

Abstract
Muscarinic cholinergic signaling plays an essential role in the control of the normal airway functions and in the development of pulmonary pathologies including asthma. In this paper we demonstrate that the airways of mice deficient in a cAMP-specific phosphodiesterase (PDE4D) are no longer responsive to cholinergic stimulation. Airway hyperreactivity that follows exposure to antigen was also abolished in PDE4D−/− mice, despite an apparently normal lung inflammatory infiltration. The loss of cholinergic responsiveness was specific to the airway, not observed in the heart, and was associated with a loss of signaling through muscarinic receptors with an inability to decrease cAMP accumulation. These findings demonstrate that the PDE4D gene plays an essential role in cAMP homeostasis and cholinergic stimulation of the airway, and in the development of hyperreactivity. In view of the therapeutic potentials of PDE4 inhibitors, our findings provide the rationale for novel strategies that target a single PDE isoenzyme.
Allergic asthma, affecting as many as 10% of individuals in industrialized nations, is characterized by chronic airway inflammation, airway hyperreactivity (AHR), and mucosal edema, resulting in bronchoconstriction and episodic airway obstruction (1). Airway remodeling and altered noncholinergic, nonadrenergic neurotransmission may contribute to irreversible airway obstruction and reduction of pulmonary function. Current treatments for asthma are not satisfactory, and few strategies are available for disease prevention. Given the high prevalence of this disease, improved and more effective therapeutic interventions are highly desirable.
Many of the events and mechanisms involved in asthma pathogenesis are inhibited by the activation of the cyclic nucleotide-signaling pathway (2). Thus, an increase in intracellular cAMP interferes with lymphocyte, eosinophil, and mast cell activation, and blocks cytokine production, cell replication, and cell chemotaxis to sites of inflammation. In addition, activation of the cAMP signaling pathway in airway smooth muscle cells promotes relaxation and blocks smooth muscle cell replication (3), thus preventing the airway remodeling observed in chronic asthmatic patients. Genetic studies show that defective β2-adrenergic receptor signaling, which results in reduced cAMP levels, is associated with increased AHR and nocturnal asthma (4–6). Similarly, aberrant enhanced expression of phosphodiesterases (PDEs), which degrade and inactivate cAMP (7), may also be associated with atopy (8), although this concept has been recently challenged (9).
In line with the inhibitory function of cAMP, inhibitors of PDEs block many of the symptoms of asthma by increasing cAMP levels in the airway and in inflammatory cells (2, 10). Thus, manipulation of the PDE activity in the airway is viewed as a strategy of considerable therapeutic potential for the treatment of asthma. Unfortunately, it has been extremely difficult to assign a particular function to any individual PDE in view of the complexity of the PDE system. Phosphodiesterases segregate into at least 10 genetically distinct families of isoenzymes (11), with PDE4 family members characterized by high affinity for cAMP and selective sensitivity to the antidepressant rolipram. Four PDE4 genes (PDE4A, -B, -C, -D) are present in mammals, and each gene encodes several different proteins (12, 13), many of them being expressed in inflammatory cells and in the airway (14, 15).
To determine the specific role of PDEs in the pathophysiology of asthma, we generated mice with targeted disruption of one of the PDE4 genes (PDE4D) (16) and examined the induction of airway inflammation and AHR. We found that the PDE4D−/− mice, when immunized and challenged in the lung with antigen, were markedly resistant to the development of methacholine-induced AHR because of reduced muscarinic receptor function. Surprisingly, however, the PDE4D−/− mice developed significant Th2-driven inflammatory responses in the lung to a similar degree as did control mice. These studies demonstrate a crucial role of PDE4D in the control of smooth muscle contraction and muscarinic cholinergic receptor signaling, but not in the control of airway inflammation. More importantly, they provide a rationale for developing new therapies for asthma that target a single PDE4 isoenzyme.
Materials and Methods
Mice.
Generation of PDE4D homozygous null mice has been described (16). Mice used in all experiments were 6–18 weeks of age and had the same mixed genetic background (C57BL/6 × 129/Ola).
PDE Assay.
Lung tissue dissected from animals was homogenized as described (17). Aliquots of the homogenates were assayed for total PDE activity as well as rolipram-insensitive PDE activity in the absence or presence of 10 μM rolipram. The PDE assay was performed according to the method of Thompson and Appleman (18) as detailed previously (19). The rolipram-sensitive activity (PDE4 activity) was obtained by subtracting the rolipram-insensitive activity from the total activity. Protein concentration was determined by using the Bradford method (20).
Membrane Preparation.
Lung tissue from adult mice was dissected and quickly frozen. To prepare membranes, the frozen tissue was pulverized, followed by homogenization with a Dounce homogenizer (30 strokes) in a buffer containing 20 mM Tris-Cl (pH 7.8), 50 mM sodium fluoride, 0.2 mM EGTA, 1 mM EDTA, 5 mM 2-mercaptoethanol, 50 mM benzamidine, 0.5 μg/ml leupeptin, 0.7 μg/ml pepstatin, 4 μg/ml aprotinin, 10 μg/ml soybean trypsin inhibitor, 2 mM PMSF, 10 mM pyrophosphate. The cell homogenates were spun at 200 × g for 10 min, and the resulting supernatant was further centrifuged at 30,000 × g for 10 min. The pellet was washed once with the homogenization buffer, followed by centrifugation at 30,000 × g for 10 min. The final membrane pellet was resuspended in 10 mM Tris-Cl (pH 7.5) at a concentration of 1–2 μg/μl of protein.
