

Abstract
Craniospinal radiation therapy (CSRT) combined with chemotherapy results in significant endocrine morbidity. Between 1987 and 1990, a trial using 18 Gy was conducted to treat 10 young children with medulloblastoma. There were 7 survivors. We compared the endocrine outcome in these children (group 18 Gy) to that of a comparable group treated with conventional doses of CSRT that ranged from 23 to 39 Gy (group CD). Both groups had an identical history of chemotherapy and tumor stage and were treated with recombinant growth hormone therapy (rhGH). The mean age of group 18 Gy at diagnosis was 4.0 years, and rhGH treatment was initiated in 6 children at age 9.2 years. Group CD (12 children) was diagnosed at a mean age of 5.8 years and rhGH started in 11 children at a mean age of 9.6 years. The dose of rhGH used in both groups was identical (0.3 mg/kg/wk). For group 18 Gy, adult heights and sitting heights (a mean standard deviation score of −1.01 ± 1.11 and −1.62 ± 1.16, respectively) were statistically greater (P < 0.05) than those for group CD (mean standard deviation score of −2.04 ± 0.83 and −3.16 ± 1.43, respectively). Moreover, adult heights of group 18 Gy were not different from midparental heights, unlike group CD, whose adult heights were less than midparental heights (P < 0.0001). Of other endocrine sequelae, 10 patients of the CD group were hypothyroid, 3 had adrenal insufficiency, 3 had hypogonadism, and 2 had early puberty. In contrast, within group 18 Gy, only 1 was hypothyroid (P = 0.006) and 1 had early puberty. We conclude that endocrine morbidity was significantly reduced with 18 Gy CSRT in young children with medulloblastoma.
Craniospinal radiation therapy (CSRT)3 and chemotherapy for medulloblastoma or other malignancies can result in significant endocrine sequelae in long-term survivors, among which are growth failure, early or delayed puberty, thyroid dysfunction, and adreno-corticotropin insufficiency (Collet-Solberg et al., 1997; Costin, 1988; Dacou-Voutetakis et al., 1993; Marx et al., 1999; Moshang, 1997; Ricardi et al., 2001; Shalet et al., 1977). Poor growth is one of the most common endocrine complications in survivors, with an estimated incidence of 75% to 100% in children treated with CSRT and chemotherapy (Clarson and Del Maestro, 1999; Moshang and Grimberg, 1996; Packer et al., 2001; Rappaport and Brauner, 1989). Despite growth hormone therapy (GHRT), children with medulloblastoma have markedly reduced adult heights, with the impairment of growth significantly greater in children with earlier disease onset (Costin, 1988; Kanev et al., 1991; Leiper, 1990; Noorda et al., 2001; Shalet and Brennan, 1998).
Growth hormone deficiency (GHD) can be correlated to the total dosage of cranial irradiation used, although dosing fractions, timing or frequency of radiation doses, and age of recipient can all impact on the degree of GHD (Merchant et al., 2002; Ogilvy-Stuart and Shalet, 1995; Rappaport and Brauner, 1989; Schmiegelow et al., 2000). However, as suggested by a study of patients with leukemia treated with cranial irradiation, a total dose of cranial irradiation of more than 27 Gy will often cause impairment in both spontaneous growth hormone (GH) secretion and provocative GH response (Shalet, 1993). The addition of chemotherapy to radiation for treatment of medulloblastoma clearly impacts growth to a substantially greater extent than radiation alone, and adult heights, despite GHRT, are significantly smaller (Olshan et al., 1992; Ogilvy-Stuart and Shalet, 1995).
Prior to 1991, at the Children’s Hospital of Philadelphia, patients with medulloblastoma with M0 (no metastasis) disease were treated with 36 Gy of CSRT. From 1987 through 1990, a protocol using a decreased dose of CSRT (18 Gy), but identical local radiotherapy and chemotherapy, was used to treat very young children with medulloblastoma (Goldwein et al., 1993). This protocol was developed in an effort to improve survival in this group while minimizing the effects of radiation on brain development. We reviewed the endocrine outcome of the 7 surviving patients who are now near adult height and compared their height outcome and endocrine sequelae to similar children with medulloblastoma treated with conventional doses of CSRT and chemotherapy.
