

Abstract
Aims
To obtain comprehensive pharmacokinetic and pharmacodynamic data for artesunate (ARTS) and its active metabolite dihydroartemisinin (DHA) following i.v. and oral administration of ARTS to patients with acute, uncomplicated falciparum malaria.
Methods
Twenty-six Vietnamese patients with falciparum malaria were randomized to receive either i.v. ARTS (120 mg; group 1) or oral ARTS (100 mg; group 2), with the alternative preparation given 8 h later in an open crossover design. Mefloquine (750 mg) was administered at 24 h. Plasma concentrations of ARTS and DHA were determined by h.p.l.c. assay. Pharmacokinetic parameters were calculated by non-compartmental methods. The time to 50% parasite clearance (PCT50) was calculated by linear interpolation of parasite density determinations. Linear least squares and multiple linear regression analyses were used to evaluate pharmacokinetic-pharmacodynamic relationships.
Results
Following i.v. bolus, ARTS had a peak concentration of 29.5 μm (11 mg l−1), elimination t1/2 = 2.7 min, CL = 2.33 l h−1 kg−1 and V = 0.14 l kg−1. The Cmax for DHA was 9.3 μm (2.64 mg l−1), t1/2 = 40 min, CL = 0.75 l h−1 kg−1 and V = 0.76 l kg−1. Following oral ARTS, relative bioavailability of DHA was 82%, Cmax was 2.6 μm (0.74 mg l−1), t1/2 = 39 min, and MAT = 67 min. Overall, the PCT50 and fever clearance time (FCT) were 6.5 h and 24 h, respectively. There was no correlation between PCT50 or FCT and AUC, Cmax or MRT for DHA.
Conclusions
Despite rapid clearance of ARTS and DHA in patients with uncomplicated falciparum malaria, prompt parasite and fever clearance were achieved. High relative bioavailability of DHA following oral ARTS administration, and clinical outcomes comparable with those after i.v. ARTS, support the use of the oral formulation in the primary care setting.
Keywords: artesunate, dihydroartemisinin, falciparum malaria, pharmacokinetics, pharmacodynamics, bioavailability
Introduction
Artemisinin and its semi-synthetic derivatives are potent, well-tolerated antimalarial drugs that have become first-line therapy in many tropical countries [1–3]. Artesunate (ARTS), a water-soluble derivative that can be administered as an intravenous (i.v.) or intramuscular (i.m.) injection, is the most promising alternative to i.v. quinine for the treatment of severe malaria [2]. Recent studies of clinical efficacy have shown that ARTS clears parasites more rapidly than quinine [2–6], but improved survival rates have not been demonstrated and large-scale randomised, comparative trials are awaited.
Despite encouraging clinical results, the paucity of pharmacokinetic and pharmacodynamic data has meant that dosage regimens for artemisinin derivatives remain empirical [2–5]. We have shown previously, in six patients with uncomplicated falciparum malaria, that elimination of ARTS and its active metabolite dihydroartemisinin (DHA) is essentially complete 6 h after an i.v. injection of ARTS [7]. This finding is consistent with results from earlier volunteer studies in adults [8, 9] and with a recent report of DHA pharmacokinetics determined by bioassay following oral ARTS therapy in Vietnamese children [10].
Pharmacokinetic data for oral ARTS in patients with malaria, particularly bioavailability, are vital for rational design of dosage regimens but have not been reported previously. In recent years, efforts to improve clinical outcome have seen doses of ARTS increase from 1–2 mg kg−1 day−1 [11–13] to 4–12 mg kg−1 day−1 [14–17]. In view of concerns regarding animal and in vitro neurotoxicity of the artemisinin drugs [18–20], escalation in total daily dose may be imprudent in the absence of reliable pharmacokinetic data.
The present study provides comprehensive pharmacokinetic and pharmacodynamic data for ARTS and its active metabolite DHA following i.v. and oral administration of ARTS to Vietnamese patients with acute, uncomplicated falciparum malaria.