Immunization Protocol.
Ovalbumin (OVA; 50 μg) complexed with aluminum potassium sulfate (alum) was administered i.p. on days 1 and 14, and intranasally (i.n.) (50 μg OVA in 50 μl of PBS) on days 14, 25, 26, and 27. Control mice received i.p. injections of alum alone and intranasal PBS. AHR to inhaled methacholine and serotonin was measured 24 h after the last i.n. dose of OVA (day 28). Bronchoalveolar lavage (BAL) and lung fixation were performed the following day (day 29).
Measurement of Airway Responsiveness.
Airway responsiveness was assessed by methacholine- or serotonin-induced airflow obstruction from conscious mice placed in a whole-body plethysmograph (model PLY 3211, Buxco Electronics, Troy, NY). Pulmonary airflow obstruction was measured by Penh by using the following formula: Penh = [(Te/RT) − 1] × (PEF/PIF), where Penh = enhanced pause (dimensionless), Te = expiratory time, RT = relaxation time, PEF = peak expiratory flow (ml/s), and PIF = peak inspiratory flow (ml/s) (21). Enhanced pause (Penh), minute volume, tidal volume, and breathing frequency were obtained from chamber pressure, measured with a transducer (model TRD5100) connected to preamplifier modules (model MAX2270) and analyzed by system XA software (model SFT 1810). Measurements of methacholine or serotonin responsiveness were obtained by exposing mice for 2 min to NaCl 0.9% (Portable Ultrasonic, 5500D, DeVilbiss Health Care, Sommerset, PA), followed by incremental doses of aerosolized methacholine (2.5–40 mg/ml) or one dose of serotonin (10 mg/ml) and monitoring Penh. Results were expressed for each methacholine or serotonin concentration as the percentage of baseline Penh values after 0.9% NaCl exposure.
Collection of BAL Fluid and Lung Histology.
Animals were injected i.p. with a lethal dose of phenobarbital (450 mg/kg). The trachea was cannulated, and the lung was then lavaged with 0.8 ml of PBS three times and the fluid pooled. Cells in the lavage fluid were counted by using a hemocytometer, and BAL cell differentials were determined on slide preparations stained with Hansel Stain (Lide Laboratories, Florissant, MO). At least 200 cells were differentiated by light microscopy based on conventional morphologic criteria. In some animals, no BAL was performed but lungs were removed, washed with PBS, and fixed in 10% formalin. Sections were routinely processed and embedded in paraffin wax, cut at 4 μm and stained with hematoxylin and eosin.
Restimulation of Spleen Cells in Vitro.
Spleens were removed and depleted of resting B cells by adherence to goat anti-mouse Ig-coated plates. Spleen cells (4 × 105) were restimulated in vitro with OVA in DMEM (Life Technologies), supplemented as described (22), and contained 50 μM 2-mercaptoethanol and 10% FCS (HyClone). Cells were cultured in 96-well microtiter plates in 200 μl medium. Supernatants were harvested after 4 days for determination of IL-4 levels.
IL-4 ELISA.
mAbs specific for IL-4 were purified from ascites of hybridomas BVD4-1D11, BVD6-24G2, generously provided by M. Howard (DNAX Research Institute, Palo Alto, CA). The amount of IL-4 in culture supernatants was measured by ELISAs as described (23). The Ab pairs used were as follows, listed by capture/biotinylated detection: IL-4, 11B11/BVD6-24G2. The standards were recombinant IL-4 curves generated in 1:2 dilutions from 500 to 31.2 pg/ml.
Measurement of Anti-OVA IgE Abs.
Mice were bled at the time of sacrifice and the OVA-specific Ab was measured by using a modified antigen-specific ELISA. Determination of OVA-specific IgE was performed by ELISA by using rat anti-mouse IgE mAb EM95 (5.0 μg/ml) to coat plates. After the samples were applied and incubated overnight, plates were washed and biotinylated OVA (10 ng/ml) was added. Two hours later, plates were washed and horseradish peroxidase-conjugated streptavidin (Southern Biotechnology Associates) was added. Plates were developed with O-phenylene diamine substrate, and the OD was determined at 492 nm.
Muscarinic Receptor Assay.
The muscarinic receptor assay was carried out in 10 mM Tris-Cl (pH 7.5) by adding the membrane suspension (100–200 μg protein) to polypropylene tubes containing [3H]quinuclidinyl benzilate (QNB) (NEN-DuPont) at concentrations ranging from 6.25 to 400 pM. The final assay volume was 5 ml. Nonspecific binding was determined by including 1 μM atropine in the assay. After a 60-min incubation at 37°C the reaction was terminated by the addition of 5 ml of wash buffer (37°C) consisting of 10 mM Tris-Cl (pH 7.5) and 145 mM NaCl, followed by immediate filtration through Whatman GF/B glass-microfiber filters. Each filter was washed twice with 5 ml of wash buffer (37°C), then placed in 3-ml scintillation mixture (Econo-Safe, Research Products International), and counted on a beta counter (Beckman LS5000TD model).