Patients and Methods
Patients
A total of 10 patients diagnosed with medulloblastoma, M0 disease, were treated with 18 Gy of CSRT and chemotherapy. The Institutional Review Boards of the Children’s Hospital of Philadelphia and the Hospital of the University of Pennsylvania approved this protocol. Three of the patients died from the malignancy. Seven patients (age range, 1.6–5.2 years at time of diagnosis) survived and constitute the study group (designated as “group 18 Gy”).
All cases of children diagnosed with medulloblastoma at Children’s Hospital of Philadelphia were reviewed to define a comparison group treated with conventional doses of CSRT. Those who met the following selection criteria composed the comparison group: (1) children who were diagnosed with medulloblastoma between 1983 and 1994 (excluding those diagnosed more than 4 years prior to or after the low-dose trial period in order to avoid significant cancer therapeutic protocol deviation); (2) patients who received both conventional doses of CSRT and chemotherapy; (3) age at irradiation less than 8 years; (4) M0 stage; and (5) adult height reached at the last clinical visit. There were a total of 31 children who received conventional doses of CSRT and chemotherapy during that period. Thirteen died, 5 had not yet reached adult height, and 1 patient was lost to follow-up. There were a total of 12 patients (age range, 2.17–7.58 years) who met the above selection criteria and constitute the comparison group (designated as the conventional dose group, or “group CD”).
Before 1991, children with nondisseminated medulloblastoma were treated with 36 Gy of CSRT. After 1991, children between the ages of 2 and 5 years were treated with 23 to 25 Gy of CSRT because of concerns related to neurotoxicity. Thus, the doses of CSRT ranged between 23 Gy and 39 Gy for the children within group CD. Other than the dose of CSRT, the radiation doses to the posterior fossa (53 Gy ± 2.89), hypothalamus (38 Gy ± 1.63), and pituitary (41 Gy ± 1.63) were similar for both groups. All of the patients received identical chemotherapy of 8 cycles of the 3-drug regimen of CCNU (lomustine), vincristine, and cisplatin, with 1 patient receiving an extra cycle.
The 2 groups were not statistically different in age at diagnosis and treatment of medulloblastoma, age at initiation of GHRT, years elapsed before GHRT initiation, duration of GHRT, and mean age at final adult height (Table 1).
Table 1.
Time frame of growth hormone deficiency diagnosis and growth hormone therapy for both groups
|
CD group
|
18 Gy group
|
|||||
|---|---|---|---|---|---|---|
| N | Mean (years) | SD | N | Mean (years) | SD | |
| Age at Diagnosis | 12 | 5.96 | 1.53 | 7 | 3.62 | 1.43 |
| Age at GHRT | 11 | 9.61 | 1.74 | 6 | 9.17 | 2.1 |
| Duration of GHRT | 9 | 6.89 | 2.16 | 5 | 5.81 | 1.47 |
| Age at Last Visit | 12 | 17.55 | 2.86 | 7 | 15.96 | 1.87 |
Abbreviations: CD, conventional dose; GHRT, growth hormone therapy.
Methods
The diagnosis of GHD was based upon (a) a decelerating growth velocity and failure to respond appropriately (peak GH concentration <8 μg/liter) to GH secretagogues (combined arginine and clonidine provocative test) or (b) low growth factors in association with low overnight GH neurosecretory profiles. All patients within group 18 Gy were diagnosed with GHD. However, only 6 of them received GHRT (5 males and 1 female). GHRT was discontinued after a year for 1 patient because of a stroke. One patient did not have GHRT following parental choice. The data from the 6 patients treated with recombinant growth hormone therapy (rhGH) were used to study height outcome. Of the 12 children in group CD, 11 were diagnosed with GHD and were treated with GH. One did not have GHD. The number of group CD CSRT survivors, the comparison group for height outcome, was therefore limited to the 11 patients with GHD who were treated with rhGH. The dose of rhGH for treatment in both groups was 0.3 mg/kg/week.