Methods
Patients
Twenty-six patients with uncomplicated falciparum malaria were recruited from the Bao Loc region of Lam Dong Province in Vietnam in 1996, either following admission to Bao Loc Hospital or on referral from neighbouring primary care health facilities. Most were farmers or rural labourers. The diagnosis was confirmed by microscopic examination of thick and thin blood films, and a complete clinical assessment including drug history was completed. Patients were excluded if they had impaired consciousness, jaundice (serum bilirubin >50 μmol l−1), renal impairment (serum creatinine >250 μmol l−1 after rehydration), anaemia (venous haematocrit <20%), hyperparasitaemia (>150 000 asexual forms per μl whole blood from thick film analysis) or if informed consent could not be obtained. Patients were not recruited if they had been treated with ARTS or DHA in the previous 8 h, artemisinin in the previous 12 h, or artemether in the previous 24 h. These criteria were based on the known pharmacokinetic data at the time, and ensured that patients were excluded for a period at least five times the elimination half-life of the drug (40 min for DHA, 2.2–2.3 h for artemisinin and 4.2 h for artemether [3]). If the patient's condition deteriorated significantly during the study, the attending physician was to withdraw the patient and start appropriate resuscitation and supportive treatment. Patients were advised that they could withdraw from the study at any stage without prejudice to their continuing care. The study was approved by the Ministry of Health, Vietnam, and the University of Western Australia Human Rights Committee.
Study design and procedures
Patients were randomized (pre-determined schedule) to receive either i.v. ARTS (120 mg diluted in 10 ml 5% w/v dextrose and given as a bolus over 2 min; group 1) or oral ARTS (100 mg as 2 × 50 mg tablets; group 2), with the alternative preparation given 8 h later in an open crossover design. Both ARTS formulations were obtained from the Guilin No. 2 Pharmaceutical Factory, Guangxi, China. A single dose of mefloquine (750 mg) was administered 24 h after admission to the study.
Venous blood samples (15 ml immediately prior to dosing and 3 ml thereafter) were obtained from the arm opposite to that used for drug administration. In the case of i.v. ARTS, sampling times were 0, 5, 7, 9, 12, 15, 20, 30, 45, 60, 90 min and 2, 3, 4 and 8 h after dosing. For oral ARTS, sampling was at 0, 15, 30, 45, 60, 75, 90, 105 min and 2, 3, 4, 6 and 8 h. Blood was collected into fluoride-oxalate tubes and chilled immediately to prevent degradation of ARTS by plasma esterases. Samples were centrifuged within 30 min to minimise haemolysis and the separated plasma was stored below −20° C until analysis. Thick and thin blood films were prepared from the hourly samples, and 4 hourly thereafter, until parasite clearance. Vital signs, including oral temperature and urine output, were monitored every 4 h. Patients were discharged when afebrile and aparasitaemic.
Pharmacokinetic and pharmacodynamic analysis
ARTS injection (powder) and tablets were weighed and assayed by h.p.l.c. [7]. The mean (95% CI) content of ARTS in the vials for injection (n = 6) was 59.2 (55.1, 63.3) mg. Since the 95% CI encompassed the stated content (60 mg), pharmacokinetic parameters were calculated from the nominal strength of the injection. The mean (95% CI) weight and content of the ARTS tablets (n = 5) were 255 (248, 262) mg and 44.8 (42.8, 46.7) mg, respectively. Pharmacokinetic parameters following oral ARTS were calculated from the assayed potency (45 mg) rather than the nominal content (50 mg). DHA, the principal degradation product of ARTS [21], was not detected in any of the tablets.
Plasma samples were assayed by h.p.l.c. [7]. The between-run coefficients of variation for h.p.l.c. analysis for ARTS were 8.2% and 7.5% at 1060 nm and 4240 nm respectively, and 7.1% and 11.3% for DHA at 900 nm and 3620 nm respectively. Stability of ARTS (780 and 4560 nm) and DHA (1060 and 6160 nm) in plasma has been assessed for up to 8 months at −25° C and found to be within ±7.6% of replicate samples stored at −80° C.