In Vitro cAMP Responses to Isoproterenol and Carbachol.
Lung tissue dissected from adult mice was sliced into pieces with a dimension of ≈3 × 2 × 1 mm. The tissue slices were incubated in 1 ml of Hepes-buffered MEM at 37°C for 15 min in the absence or presence of test drugs, including 20 μM isoproterenol alone or a combination of 20 μM isoproterenol and 10 μM carbachol. The incubation was terminated by the addition of trichloroacetic acid to a final concentration of 5%. The tissue was homogenized with a Dounce homogenizer (30 strokes) before centrifugation at full speed in a microcentrifuge at 4°C for 30 min. The supernatants were extracted five times with water-saturated ether and heated to 60°C for 5 min to remove residual ether. cAMP content was then measured by RIA as described by Harper and Brooker (24).
Results
Absence of PDE4D Activity in the Lung of PDE4D−/− Mice.
To investigate the expression of PDE4 in the lung of the PDE4D−/− mice, phosphodiesterase activity was measured in the absence or presence of PDE4 inhibitor rolipram. A 50% reduction in total PDE activity was observed in the lung from PDE4D−/− mice when compared with wild-type mice (Fig. 1). This reduction was caused by a loss of PDE4, because the decrease in PDE activity was associated with loss of sensitivity to rolipram. The absence of PDE4D expression in the lung of the PDE4D−/− mice was confirmed by immunoprecipitation with a PDE4D-selective Ab (M3S1) (25). This Ab immunoprecipitated 49.86 pmol/min of activity from the PDE4D+/+ mice, whereas negligible activity (<0.1 pmol/min) was recovered from the PDE4D−/− mice. Thus, inactivation of the PDE4D gene causes a significant reduction in the PDE activity expressed in the lung with no significant compensation by other PDEs. These findings also indicated that PDE4D is the predominant PDE4 isoform expressed in the lung.
Figure 1.

Absence of PDE4D activity in the lung of PDE4D−/− mice. Lung tissue was dissected from PDE4D+/+, PDE4D+/−, and PDE4D−/− mice and homogenized as described (16). Aliquots of the homogenates were assayed for PDE activity in the absence (total) or presence (RI) of 10 μM rolipram. Data are the mean ± SEM (n = 3–8 mice/genotype, P < 0.05 determined by Student's t test). The number of mice in each group is reported above the bars.
Absence of Allergen-Induced AHR in PDE4D−/− Mice.
To determine whether the absence of PDE4D activity protected against the development of allergen-induced AHR, PDE4D−/− mice were sensitized i.p. and challenged i.n. with OVA. AHR was then measured in a whole-body plethysmography after inhalation of increasing amounts of methacholine. Fig. 2A demonstrates that although the OVA-immunized control PDE4D+/+ mice exhibited strong AHR, the PDE4D−/− mice showed no signs of AHR. More importantly, challenge with a very high concentration of methacholine (120 mg/ml), which induced strong bronchospasm in naive, nonimmunized PDE4D+/+ mice, did not induce any bronchoconstriction in the PDE4D−/− mice (Fig. 2B). Absence of a methacholine response was observed in all 28 PDE4D−/− mice tested, whereas the heterozygous mice displayed a wild-type phenotype (12 PDE4D+/− mice and 18 PDE4D+/+ mice; data not shown). Both PDE4D+/+ and PDE4D−/− mice developed comparable bronchoconstriction after inhalation of 5-hydroxytryptamine (serotonin), indicating that smooth muscle function in the lungs of PDE4D−/− mice was not impaired (Fig. 3). In addition, pretreatment of wild-type mice with rolipram (1 μg/kg) decreased the methacholine-induced airway resistance (data not shown), suggesting that pharmacological inhibition of PDE4 produces an effect similar to that observed with the inactivation of the PDE4D gene.
Figure 2.

AHR in PDE4D+/+ and PDE4D−/− mice. (A) Concentration dependence of the methacholine response. PDE4D+/+ and PDE4D−/− mice were immunized with OVA (50 μg) in alum i.p. on days 0 and 14, and i.n. (50 μg OVA in 50 μl PBS) on days 14, 25, 26, and 27. Control mice received alum i.p. and normal saline i.n.. One day after the last i.n. challenge with OVA, AHR in response to increasing concentrations of methacholine was measured from conscious mice placed in a whole-body plethysmograph as detailed in Materials and Methods. Data are expressed as percent above baseline (mean ± SEM; n = 6 for each data point). (B) Airway response to high concentrations of methacholine in nonimmunized mice. AHR was measured in PDE4D+/+ and PDE4D−/− mice after challenge with a single dose of methacholine (120 mg/ml) (n = 5).
Figure 3.

Airway response to serotonin in PDE4D+/+ and PDE4D−/− mice. Mice were immunized with OVA following the protocol described in Fig. 2. One day after the last i.n. challenge with OVA, AHR in response to increasing concentrations of serotonin was measured in conscious mice. Data are expressed as percentage above baseline (mean ± SEM; n = 8).