In both groups, anthropometric measurements were made in an endocrinology clinic by trained observers to determine standing height and sitting height measurements, using Harpenden stadiometers. The information for midparental heights was obtained by report. The standing height and sitting height data were transformed to a standard deviation score (SDS) by using adult height values for the U.S. population (means and SD) provided by the National Center for Health Statistics of the Centers for Disease Control (CDC/NCHS, 2002) and recent data for sitting height (Dangour et al., 2002). The use of standard deviation scores allows for analysis of height in order to correct for gender-related differences.
Standard clinical and biochemical measures diagnosed other endocrine disorders. Standard replacement doses for other hormone deficiencies were used for those patients with multiple hormone deficiencies.
The age at diagnosis of medulloblastoma, the age at GH treatment, the age at final adult height, the intervals between medulloblastoma diagnosis and GHRT, and the duration of GHRT were analyzed by one-way analysis of variance and post-tested with Newman-Keul’s multiple column pair comparisons. The height measurement at last clinic visit was used for all the subjects. A paired t-test was applied when final standing height was compared to the midparental height. Statistical comparisons (analysis of variance) were also made among the mean adult standing height, mean sitting height, and midparental height within each group and between the groups. The effect of age at onset of CSRT as to adult standing height outcome was determined by linear regression analysis. Because of the small sample size, a Fisher exact test was applied to the information pertaining to outcome of other endocrine sequelae. The significant level for all comparisons was 0.05.
Results
Height Outcome
For group 18 Gy, adult heights achieved (mean SDS −1.01 ± 1.11) were statistically taller (P = 0.0384) than heights achieved for group CD (mean SDS −2.04 ± 0.77). Moreover, adult heights for patients in group 18 Gy were not statistically different from midparental heights, unlike group CD, whose adult heights were less than midparental heights (P < 0.0001), as shown in Fig. 1. There was no difference between the 2 groups for the midparental height.
Fig. 1.
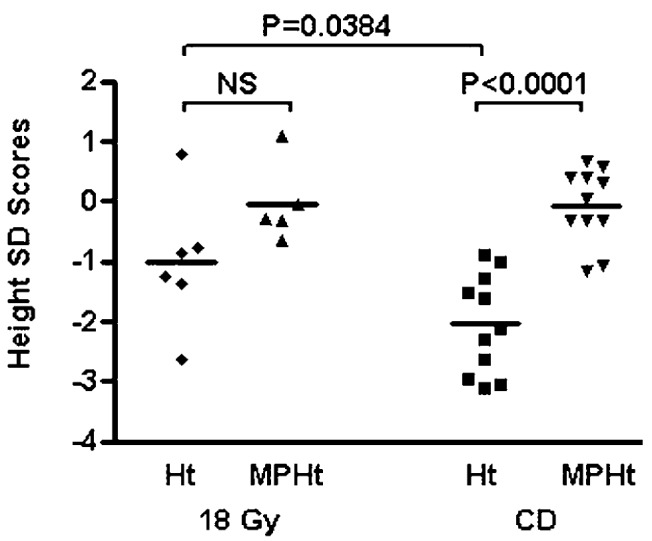
Comparison of adult heights and midparental heights (SD scores). Mean adult height for group CD was −2.04 (n = 11). The mean adult height for group 18 Gy was −1.01 (n = 6). The bars represent mean height SD score. Abbreviations: Ht, height; MPHt, midparental height; NS, not significant.
The sitting heights were available for 15 patients (10 of 11 in group CD and 5 of 6 in group 18 Gy), and the results are shown in Fig. 2. In comparison with the mean value for the general population, the sitting heights in both groups were impaired, with mean SDS of −1.62 ± 1.16 for group 18 Gy and −3.16 ± 1.43 for group CD. Consistent with standing height results, there was a significant difference in sitting heights for the 2 groups (P < 0.05).
Fig. 2.