Pharmacokinetic parameters AUC(0, ∞), λz, t1/2, MRT, CL, V, Cmax and tmax) were determined from the plasma concentration-time data using non-compartmental analysis [22]. Bioavailability was calculated as F = (AUCoral/ AUCi.v.) × (Dosei.v./Doseoral), with correction for tablet potency. Pharmacokinetic parameters derived for DHA assume complete bioconversion from ARTS.
Blood films were stained (Giemsa) within 12 h of preparation and examined by a single microscopist (NPT). Thick films were used to determine parasite density in all patients. The number of asexual parasites per μl of whole blood was determined by counting the number of white cells (WBC) in high power fields containing a total of 500 parasites where the ratio of parasites/WBC was more than one, or the number of parasites per 1000 WBC where the ratio of parasites/WBC was less than unity. Parasitaemia was calculated as the product of the parasite/WBC ratio and WBC count.
Multiple thick films (n = 4 to 5) were obtained from randomly selected patients at random times and examined in the same way to determine the precision of parasite counting. Seventy-six sets of thick films were obtained, with parasite densities ranging from 100 to 177 000 parasites per μl. The median (interquartile range) coefficient of variation of the parasite counts was 5.1% (3.7, 8.7%).
The time to reach 50% of the original parasite count (PCT50) was determined by simple linear interpolation of the parasite count–time data. Fever clearance time (FCT) was taken to be the first oral temperature <37.5° C.
Statistical analysis
The study was designed to detect a 30% difference (80% power at P < 0.05) in the principal parameters of interest, AUC and CL, and PCT50. Differences between means were analyzed by Student's t-test or by the Mann-Whitney Rank Sum Test, as appropriate, and the Kolmogorov-Smirnov test was used to determine normality of the data [23]. Linear least squares and best subsets multiple regression analyses were used to evaluate relationships between pharmacokinetic (AUC, Cmax, MRT) and pharmacodynamic (PCT50, FCT) parameters.
Results
Clinical course
Twenty-eight patients with falciparum malaria were admitted to Bao Loc Hospital during the study period. Two had severe malaria and were ineligible for recruitment into the present study. All other patients satisfied the inclusion criteria. Details of the patients in groups 1 and 2 are given in Table 1. There were no significant differences in any demographic characteristic. Consistent with the lack of complications of malaria, biochemical variables in all patients were within normal ranges. In almost all subjects, the body mass index (BMI) was below the normal range reported in developed countries (20–25 kg m−2), with values typically indicative of ‘mild starvation’ (18–20 kg m−2) [24].
Table 1.
Demographic data. Group 1 patients received i.v. and oral artesunate at 0 and 8 h, respectively. The order was reversed for group 2. Data are presented as means (95% CI) or mediansa (interquartile rangeb), as appropriate. There were no significant differences between groups 1 and 2 for any parameter (Student's t-test or Mann-Whitney Rank Sum Test, as appropriate).

Five patients had received unspecified antimalarial therapy prior to admission. Two had taken an unknown medication 24 h prior to admission, two had received treatment at least 1 week prior to admission, and the fifth had taken ARTS 11 days prior to admission and an unknown medication 1 week prior to admission. Some of the unknown medications may have been antipyretic or antiemetic drugs but ARTS, chloroquine and mefloquine are available in the region. Two patients were excluded from the pharmacodynamic analysis due to low initial parasite densities (1048 and 3850 per μl, respectively) and variable subsequent counts.
Pharmacokinetic analysis
i) Artesunate
Pharmacokinetic data are summarized in Table 2 and presented in Figure 1a. Following i.v. administration, ARTS had a mean extrapolated peak concentration of 29.5 μm (11 mg l−1) and a mean t1/2 of 2.7 min. Following oral therapy, plasma concentrations of ARTS were above the assay limit of sensitivity (130 nm; 50 μg l−1) in only 13 of the 26 patients. From these limited data, the bioavailability of ARTS was estimated to be 15%. Median values for CL (2.33 l h−1 kg−1) and V (0.14 l kg−1) are presented because the data were not normally distributed.