Pulmonary Allergic Inflammation in PDE4−/− Mice Is Normal.
The lack of allergen-induced airway reactivity elicited by methacholine in PDE4D−/− mice was not caused by a failure to immunologically sensitize the mice. Fig. 4 shows that following sensitization and challenge with OVA, significant airway inflammation occurred in both PDE4D−/− and PDE4D+/+ mice. The lungs of both mice contained dense peribronchiolar and perivascular infiltrates consisting of lymphocytes, eosinophils, and some neutrophils (Fig. 4) and comparable mucus accumulation in the airway. These histological findings were confirmed by analysis of the BAL fluid of the animals, which showed comparable numbers of cells and a comparable distribution of the different cell types (Fig. 5). In particular, there was no significant difference between the percentages of eosinophils detected in the BAL fluid of OVA-immunized PDE4D+/+ and PDE4D−/− mice. Both PDE4D−/− and PDE4D+/+ mice possessed comparable levels of serum OVA-specific IgE. In addition, splenic T cells from both PDE4D−/− and PDE4D+/+ mice proliferated to the same extent and produced comparable amounts of IL-4 in response to OVA challenge (Table 1). These results demonstrated that inactivation of the PDE4D gene had no impact on the immunological response to OVA in these animals, and that the loss of AHR observed in the PDE4D−/− mice was not caused by a decrease in pulmonary allergic inflammation.
Figure 4.

Histopathology of lung from PDE4D+/+ and PDE4D−/− mice sensitized to OVA. (A) Histology of lung derived from immunized wild-type mice. Sections show dense perivascular and peribronchiolar mixed inflammatory cell infiltrates composed predominantly of lymphocytes and eosinophils. Intraluminal mucous accumulation is noted [hematoxylin and eosin (H&E), ×200]. (B) PDE4D−/− mice. The lungs of PDE4D−/− mice show similar histopathologic changes (H&E, ×200).
Figure 5.

Immune response to OVA immunization in PDE4D+/+ and PDE4D−/− mice. Cellular composition of the BAL fluid from PDE4D+/+ and PDE4D−/− mice. Mice were immunized i.p. and i.n. with OVA as described in Fig. 2. Two days after the last immunization with OVA, the mice were bled and killed and the BAL was performed as detailed in Materials and Methods. BAL was performed with three aliquots of 0.8 ml serum-free medium. The total number of different types of leukocytes was determined from Hansel-Stain slide preparations of BAL fluid. The data are expressed as cells recovered ± SD/mouse of each cell type, based on differentials of 200 cells (n = 12 mice in each group). Mo, macrophages; Lym, lymphocytes; Eos, eosinophils; Neu, neutrophils.
Table 1.
Splenocyte proliferation, and IL-4 and IgE production in PDE4D+/+ and PDE4D−/− mice immunized with OVA
| Parameter | PDE4D+/+ mice
|
PDE4D−/− mice
|
||
|---|---|---|---|---|
| Control | OVA | Control | OVA | |
| Proliferation* | 2,017 ± 479 | 35,427 ± 3,120 | 1,512 ± 354 | 36,577 ± 2,994 |
| IL-4 (pg/ml)* | <31 | 393 ± 18 | <31 | 430 ± 22 |
| IgE (μg/ml)† | <0.001 | 414 ± 120 | <0.001 | 401 ± 84 |
Spleens were removed from OVA-primed mice, and spleen cells were cultured at 5 × 105 cells/well with or without 100 μg/ml OVA. Proliferation of OVA-specific cells was assessed by incorporation of [3H]thymidine in triplicate cultures over the last 18 h of a 4-day culture. IL-4 content in supernatants was determined after 4 days of culture by ELISA. Data are the mean of triplicate determinations.
Serum IgE Ab levels were determined by ELISA. Data are the mean of triplicate determinations on ≥8 mice ± SD.
Reduced Muscarinic Receptor Responses in PDE4D−/− Mice.
Pulmonary tissue from both PDE4D+/+ and PDE4D−/− mice bound similar amounts of [3H]QNB indicating comparable expression of muscarinic receptor-binding activity (Bmax, fmol/mg protein +/+, 31.82 ± 0.6; +/−, 32.53 ± 2.1.2; −/−, 31.95 ± 4.20) (Fig. 6). In PDE4D+/+, PDE4D+/−, and PDE4D−/− mice, binding was saturable with a calculated Kd of 20–30 pM QNB. Despite a normal expression of muscarinic receptors, signaling through these receptors was significantly reduced in the PDE4D−/− mice. In lung preparations from PDE4D−/− mice, increased cAMP levels induced with isoproterenol were not reduced by incubation with the cholinergic agonist carbachol, whereas this agonist significantly inhibited the isoproterenol-induced cAMP accumulation in PDE4D+/+ or PDE4D+/− mice (Fig. 7). Moreover, isoproterenol responses were usually higher in the −/− mice when compared with the control (data not shown). Because the PDE4D−/− mice responded to i.v. administration of methacholine with bradycardia to the same degree as the wild-type littermates (data not shown), muscarinic cholinergic signaling does not appear to be affected in the heart. Moreover, the rolipram-sensitive activity in the heart was only slightly reduced in the PDE4D−/− mice, indicating that PDE4D activity is expressed at a low level in the heart or that other PDEs have compensated for the loss of PDE4D (data not shown).