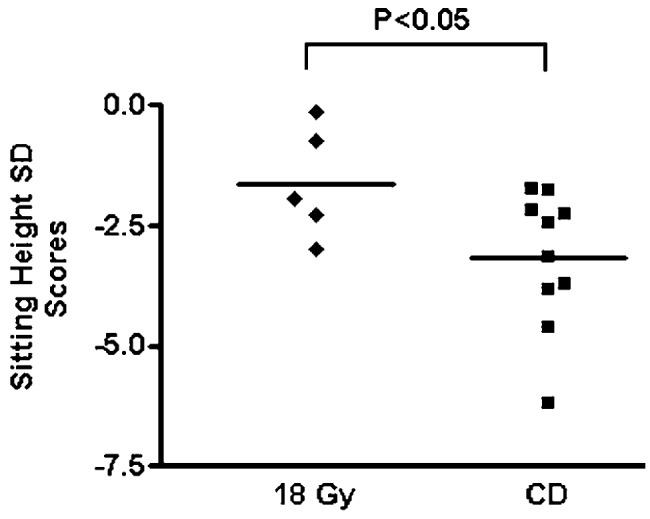
Sitting height. Mean sitting height SD scores for group 18 Gy (n = 5) and group CD (n = 10) are compared. The bars represent mean sitting height SD score.
Relationship Between Age at Onset of Therapy and Final Height
When the ages at onset of CSRT and chemotherapy and height outcome were analyzed, the variables observed were positively correlated in the CD group with correlation γ2 of 0.3957 (P = 0.0381). The younger the patients were diagnosed and treated with CSRT, the shorter the adult height achieved, as previously documented (Noorda et al., 2001). However, there was no such correlation for children treated with 18 Gy.
Other Endocrine Sequelae
Of other endocrine sequelae, there was a significantly higher incidence of hypothyroidism for group CD (P = 0.006). As well, there was an increase in adrenal insufficiency, requiring corticosteroid replacement therapy. Two males received testosterone replacement therapy, and one female received estrogen replacement therapy for hypogonadism. Two children also required gonadotropic release hormone agonist therapy to treat early puberty. In contrast, group 18 Gy had only 1 of 7 children with early puberty (Table 2).
Table 2.
Other endocrine sequelae in children with medulloblastoma treated with either conventional doses or 18 Gy of CSRT and chemotherapy
| CD n | 18 Gy n | P | |
|---|---|---|---|
| Hypothyroidism | 10 | 1 | 0.00627 |
| Hypogonadism | 3 | 0 | NS |
| Precocious puberty | 2 | 1 | NS |
| Adrenal insufficiency | 3 | 0 | NS |
Abbreviations: CD, conventional dose; NS, not significant.
Discussion
Medulloblastoma is the most common malignant brain tumor of childhood (Packer et al., 1999; Whelan et al., 1998). It can occur in any age group, but the peak incidence is between the ages of 3 and 7 years. The most common treatment is CSRT with a local boost to the primary tumor site, combined with chemotherapy. The conventional dose of CSRT in the time period studied, for patients without disseminated disease at diagnosis, was 36 Gy. Only since the mid-1990s, because of concerns over excessive neurocognitive toxicities, have lower doses (23.4 Gy) coupled with chemotherapy been used and has reasonable efficacy as regards disease control been shown (Packer et al., 1999).
It has been well documented that with this amount of radiation (24 – 36 Gy), most children suffer some degree of GH insufficiency, with incidence estimates as high as 75% to 100% (Noorda et al., 2001; Packer et al., 2001; Rappaport and Brauner, 1989; Shalet and Brennan, 1998). It is estimated that CSRT causes a mean height loss of 1.4 SD, and resultant adult height outcomes with SDS of −2.7 to −5 SD, despite GHRT (Sulmont et al., 1990; Ogilvy-Stuart and Shalet, 1995; Ranke and Price, 1999). The relative risk of attaining an adult height below the third percentile was 6 times increased after CSRT (Noorda et al., 2001). Several studies have shown that GHRT does improve linear growth, but adult height remains significantly less than expected, especially when compared with midparental height and when CSRT is combined with chemotherapy (Clayton et al., 1988; Darendeliler et al., 1990; Ogilvy-Stuart and Shalet, 1995; Sulmont et al., 1990).