Table 2.
Pharmacokinetic parameters for artesunate and dihydroartemisinin following intravenous (120 mg; 312.5 μmol) and oral (100 mga; 260.4 μmol) administration of artesunate. Data are given as means (95% CI) or mediansb (interquartile rangec), as appropriate. There were no significant differences between groups 1 and 2 for any parameter (Student's t-test or Mann-Whitney Rank Sum Test, as appropriate), except ARTS t1/2 after i.v. administration (see text).
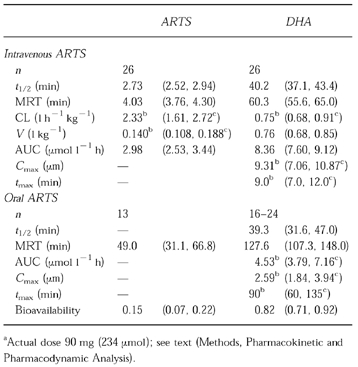
Figure 1.
Plasma concentration-time profile for artesunate (▴) and dihydroartemisinin (•) following 312.5 μmol (120 mg) i.v. dose (a) of artesunate to 26 Vietnamese patients, and for dihydroartemisinin (○) following 260.4 μmol (100 mg) oral dose (b) of artesunate to 19 Vietnamese patients. Data are shown as mean+s.d.
ii) Dihydroartemisinin
After i.v. ARTS administration, the median peak concentration of DHA was 9.3 μm (2.64 mg l−1), and mean elimination t1/2 was 40 min (Table 2 and Figure 1a). Extrapolation of these data suggest that the plasma concentration would have been less than the in vitro EC50 (1 nm [25, 26]) after 9 h. The CL (0.75 l h−1 kg−1) and V (0.76 l kg−1) data assume complete conversion of ARTS to DHA [27]. These parameters were not calculated from plasma concentration-time data after oral dosing.
Valid pharmacokinetic data for DHA were obtained from 24 of the 26 patients after oral ARTS (Table 2 and Figure 1b). Plasma concentration-time data for the other two patients were insufficient for comprehensive analysis. In seven patients, t1/2 could not reliably be estimated from the data and t1/2 from the paired i.v. administration was used in the determination of oral AUC and MRT. AUC and Cmax were not normally distributed; median (range) AUC and Cmax for DHA were 4.53 (2.67, 10.89) μmol l h−1 and 2.59 (1.25, 8.26) μm, respectively. Mean relative bioavailability of DHA (n = 24), which also assumes complete conversion of ARTS to DHA before any subsequent metabolism, was 82%. The mean absorption time (MAT = MRToral-MRTi.v.) was 67 min and, assuming first order kinetics, the absorption rate constant (ka = 1/MAT) was 0.89 h−1.
iii) Time-dependent changes
Apart from t1/2 for ARTS, the pharmacokinetic parameters for both ARTS and DHA were independent of the order of administration of i.v. and oral ARTS. Mean (95% CI) t1/2 of ARTS in group 1 (2.5 (2.3, 2.7) min) was significantly shorter than in group 2 (3.0 (2.6, 3.3) min; P = 0.04).
Pharmacodynamic analysis
Mean parasite clearance curves for groups 1 and 2, expressed as a percentage of the original density, are presented in Figure 2. At 8 h, the mean parasite densities for groups 1 and 2 were respectively 36% and 48% of the original counts. PCT50 was less than 8 h for 67% (16/24) of the patients. Only three patients (13%) had a PCT50greater than 10 h. Median FCT was 24 h in both groups (Table 1). There was no correlation between PCT50 or FCT and AUC, Cmax or MRT.
Figure 2.