Figure 6.

Muscarinic cholinergic receptor expression in lungs from wild-type and PDE4D−/− mice. Lungs were excised from PDE4D+/+ and PDE4D−/− mice and homogenized in homogenization buffer. The particulate fraction was prepared by centrifugation as detailed in Materials and Methods. Aliquots of the resuspended pellet were incubated for 60 min with increasing concentrations of [3H]QNB. Nonspecific binding was determined by incubation in the presence of 1 μM atropine. After 60 min at 37°C, free and bound antagonist was separated by rapid filtration on glass-microfiber filters. Each point is the mean of triplicate observations. A representative experiment of the two performed is reported.
Figure 7.
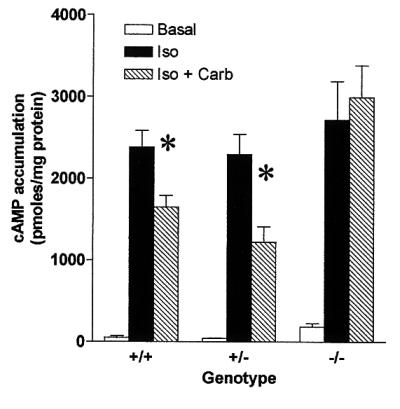
cAMP accumulation in lung tissue of PDE4D+/+ and PDE4D−/− mice. Slices of lung tissue from PDE4D+/+, PDE4D+/−, and PDE4D−/− mice were preincubated for 15 min in MEM tissue culture medium containing 10 mM Hepes (pH 7.4). After preincubation, 20 μM isoproterenol in the absence or presence of 10 μM carbachol was added to the tubes. Control samples received vehicle only. Incubation was terminated after 15 min by the addition of trichloroacetic acid to a final concentration of 5% (vol/vol). The tissue slices were homogenized and extracts were clarified by centrifugation. The concentration of cAMP in the acid extracts was measured by RIA after acetylation of the samples. Concentrations were corrected for the amount of protein in the extracts. Each point is the mean ± SE of five mice each assayed in triplicate. ∗, P < 0.05
Discussion
By using a mouse model with a targeted deletion of the PDE4D gene, we demonstrated that inactivation of a single PDE isoform profoundly affects the cholinergic regulation of the airway. The loss of PDE4D, one of the four PDE4 genes present in mammals, resulted in a greatly decreased airway response to methacholine in both naive mice and mice sensitized and challenged in the airway with antigen. The loss of PDE4D, although greatly reducing AHR, had no effect on the allergen-induced inflammatory response in the airway. Moreover, cholinergic regulation of intracellular cAMP was impaired in the lung of the PDE4D−/− mice. Because AHR, defined as the increased response to acetylcholine agonists, correlates strongly with clinical expression of asthma (26), the lack of responses to methacholine in PDE4D−/− mice suggests that inactivation of PDE4D in human would specifically diminish airway reactivity. These findings demonstrate an essential role of PDE4D in the development of AHR through the control of muscarinic cholinergic responses, and provide an experimental rationale for generating PDE4 isoform-selective inhibitors for the treatment of asthma or other chronic pulmonary inflammatory diseases.
Because four PDE4 genes are present in mammals, the lung phenotype of the PDE4D-deficient mice demonstrates that this gene plays a nonredundant role in cAMP homeostasis. Ablation of a single PDE4 gene causes a significant reduction in PDE activity and an increase in resting and stimulated cAMP levels in the lung, indicating that other PDE4s, or isoenzymes belonging to other PDE families, are not up-regulated and cannot compensate for the loss of PDE4D. Thus, this finding supports the concept that the PDE4D gene serves unique, nonoverlapping functions in cell signaling (16). However, further studies are necessary to determine whether the PDE4 genes themselves are differentially regulated, with distinct promoters controlling cell-specific expression, and/or whether the PDE4 proteins have nonoverlapping functions and are therefore nonequivalent.
Our data demonstrate that inactivation of PDE4D either by pharmacological manipulation or by inactivation of the corresponding gene causes a decrease in airway resistance induced by methacholine. In a similar fashion, genetic inactivation of PDE4D obliterate the muscarinic receptor inhibition of cAMP accumulation. Taken together, these findings strongly argue for a crucial role of PDE4D in muscarinic cholinergic signaling in the lung.
Five genetically and pharmacologically distinct muscarinic receptor subtypes have thus far been identified (27, 28). M1, M3, and M5 receptors are coupled to phosphoinositol turnover through Gq/11, whereas M2 and M4 receptors are coupled through Gi/o to adenylyl cyclase inhibition or to K channel activation (28). M1, M2, and M3 receptors are the subtypes expressed in the lung and are differentially distributed in the airways, with M1 receptors in the alveolar walls, parasympathetic ganglia, and submucosal glands, and M2 and M3 in the smooth muscle in the small airways (29, 30). Although most of the muscarinic receptors are expressed postjunctionally in the lung, M2 are also presynaptic, functioning as autoreceptors involved in the control of acetylcholine release from postganglionic cholinergic nerves (31).