The present study showed a severely diminished adult height outcome for group CD, with the adult heights significantly less than the midparental heights, similar to the above-cited studies. However, in the children treated with 18 Gy CSRT and chemotherapy, despite their younger ages, the resultant adult height was significantly greater, with a mean height SDS of −1.01 (range from 0.79 to −2.67). As well, their spinal growth was not as severely affected, as demonstrated by their significantly greater sitting heights. Additionally, their adult heights were not different from their midparental heights. These findings are especially notable given that the trial to use 18 Gy CSRT was specifically designed for young children less than 6 years of age. The correlation between the age at irradiation and significantly smaller adult height is well known (Helseth et al., 1999; Kiltie et al., 1997; Ogilvy-Stuart and Shalet, 1995; Probert et al., 1973; Schriock et al., 1991; Shalet et al., 1987, 1992). Unlike the previous reports that indicate worsening adult height outcomes with CSRT to younger children, there was, in fact, a better height outcome and no correlation with age of diagnosis and adult height outcome with 18 Gy CSRT.
Disproportionate growth is well known to occur after CSRT, with sitting height SDS of −3.0 to −3.4 (Adan et al., 2000; Burns et al., 1981; Kiltie et al., 1997; Ogilvy-Stuart and Shalet, 1995). The present study showed that the sitting heights in both groups were significantly smaller than the mean value for the general population, and there was no difference between the sitting heights achieved for group CD and the mean sitting height values of previous studies. However, there was a 1.5 SD difference between the 2 groups (−1.62 for group 18 Gy and −3.16 for CD), consistent with lesser damage to spinal growth with lower dosing of CSRT.
Although the number of patients studied was small, the 2 groups were well matched in most respects other than dose of CSRT. Nevertheless, other endocrine sequelae occurred less frequently for those patients who received 18 Gy CSRT. This was especially true for hypothyroidism, which occurred in 10 of 11 patients in the CD group and only 1 of 7 patients within group 18 Gy.
The clinical trial with 18 Gy CSRT was attempted briefly, but because of initial concern over loss of disease control, the study was halted in 1990. Follow-up of these patients showed continued survival, improved cognitive function (Goldwein et al., 1996), and now, in this report, improved endocrine outcome. Larger studies are required to know whether a CSRT dose of 18 Gy in young children will provide a survival rate comparable to that of the present treatment regimens (Packer et al., 2001). It is possible that such a dose will provide comparable survival rates with lower endocrine sequelae because, in nondisseminated medulloblastoma patients treated with reduced-dose CSRT (23.4 Gy) and adjuvant chemotherapy, there was a 5-year progression-free survival rate of 79% ± 7% (Whelan et al., 1998).
In conclusion, 18 Gy CSRT in very young children with medulloblastoma resulted in a significantly better height outcome than that observed in patients treated with higher doses of CSRT. Furthermore, there was less hypothyroidism and possibly less adrenal insufficiency and hypogonadism following low-dose CSRT. Given the results of more recent trials, the concept of what represents “conventional” CSRT is in flux, and in recent studies (Packer et al., 1999), 24 Gy of CSRT with unchanged primary site doses of radiotherapy (54–56 Gy) and adjuvant chemotherapy may have improved the quality of life in long-term survivors. Reduction of CSRT may have beneficial effects on cognition and endocrine function but also may have a potential risk of increasing disease relapse (Thomas et al., 2000). Studies prospectively comparing survival, endocrine status, and cognitive outcome in those receiving lower doses of CSRT and chemotherapy or other treatment approaches seem warranted.
Acknowledgment
We thank Sarah Homan for data preparation.
Footnotes
This paper was presented in part at the 6th Joint International Meeting of the Lawson Wilkins Pediatric Endocrine Society and the European Society of Paediatric Endocrinology, held in Montreal, Canada, July 5–10, 2001, and at the meeting of the Pediatric Academic Societies held in Baltimore, Md., May 4–7, 2002.
Dr. Xu was supported by a Pharmacia-funded fellowship training grant from the Lawson Wilkins Pediatric Endocrine Society and by training support from Eli Lilly, Inc.
Abbreviations used are as follows: CD, conventional dose; CSRT, craniospinal radiation therapy; GH, growth hormone; GHD, growth hormone deficiency; GHRT, growth hormone therapy; rhGH, recombinant growth hormone therapy; M0, no metastasis; SDS, standard deviation score.