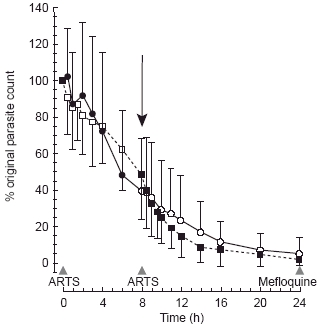
Parasite clearance curves, as a percentage of the original parasite count, for group 1 (i.v. • at 0, oral ○ at 8 h; data are mean+s.d.) and group 2 (oral ARTS □ at 0, i.v. ▪ at 8 h; data are mean−s.d.). The arrow (↓) highlights the time at which the second dose (by alternative route) was administered.
Discussion
The pharmacokinetic data obtained from the present group of Vietnamese patients treated with ARTS for uncomplicated falciparum malaria are the most comprehensive reported to date [7–10, 28, 29]. Intravenous administration of ARTS produced plasma concentration profiles which confirm the relatively short half-lives of both ARTS and DHA and suggest little inter-individual variability in disposition (Figure 1a). When ARTS was given orally, plasma concentration profiles were more variable. The mean absolute bioavailability of ARTS was low, but the mean relative bioavailability of DHA was more than 80%.
In a previous study of six Vietnamese patients with uncomplicated malaria given 120 mg ARTS i.v. [7], we used a blood sampling protocol which did not include sufficient time points immediately after dosing to allow an accurate determination of ARTS pharmacokinetic parameters. Nevertheless, t1/2 was estimated to be 3.5 min, a figure consistent with both the present results and the 2–5 min t1/2 reported previously in healthy Chinese volunteers [8, 9]. We were able to determine the elimination t1/2 (34 min) for DHA more accurately in our original study and estimated Cmax to be at least 7 μm (2 mg l−1) [7]. The results were comparable with those of the larger sample of patients in the present study (t1/2 = 40 min; Cmax = 9.3 μm) and to t1/2 reported in the Chinese volunteer studies (48 min [8]). In contrast to these data, preliminary results from patients with acute malaria reported by Benakis et al. [28] suggest that the t1/2 for ARTS and DHA following i.m. administration are significantly longer at 29 and 95 min, respectively. These values are inconsistent with our data and with all previous studies, including a more recent volunteer study by the same group [29]. The only possible explanation for this inconsistency is that ARTS and DHA clearance are absorption rate dependent following i.m. administration.
Previously published data on the pharmacokinetics of oral ARTS are restricted to studies in healthy volunteers [29] and in children with falciparum malaria [10]. In a study by Benakis et al. [29], six adult volunteers were given oral ARTS but, due to low and variable plasma concentrations, pharmacokinetic parameters for ARTS were not determined. The mean elimination t1/2 of DHA (39 min [29]) was identical to that in the present patients. However, the Cmax (0.57 mg l−1; 2029 nm) and AUC (0.74 μg ml−1 h; 2.61 μmol l−1 h) after a 200 mg dose were substantially lower than those in the present study (2590 nm and 4.53 μmol l−1 h, respectively) where a dose of only 100 mg ARTS was administered. This represents an average 3.5-fold greater bioavailability in the present study. The discrepancy may be explained by the lower body weights of our patients (49.5 kg) compared with those of the healthy volunteers (78 kg) [29]. Dehydration and a higher relative bioavailability of the formulation used in the present study may also have contributed. The absorption (biotransformation) t1/2 reported by Benakis et al. (20 min [29]) was substantially shorter than that in our patients (47 min). Furthermore, tmax and MRT (30 and 75 min respectively [29]) were shorter than our values of 90 and 127 min respectively. These differences could be related to factors such as the dissolution characteristics of the dose formulations [30], reduction in gastric motility [30] or impairment of splanchnic perfusion which has been reported in patients with malaria [31].