Our studies demonstrate that an absence of cAMP inhibition is not associated with a detectable loss of muscarinic receptor expression as measured by [3H]QNB binding. Although loss of a minor pool of receptors cannot be discounted, it is likely that the impaired muscarinic cholinergic response is due to a postreceptor event. The common tenet is that acetylcholine inhibits cyclase activity and cAMP accumulation via interaction with M2 or M4 receptors coupled to activation of Gi. The reason why ablation of PDE4 activity in the mouse lung interferes with this response is at present unclear. In contrast to our findings, several studies have shown that a decrease in cAMP accumulation by M2 receptors is observed in the presence of PDE inhibitors (32, 33).
It is generally accepted that acetylcholine stimulates smooth muscle contraction primarily via the M3 receptor. Conversely, the M2 receptor role in airway contractility is still a matter of debate despite the fact that it constitutes more than 60% of the total muscarinic receptors present in the airway (34). In mice deficient in a M2 receptor, tracheal smooth muscle still contracts in response to muscarinic cholinergic agonists, albeit with a decrease in sensitivity (35). Thus, the prevailing view is that while contraction is primarily due to M3 signaling, M2 receptors play an ancillary role by preventing relaxation induced by β-receptor stimulation. In view of the above findings, it is unlikely that the reduced M2 muscarinic cholinergic inhibition of cAMP accumulation in the PDE4D−/− mice is, per se, the cause of the impaired airway contractility. One can propose two hypotheses to reconcile the inactivation of PDE4D with the loss of acetylcholine-induced contraction. It is possible that the absence of PDE4D leads to an increase in sensitivity of the lung to β-adreneregic stimulation, which in turn causes an increase in cAMP and relaxation. Under these conditions of elevated cAMP, muscarinic signaling cannot overcome the β-adrenergic-induced relaxation. Alternatively, PDE4D may be directly involved in M3 signaling. Work done in 132N11 cell lines and dog thyroid slices shows that muscarinic cholineric inhibition of cAMP accumulation depends on M1/M3 receptors and involves the activation of a calmodulin-regulated PDE (PDE1) (36, 37). However, this PDE has properties different from those of PDE4D. In addition, it has been proposed that α1-adrenergic receptors, which use a signaling pathway similar to that of M3 receptors, may also signal in the heart via activation of a PDE (38).
Our studies with bronchoconstrictive agents other than acetylcholine agonists indicate that the serotonin response in the PDE4D−/− mice is not affected. This finding excludes the possibility that nonspecific cellular or anatomical changes in the airway are the cause of failure to constrict in response to muscarinic agonists. Because of this divergent sensitivity to PDE4 inactivation, it can be concluded that the serotonin and muscarinic pathways controlling contractility do not overlap. Such a dissociation has been observed in several other instances. In the endothelin-1 heterozygous knockout mice, an increased response to methacholine is not accompanied by an increased response to serotonin in the airways (39). Moreover, airway response to serotonin and to acetylcholine is inherited independently in the mouse (40), again suggesting that two distinct pathways mediate the effect of the two neurotransmitters.
In addition to providing insight on the physiological role of PDE4s, the genetic manipulation of the PDE genes consolidates the pharmacological tenet that PDE4 inhibition improves the asthma symptoms. After a methacholine challenge in naive and allergen-sensitized PDE4D−/− mice, bronchoconstriction was suppressed. More importantly, the AHR could be dissociated from the allergen-induced inflammatory response. All measured parameters of inflammation studied indicated that the antigen sensitization and inflammatory cell response were normal in PDE4D−/− mice. In view of the well-established effect of PDE4 inhibition on inflammatory responses (10), we conclude that inhibition of PDE4s, other than PDE4D, must play a major role in the suppression of inflammation. Thus, the impact of different PDE4s on the different asthma manifestations could be differentiated. These differences could not have been detected with available PDE4 inhibitors because they are largely nonselective. Taking into consideration possible species differences, we predict that inhibition of PDE4B or PDE4A is the most efficacious strategy to block the inflammatory responses in asthma, whereas PDE4D inhibitors may predominantly affect the bronchoconstrictive component. PDE4C has not been detected in inflammatory cells (41). A third generation of inhibitors selective for each PDE4 may therefore have increased selectivity while providing additional advantages. PDE4D is expressed at a high level in the area postrema of the hindbrain (42), and it has been suggested that it may be the enzyme responsible for the emetic effects often experienced with PDE4 inhibition. Thus, targeting PDE4s other than PDE4D may still demonstrate efficacy toward inflammation without producing significant emetic side effects.