References
- Adan L, Sainte-Rose C, Souberbielle JC, Zucker JM, Kalifa C, Brauner R. Adult height after growth hormone (GH) treatment for GH deficiency due to cranial irradiation. Med Pediatr Oncol. 2000;34:14–19. doi: 10.1002/(sici)1096-911x(200001)34:1<14::aid-mpo3>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- Burns EC, Tanner JM, Preece MA, Cameron N. Growth hormone treatment in children with craniopharyngioma: Final growth status. Clin Endocrinol. 1981;14:587–595. doi: 10.1111/j.1365-2265.1981.tb02969.x. [DOI] [PubMed] [Google Scholar]
- CDC/NCHS. Centers for Disease Control, National Center for Health Statistics (2002) 2000 CDC Growth Charts: United States. Available at http://www.cdc.gov/growthcharts
- Clarson CL, Del Maestro RF. Growth failure after treatment of pediatric brain tumors. Pediatrics. 1999;103:E37. doi: 10.1542/peds.103.3.e37. [DOI] [PubMed] [Google Scholar]
- Clayton PE, Shalet SM, Price DA, Addison GM. Growth and growth hormone responses to oxandrolone in boys with constitutional delay of growth and puberty (CDGP) Clin Endocrinol. 1988;29:123–130. doi: 10.1111/j.1365-2265.1988.tb00254.x. [DOI] [PubMed] [Google Scholar]
- Collet-Solberg PF, Sernyak H, Satin-Smith M, Katz LL, Sutton L, Molloy P, Moshang T., Jr Endocrine outcome in long-term survivors of low-grade hypothalamic/chiasmatic glioma. Clin Endocrinol. 1997;47:79–85. doi: 10.1046/j.1365-2265.1997.2211032.x. [DOI] [PubMed] [Google Scholar]
- Costin G. Effects of low-dose cranial radiation on growth hormone secretory dynamics and hypothalamic-pituitary function. Am J Dis Child. 1988;142:847–852. doi: 10.1001/archpedi.1988.02150080053022. [DOI] [PubMed] [Google Scholar]
- Dacou-Voutetakis C, Kitra V, Grafakos S, Polychronopoulou S, Drakopoulou M, Haidas S. Auxologic data and hormonal profile in long-term survivors of childhood acute lymphoid leukemia. Am J Pediatr Hematol Oncol. 1993;15:277–283. [PubMed] [Google Scholar]
- Dangour AD, Schilg S, Hulse JA, Cole TJ. Sitting height and subischial leg length centile curves for boys and girls from Southeast England. Ann Hum Biol. 2002;29:290–305. doi: 10.1080/03014460110085331. [DOI] [PubMed] [Google Scholar]
- Darendeliler F, Livesey EA, Hindmarsh PC, Brook CG. Growth and growth hormone secretion in children following treatment of brain tumours with radiotherapy. Acta Paediatr Scand. 1990;79:950–956. doi: 10.1111/j.1651-2227.1990.tb11357.x. [DOI] [PubMed] [Google Scholar]
- Goldwein JW, Radcliffe J, Packer RJ, Sutton LN, Lange B, Rorke LB, D’Angio GJ. Results of a pilot study of low-dose craniospinal radiation therapy plus chemotherapy for children younger than 5 years with primitive neuroectodermal tumors. Cancer. 1993;71:2647–2652. doi: 10.1002/1097-0142(19930415)71:8<2647::aid-cncr2820710833>3.0.co;2-s. [DOI] [PubMed] [Google Scholar]
- Goldwein JW, Radcliffe J, Johnson J, Moshang T, Packer RJ, Sutton LN, Rorke LB, D’Angio GJ. Updated results of a pilot study of low dose craniospinal irradiation plus chemotherapy for children under five with cerebellar primitive neuroectodermal tumors (medulloblastoma) Int J Radiat Oncol Biol Phys. 1996;34:899–904. doi: 10.1016/0360-3016(95)02080-2. [DOI] [PubMed] [Google Scholar]
- Helseth E, Due-Tonnessen B, Wesenberg F, Lote K, Lundar T. Posterior fossa medulloblastoma in children and young adults (0–19 years): Survival and performance. Childs Nerv Syst. 1999;15:451–455. doi: 10.1007/s003810050437. [DOI] [PubMed] [Google Scholar]