Bethell et al. [10] studied 10 Vietnamese children who were given 3 mg kg−1 oral ARTS on admission and 2 mg kg−1 day−1 for 4 days thereafter. Pharmacokinetic parameters were determined by bioassay (as DHA equivalents) of serial plasma samples. Assuming that DHA was the predominant antimalarial drug in the patients’ plasma, MRT, AUC, Cmax and tmax values of 2.61 h, 4.53 μmol l−1 h, 2.34 μm and 102 min respectively were reported [10]. These results are consistent with data from the present study where a mean dose of 2.5 mg kg−1 ARTS was administered. The only difference between the two studies was the elimination t1/2 of DHA, reported as 1 h (95% CI 0.8, 1.4) by Bethell et al. [10] and 0.66 h (0.53, 0.78) in our study. The slower clearance in the children could be due to more severe hepatic and/or renal impairment than in our patients, or there may be age-related differences in the intrinsic clearance of DHA. However, the difference in elimination t1/2 is unlikely to be of clinical significance. Furthermore, the comparability of pharmacokinetic data from the two studies suggests that DHA is the only active metabolite of ARTS [10, 27].
The present study was designed to include an evaluation of the bioavailability of ARTS and, indirectly, DHA. The 8 h crossover design minimised potential changes in pharmacokinetic parameters due to an improvement in the patient's clinical condition or the possibility of time-dependent changes in drug clearance [32]. The bioavailability of ARTS (15%) reported in this study is an overestimate of the true value since ARTS could not be detected in half of the patients. Whilst there is no information on the site(s) of metabolism of ARTS after oral administration, the gut wall and liver probably contribute most to the low absolute oral bioavailability of ARTS. Nevertheless, the high relative bioavailability of DHA (82%) indicates that the overall absorption of ARTS is high, and therefore adequate for the treatment of uncomplicated malaria. Our data indicate that a dose of 150 mg ARTS will give an AUC of DHA similar to that achieved following 120 mg i.v. ARTS. Nevertheless, delayed absorption and lower, variable peak plasma concentrations (Figure 1b) are potential limitations of oral administration.
The disposition of the artemisinin drugs in patients with malaria is not fully established [3, 5, 27]. Our data allow an assessment of the in vivo elimination of ARTS and DHA. Clearance of ARTS (approximately 120 l h−1) was significantly greater than either liver or kidney blood flow rates (80 l h−1 and 65 l h−1, respectively), suggesting that several organs and/or enzyme systems contribute to its metabolism. The high peak concentration of DHA, less than 10 min after i.v. ARTS administration, supports the hypothesis that ARTS is rapidly and completely metabolised to DHA in vivo [27]. Time-dependent changes in artemisinin clearance have been reported [32], but this phenomenon has not been demonstrated with the semi-synthetic derivatives. Nevertheless, the short duration of our study (16 h) effectively eliminated the possibility of time-dependent enzyme induction. The statistically significant difference in ARTS t1/2 between groups 1 and 2 is of negligible biological importance.
The relationship between the prompt, sustained antimalarial effect of ARTS and rapid clearance of both ARTS and DHA appears unique in antimalarial pharmacology. Peak concentrations of DHA occurred 9 min after i.v. administration of ARTS, at a time when there had been no appreciable reduction in parasite count (Figure 2). Three hours after i.v. administration of ARTS, mean plasma concentrations of DHA were near the h.p.l.c. assay limit of sensitivity (400 nm) and the parasite count had fallen by approximately 20%. At the time of administration of the second (oral) dose of ARTS, the extrapolated mean plasma DHA concentration was 2 nm (close to the in vitro EC50 [25, 26]) and the reduction in parasite count was 60%. The parasite count had fallen by more than 95% when mefloquine (15 mg kg−1) was given at 24 h. Despite rapid clearance of ARTS and DHA from the body, these observations confirm that a potent and apparently sustained antimalarial effect can be achieved with short-course ARTS therapy in many patients.
A potential advantage to the use of ARTS is reduced toxicity. Analogous to modern aminoglycoside therapy, the high peak concentration and short t1/2 of DHA means there is no accumulation of the drug and toxicity may be less likely than with the longer acting artemisinin derivatives, artemether and arteether. Neurotoxicity has been described for a range of artemisinin derivatives in cultured cells [19, 20], and also after high-dose regimens of artemether and arteether in rats and dogs [18, 20]. However, neurotoxicity has not been documented in any of the controlled studies in human volunteers or patients [3, 5]. A recent case report of a temporal association between oral ARTS and a reversible cerebellar syndrome [33] did not exclude the possibility of a post malaria neurological syndrome [34, 35].