Finally, the finding that inactivation of the PDE4D gene has a dramatic effect on AHR opens the possibility that different alleles of the PDE4 genes may contribute to the hereditary basis of asthma, or more in general, to atopic diseases. Indeed, Hanifin et al. (43) have proposed that expression of an activated PDE and the consequent decrease in cAMP in inflammatory cells may result in hyperreactivity and that this condition may be inherited. This is certainly possible in the PDE4 proteins, for it has been demonstrated that PDE4s exist in several conformations that affect the activity of the catalytic domain (44), and point mutations in a phosphorylation site of PDE4D yield a constitutively active enzyme (13). One can therefore speculate that, if alleles of a PDE4D gene encoding an activated enzyme exist in the population, they would cause a decrease in cAMP in the airway and an increased sensitivity to muscarinic cholinergic agonists, thereby causing a state of hyperreactivity. It is intriguing that the PDE4D gene maps on the long arm of chromosome 5, a region that is thought to regulate the development of AHR. In the same vein, alleles with inactivating mutations in PDE4s may decrease the responsiveness of inflammatory cells to cytokines and inhibit AHR, thus creating a genetic background unfavorable to asthma.
Acknowledgments
We acknowledge the technical assistance of Gu Zhang and Linda Lan. We are also indebted to Drs. D. Bernstein and G. Desai for the cardiac measurements, and to Dr. G. Berry for the histological analyses. This work was supported by National Institutes of Health Grants RO1 HD20788 (to M.C.) and RO1 AI26322 (to D.U).
Abbreviations
- PDE
cyclic nucleotide phosphodiesterase
- PDE4
type 4 cAMP-specific PDE
- AHR
airway hyperreactivity
- OVA
ovalbumin
- BAL
bronchoalveolar lavage
- i.n.
intranasally
- QNB
quinuclidinyl benzilate
Footnotes
This paper was submitted directly (Track II) to the PNAS office.
See commentary on page 6245.
References
- 1.Shirakawa T, Enomoto T, Shimazu S, Hopkin J M. Science. 1997;275:77–79. doi: 10.1126/science.275.5296.77. [DOI] [PubMed] [Google Scholar]
- 2.Tenor H, Schudt C. In: Anti-Inflammatory Drugs in Asthma. Sampson A P, Church M K, editors. Basel: Birkhauser; 1999. pp. 87–135. [Google Scholar]
- 3.Tomlinson P R, Wilson J W, Stewart A G. Biochem Pharmacol. 1995;49:1809–1819. doi: 10.1016/0006-2952(94)00532-q. [DOI] [PubMed] [Google Scholar]
- 4.Turki J, Pak J, Green S A, Martin R J, Liggett S B. J Clin Invest. 1995;95:1635–1641. doi: 10.1172/JCI117838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Green S A, Turki J, Innis M, Liggett S B. Biochemistry. 1994;33:9414–9419. doi: 10.1021/bi00198a006. [DOI] [PubMed] [Google Scholar]
- 6.Holgate S T. Nat Genet. 1997;15:227–229. doi: 10.1038/ng0397-227. [DOI] [PubMed] [Google Scholar]
- 7.Beavo J A. Physiol Rev. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- 8.Chan S C, Reifsnyder D, Beavo J A, Hanifin J M. J Allergy Clin Immunol. 1993;91:1179–1188. doi: 10.1016/0091-6749(93)90321-6. [DOI] [PubMed] [Google Scholar]
- 9.Gantner F, Tenor H, Gekeler V, Schudt C, Wendel A, Hatzelmann A. J Allergy Clin Immunol. 1997;100:527–535. doi: 10.1016/s0091-6749(97)70146-5. [DOI] [PubMed] [Google Scholar]
- 10.Torphy T J. Am J Respir Crit Care Med. 1998;157:351–370. doi: 10.1164/ajrccm.157.2.9708012. [DOI] [PubMed] [Google Scholar]
- 11.Conti M, Jin S-L C. Prog Nucleic Acid Res Mol Biol. 1999;63:1–38. doi: 10.1016/s0079-6603(08)60718-7. [DOI] [PubMed] [Google Scholar]
- 12.Conti M, Nemoz G, Sette C, Vicini E. Endocr Rev. 1995;16:370–389. doi: 10.1210/edrv-16-3-370. [DOI] [PubMed] [Google Scholar]
- 13.Houslay M D, Sullivan M, Bolger G B. Adv Pharmacol. 1998;44:225–342. doi: 10.1016/s1054-3589(08)60128-3. [DOI] [PubMed] [Google Scholar]
- 14.Fuhrmann M, Jahn H U, Seybold J, Neurohr C, Barnes P J, Hippenstiel S, Kraemer H J, Suttorp N. Am J Respir Cell Mol Biol. 1999;20:292–302. doi: 10.1165/ajrcmb.20.2.3140. [DOI] [PubMed] [Google Scholar]
- 15.Barnes P J. Eur Respir J. 1995;8:457–462. doi: 10.1183/09031936.95.08030457. [DOI] [PubMed] [Google Scholar]