- Kanev PM, Lefebvre JF, Mauseth RS, Berger MS. Growth hormone deficiency following radiation therapy of primary brain tumors in children. J Neurosurg. 1991;74:743–748. doi: 10.3171/jns.1991.74.5.0743. [DOI] [PubMed] [Google Scholar]
- Kiltie AE, Lashford LS, Gattamaneni HR. Survival and late effects in medulloblastoma patients treated with craniospinal irradiation under three years old. Med Pediatr Oncol. 1997;28:348–354. doi: 10.1002/(sici)1096-911x(199705)28:5<348::aid-mpo4>3.0.co;2-h. [DOI] [PubMed] [Google Scholar]
- Leiper AD. Management of growth failure in the treatment of malignant disease. Pediatr Hematol Oncol. 1990;7:365–371. doi: 10.3109/08880019009033413. [DOI] [PubMed] [Google Scholar]
- Marx M, Schoof E, Grabenbauer GG, Beck JD, Doerr HG. Effects of puberty on bone age maturation in a girl after medulloblastoma therapy. J Pediatr Adolesc Gynecol. 1999;12:62–66. doi: 10.1016/s1083-3188(00)86628-0. [DOI] [PubMed] [Google Scholar]
- Merchant TE, Goloubeva O, Pritchard DL, Gaber MW, Xiong X, Danish RK, Lustig RH. Radiation dose volume effects on growth hormone secretion. Int J Radiat Oncol Biol Phys. 2002;52:1264–1270. doi: 10.1016/s0360-3016(01)02788-2. [DOI] [PubMed] [Google Scholar]
- Moshang T. Outcomes of pediatric brain tumors as related to growth and adolescent development. Curr Opin Endocrinol Diabetes. 1997;4:80–84. [Google Scholar]
- Moshang T, Jr, Grimberg A. The effects of irradiation and chemotherapy on growth. Endocrinol Metab Clin North Am. 1996;25:731–741. doi: 10.1016/s0889-8529(05)70350-9. [DOI] [PubMed] [Google Scholar]
- Noorda EM, Somers R, van Leeuwen FE, Vulsma T, Behrendt H. The Dutch Late Effects Study Group. Adult height and age at menarche in childhood cancer survivors. Eur J Cancer. 2001;37:605–612. doi: 10.1016/s0959-8049(00)00438-x. [DOI] [PubMed] [Google Scholar]
- Ogilvy-Stuart AL, Shalet SM. Growth and puberty after growth hormone treatment after irradiation for brain tumors. Arch Dis Child. 1995;73:141–146. doi: 10.1136/adc.73.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olshan JS, Gubernick J, Packer RJ, D’Angio DJ, Goldwein JW, Willi SM, Moshang T., Jr The effects of adjuvant chemotherapy on growth in children with medulloblastoma. Cancer. 1992;70:2013–2017. doi: 10.1002/1097-0142(19921001)70:7<2013::aid-cncr2820700734>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Packer RJ, Goldwein J, Nicholson HS, Vezina LG, Allen JC, Ris MD, Muraszko K, Rorke LB, Wara WM, Cohen BH, Boyett JM. Treatment of children with medulloblastomas with reduced-dose craniospinal radiation therapy and adjuvant chemotherapy: A Children’s Cancer Group study. J Clin Oncol. 1999;17:2127–2136. doi: 10.1200/JCO.1999.17.7.2127. [DOI] [PubMed] [Google Scholar]
- Packer RJ, Boyett JM, Janss AJ, Stavrou T, Kun L, Wisoff J, Russo C, Geyer R, Phillips P, Kieran M, Greenberg M, Goldman S, Hyder D, Heideman R, Jones-Wallace D, August GP, Smith SH, Moshang T., Jr Growth hormone replacement therapy in children with medulloblastoma: Use and effect on tumor control. J Clin Oncol. 2001;19:480–487. doi: 10.1200/JCO.2001.19.2.480. [DOI] [PubMed] [Google Scholar]
- Probert JC, Lederman M, Bagshaw MA. Medulloblastoma—treatment and prognosis. A study of seventeen cases in ten years. Calif Med. 1973;118:14–17. [PMC free article] [PubMed] [Google Scholar]
- Ranke, M.B., and Price, D.A. (1999) Growth hormone treatment in children with malignant cranial tumors in KIGS. In: Ranke, M.B., and Wilton, P. (Eds.) Growth Hormone Therapy in KIGS: 10 Years’ Experience. Heidelberg: Johann Ambrosius Barth Verlag, 159–174.
