD. Functions of the neurotrophins during nervous system development: What the knockouts are teaching us. Cell. 1994;77:627–638. doi: 10.1016/0092-8674(94)90048-5. [DOI] [PubMed] [Google Scholar]
- Soffietti R, Ruda R, Mutani R. Management of brain metastases. J Neurol. 2002;249:1357–1369. doi: 10.1007/s00415-002-0870-6. [DOI] [PubMed] [Google Scholar]
- Steck, P.A., and Nicolson, G.L. (1993) Metastases to the central nervous system. In: Levine, A.J., and Schmidek, H.H. (Eds.), Molecular Genetics of Nervous System Tumors, Chapter 33. New York: Wiley-Liss, pp. 371–379.
- Stephens RM, Loeb DM, Copeland TD, Pawson T, Greene LA, Kaplan DR. Trk receptors use redundant signal transduction pathways involving SHC and PLC-gamma 1 to mediate NGF responses. Neuron. 1994;12:691–705. doi: 10.1016/0896-6273(94)90223-2. [DOI] [PubMed] [Google Scholar]
- Taylor LK, Swanson KD, Kerigan J, Mobley W, Landreth GE. Isolation and characterization of a nerve growth factor-regulated Fos kinase from PC12 cells. J Biol Chem. 1994;269:308–318. [PubMed] [Google Scholar]
- Toyoshima M, Nakajima M. Human heparanase. Purification, characterization, cloning, and expression. J Biol Chem. 1999;274:24153–24160. doi: 10.1074/jbc.274.34.24153. [DOI] [PubMed] [Google Scholar]
- Verdi JM, Birren SJ, Ibanez CF, Persson H, Kaplan DR, Benedetti M, Chao MV, Anderson DJ. p75LNGFR regulates Trk signal transduction and NGF-induced neuronal differentiation in MAH cells. Neuron. 1994;12:733–745. doi: 10.1016/0896-6273(94)90327-1. [DOI] [PubMed] [Google Scholar]
- Vlodavsky I, Friedmann Y. Molecular properties and involvement of heparanase in cancer metastasis and angiogenesis. J Clin Invest. 2001;108:341–347. doi: 10.1172/JCI13662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlodavsky I, Friedmann Y, Elkin M, Aingorn H, Atzmon R, Ishai-Michaeli R, Bitan M, Pappo O, Peretz T, Michal I, Spector L, Pecker I. Mammalian heparanase: Gene cloning, expression and function in tumor progression and metastasis. Nat Med. 1999;5:793–802. doi: 10.1038/10518. [DOI] [PubMed] [Google Scholar]
- Volonté C, Greene LA. Nerve growth factor-activated protein kinase N. Characterization and rapid near homogeneity purification by nucleotide affinity-exchange chromatography. J Biol Chem. 1992;267:21663–21670. [PubMed] [Google Scholar]
- Volonté C, Ross AH, Greene LA. Association of a purine-analogue-sensitive protein kinase activity with p75 nerve growth factor receptors. Mol Biol Cell. 1993;4:71–78. doi: 10.1091/mbc.4.1.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Bartheld CS, Kinoshita Y, Prevette D, Yin QW, Oppenheim RW, Bothwell M. Positive and negative effects of neurotrophins on the isthmo-optic nucleus in chick embryos. Neuron. 1994;12:639–654. doi: 10.1016/0896-6273(94)90219-4. [DOI] [PubMed] [Google Scholar]
- Walch ET, Albino AP, Marchetti D. Correlation of overexpression of the low-affinity p75 neurotrophin receptor with augmented invasion and heparanase production in human malignant melanoma cells. Int J Cancer. 1999;82:112–120. doi: 10.1002/(sici)1097-0215(19990702)82:1<112::aid-ijc19>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- Wilkin GP, Marriott DR, Cholewinski AJ. Astrocyte heterogeneity. Trends Neurosci. 1990;13:43–46. doi: 10.1016/0166-2236(90)90065-i. [DOI] [PubMed] [Google Scholar]
- Wolff RA, Dobrowsky RT, Bielawska A, Obeid LM, Hannun YA. Role of ceramide-activated protein phosphatase in ceramide-mediated signal transduction. J Biol Chem. 1994;269:19605–19609. [PubMed] [Google Scholar]
- Yaar M, Gilchrest BA. Human melanocyte growth and differentiation: A decade of new data. J Invest Dermatol. 1991;97:611–617. doi: 10.1111/1523-1747.ep12482985. [DOI] [PubMed] [Google Scholar]
- Yaar M, Eller MS, DiBenedetto P, Reenstra WR, Zhai S, McQuaid T, Archambault M, Gilchrest BA. The trk family of receptors mediates nerve growth factor and neurotrophin-3 effects in melanocytes. J Clin Invest. 1994;94:1550–1562. doi: 10.1172/JCI117496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagishita M, Hascall VC. Cell surface heparan sulfate proteoglycans. J Biol Chem. 1992;267:9451–9454. [PubMed] [Google Scholar]
- Yano S, Shinohara H, Herbst RS, Kuniyasu H, Bucana CD, Ellis LM, Davis DW, McConkey DJ, Fidler IJ. Expression of vascular endothelial growth factor is necessary but not sufficient for production and growth of brain metastasis. Cancer Res. 2000;60:4959–4967. [PubMed] [Google Scholar]
- Yoshida K, Gage FH. Cooperative regulation of nerve growth factor synthesis and secretion in fibroblasts and astrocytes by fibroblast growth factor and other cytokines. Brain Res. 1992;569:14–25. doi: 10.1016/0006-8993(92)90364-f. [DOI] [PubMed] [Google Scholar]