- 16.Jin S-L C, Richard F, Kuo W-P, D'Ercole A J, Conti M. Proc Natl Acad Sci USA. 1999;96:11998–12003. doi: 10.1073/pnas.96.21.11998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jin S-L C, Bushnik T, Lan L, Conti M. J Biol Chem. 1998;273:19672–19678. doi: 10.1074/jbc.273.31.19672. [DOI] [PubMed] [Google Scholar]
- 18.Thompson W J, Appleman M M. Biochemistry. 1971;10:311–316. [PubMed] [Google Scholar]
- 19.Conti M, Toscano M V, Petrelli L, Geremia R, Stefanini M. Endocrinology. 1982;110:1189–1196. doi: 10.1210/endo-110-4-1189. [DOI] [PubMed] [Google Scholar]
- 20.Bradford M M. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 21.Hamelmann E, Schwarze J, Takeda K, Oshiba A, Larsen G L, Irvin C G, Gelfand E W. Am J Respir Crit Care Med. 1997;156:766–775. doi: 10.1164/ajrccm.156.3.9606031. [DOI] [PubMed] [Google Scholar]
- 22.Clayberger C R, DeKruyff R H, Aisenberg J, Cantor H. J Exp Med. 1983;157:19061919. doi: 10.1084/jem.157.6.1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Macaulay A E, DeKruyff R H, Umetsu D T. J Immunol. 1998;160:1694–1700. [PubMed] [Google Scholar]
- 24.Harper J F, Brooker G. J Cyclic Nucleotide Res. 1975;1:207–218. [PubMed] [Google Scholar]
- 25.Iona S, Cuomo M, Bushnik T, Naro F, Sette C, Hess M, Shelton E R, Conti M. Mol Pharmacol. 1998;53:23–32. doi: 10.1124/mol.53.1.23. [DOI] [PubMed] [Google Scholar]
- 26.Hargreave F E, Ryan G, Thomson N C, O'Byrne P M, Latimer K, Juniper E F, Dolovich J. J Allergy Clin Immunol. 1981;68:347–335. doi: 10.1016/0091-6749(81)90132-9. [DOI] [PubMed] [Google Scholar]
- 27.Wess J. Crit Rev Neurobiol. 1996;10:69–99. doi: 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- 28.Hulme E C, Birdsall N J, Buckley N J. Annu Rev Pharmacol Toxicol. 1990;30:633–673. doi: 10.1146/annurev.pa.30.040190.003221. [DOI] [PubMed] [Google Scholar]
- 29.Maeda A, Kubo T, Mishina M, Numa S. FEBS Lett. 1988;239:339–342. doi: 10.1016/0014-5793(88)80947-5. [DOI] [PubMed] [Google Scholar]
- 30.Zaagsma J, Roffel A F, Meurs H. Life Sci. 1997;60:1061–1068. doi: 10.1016/s0024-3205(97)00048-9. [DOI] [PubMed] [Google Scholar]
- 31.Costello R W, Jacoby D B, Fryer A D. Thorax. 1998;53:613–616. doi: 10.1136/thx.53.7.613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Torphy T J, Cieslinski L B. Mol Pharmacol. 1990;37:206–214. [PubMed] [Google Scholar]
- 33.Tanner L I, Harden T K, Wells J N, Martin M W. Mol Pharmacol. 1986;29:455–460. [PubMed] [Google Scholar]
- 34.Eglen R M, Watson N. Pharmacol Toxicol. 1996;78:59–68. doi: 10.1111/j.1600-0773.1996.tb00181.x. [DOI] [PubMed] [Google Scholar]
- 35.Stengel P W, Gomeza J, Wess J, Cohen M L. J Pharmacol Exp Ther. 1999;292:877–885. [PubMed] [Google Scholar]
- 36.Erneux C, Van Sande J, Miot F, Cochaux P, Decoster C, Dumont J E. Mol Cell Endocrinol. 1985;43:123–134. doi: 10.1016/0303-7207(85)90075-9. [DOI] [PubMed] [Google Scholar]
- 37.Harden T K, Evans T, Hepler J R, Hughes A R, Martin M W, Meeker R B, Smith M M, Tanner L I. Adv Cyclic Nucleotide Protein Phosphorylation Res. 1985;19:207–220. [PubMed] [Google Scholar]
- 38.Buxton I L, Brunton L L. J Biol Chem. 1985;260:6733–6737. [PubMed] [Google Scholar]
- 39.Nagase T, Kurihara H, Kurihara Y, Aoki T, Fukuchi Y, Yazaki Y, Ouchi Y. Am J Respir Crit Care Med. 1998;157:560–564. doi: 10.1164/ajrccm.157.2.9706009. [DOI] [PubMed] [Google Scholar]
- 40.Levitt R C, Mitzner W. J Appl Physiol. 1989;67:1125–1132. doi: 10.1152/jappl.1989.67.3.1125. [DOI] [PubMed] [Google Scholar]
- 41.Engels P, Fichtel K, Lubbert H. FEBS Lett. 1994;350:291–295. doi: 10.1016/0014-5793(94)00788-8. [DOI] [PubMed] [Google Scholar]
- 42.Takahashi M, Terwilliger R, Lane C, Mezes P S, Conti M, Duman R S. J Neurosci. 1999;19:610–618. doi: 10.1523/JNEUROSCI.19-02-00610.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hanifin J M, Chan S C. J Invest Dermatol. 1995;105:84S–88S. doi: 10.1111/1523-1747.ep12316116. [DOI] [PubMed] [Google Scholar]
- 44.Lim J, Pahlke G, Conti M. J Biol Chem. 1999;274:19677–19685. doi: 10.1074/jbc.274.28.19677. [DOI] [PubMed] [Google Scholar]