- Rappaport R, Brauner R. Growth and endocrine disorders secondary to cranial irradiation. Pediatr Res. 1989;25:561–567. doi: 10.1203/00006450-198906000-00001. [DOI] [PubMed] [Google Scholar]
- Ricardi U, Corrias A, Einaudi S, Genitori L, Sandri A, di Montezemolo LC, Besenzon L, Madon E, Urgesi A. Thyroid dysfunction as a late effect in childhood medulloblastoma: A comparison of hyperfractionated versus conventionally fractionated craniospinal radiotherapy. Int J Radiat Oncol Biol Phys. 2001;50:1287–1294. doi: 10.1016/s0360-3016(01)01519-x. [DOI] [PubMed] [Google Scholar]
- Schmiegelow M, Lassen S, Poulsen HS, Feldt-Rasmussen U, Schmiegelow K, Hertz H, Muller J. Cranial radiotherapy of childhood brain tumors: Growth hormone deficiency and its relation to the biological effective dose of irradiation in a large population based study. Clin Endocrinol. 2000;53:191–197. doi: 10.1046/j.1365-2265.2000.01079.x. [DOI] [PubMed] [Google Scholar]
- Shalet SM. Radiation and pituitary dysfunction. N Engl J Med. 1993;328:131–133. doi: 10.1056/NEJM199301143280211. (letter, comment) [DOI] [PubMed] [Google Scholar]
- Shalet SM, Brennan BM. Growth and growth hormone status following treatment for childhood leukaemia. Horm Res. 1998;50:1–10. doi: 10.1159/000023193. [DOI] [PubMed] [Google Scholar]
- Schriock EA, Schell MJ, Carter M, Hustu O, Ochs JJ. Abnormal growth patterns and adult short stature in 115 long-term survivors of childhood leukemia. J Clin Oncol. 1991;9:400–405. doi: 10.1200/JCO.1991.9.3.400. [DOI] [PubMed] [Google Scholar]
- Shalet SM, Beardwell CG, Twomey JA, Jones PH, Pearson D. Endocrine function following the treatment of acute leukemia in childhood. J Pediatr. 1977;90:920–923. doi: 10.1016/s0022-3476(77)80559-3. [DOI] [PubMed] [Google Scholar]
- Shalet SM, Gibson B, Swindell R, Pearson D. Effect of spinal irradiation on growth. Arch Dis Child. 1987;62:461–464. doi: 10.1136/adc.62.5.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalet SM, Crowne EC, Didi MA, Ogilvy-Stuart AL, Wallace WH. Irradiation-induced growth failure. Baillieres Clin Endocrinol Metab. 1992;6:513–526. doi: 10.1016/s0950-351x(05)80109-0. [DOI] [PubMed] [Google Scholar]
- Sulmont V, Brauner R, Fontoura M, Rappaport R. Response to growth hormone treatment and final height after cranial or craniospinal irradiation. Acta Paediatr Scand. 1990;79:542–549. doi: 10.1111/j.1651-2227.1990.tb11509.x. [DOI] [PubMed] [Google Scholar]
- Thomas PR, Deutsch M, Kepner JL, Boyett JM, Krischer J, Aronin P, Albright L, Allen JC, Packer RJ, Linggood R, Mulhern R, Stehbens JA, Langston J, Stanley P, Duffner P, Rorke L, Cherlow J, Friedman HS, Finlay JL, Vietti TJ, Kun LE. Low-stage medulloblastoma: Final analysis of trial comparing standard-dose with reduced-dose neuraxis irradiation. J Clin Oncol. 2000;18:3004–3011. doi: 10.1200/JCO.2000.18.16.3004. [DOI] [PubMed] [Google Scholar]
- Whelan HT, Krouwer HG, Schmidt MH, Reichert KW, Kovnar EH. Current therapy and new perspectives in the treatment of medulloblastoma. Pediatr Neurol. 1998;18:103–115. doi: 10.1016/s0887-8994(97)00221-x. [DOI] [PubMed] [Google Scholar]