Optimal doses and dose intervals for ARTS and DHA have not been determined. Therefore, in the absence of well controlled dose-ranging studies and valid pharmacodynamic relationships, widely-used empirical regimens remain unchallenged. Nevertheless, several recent reports [36–39] and the present study support the results of earlier studies [3, 5] that showed ARTS to be effective at doses less than 4 mg kg−1 day−1. Our pharmacokinetic data could be used to design dose-ranging studies aimed at minimising potential adverse effects whilst maximising antimalarial efficacy.
Our study found a mean PCT50 value of approximately 6.5 h following 2.4 mg kg−1 i.v. ARTS or 2 mg kg−1 oral ARTS. This is similar to the range of 4 to 10 h reported previously [13, 36, 37] following single doses of up to 4 mg kg−1 of ARTS. Moreover, we have shown that virtually all of a dose of this magnitude is cleared within 8 h. On the basis of our results and those reported in patient groups from Africa and Asia [13, 36, 37], we recommend that treatment for uncomplicated falciparum malaria commence with a dose of 2 to 4 mg kg−1 ARTS. Those patients who fail to achieve at least a 50% reduction in parasite density within 8 to 10 h of the first dose could then be given a second dose with no risk of accumulation of either ARTS or DHA. Whilst further parasite counts would guide the need for subsequent doses, short courses of ARTS therapy (2 to 3 days) supplemented with a single dose of mefloquine (15 to 25 mg kg−1) give high cure and low recrudescence rates [14–16, 36, 39].
Acknowledgments
We are indebted to Professor Trinh Kim Anh and Mr Vuong Van Chon, Cho Ray Hospital, and Dr Vo Thanh Chien, Dr Vu Nam Bien, Mrs Dang Thi Vinh Thuan and staff of the Malaria and Biochemistry & Haematology Departments, Bao Loc Hospital, for facilitating the conduct of this study.
This work was supported by a Project Grant from the National Health and Medical Research Council of Australia (T. M. E. D & K. F. I). K. T. B was a recipient of an NHMRC Dora Lush (Biomedical) Scholarship.
References
- 1.Price RN, Nosten F, Luxemburger C, et al. Effects of artemisinin derivatives on malaria transmissibility. Lancet. 1996;347:1654–1658. doi: 10.1016/s0140-6736(96)91488-9. [DOI] [PubMed] [Google Scholar]
- 2.Hien TT, White NJ. Qinghaosu. Lancet. 1993;341:603–608. doi: 10.1016/0140-6736(93)90362-k. [DOI] [PubMed] [Google Scholar]
- 3.de Vries PJ, Dien TK. Clinical pharmacology and therapeutic potential of artemisinin and its derivatives in the treatment of malaria. Drugs. 1996;52:818–836. doi: 10.2165/00003495-199652060-00004. [DOI] [PubMed] [Google Scholar]
- 4.White NJ. Clinical pharmacokinetics and pharmacodynamics of artemisinin and derivatives. Trans R Soc Trop Med Hyg. 1994;88(Suppl 1):S41–S43. doi: 10.1016/0035-9203(94)90471-5. [DOI] [PubMed] [Google Scholar]
- 5.Barradell LB, Fitton A. Artesunate. A review of its pharmacology and therapeutic efficacy in the treatment of malaria. Drugs. 1995;50:714–741. doi: 10.2165/00003495-199550040-00009. [DOI] [PubMed] [Google Scholar]
- 6.Karbwang J, Na-Bangchang K, Thanavibul A, Bunnag D, Chongsuphajaisiddhi T, Harinasuta T. Comparison of oral artesunate and quinine plus tetracycline in acute uncomplicated falciparum malaria. Bull WHO. 1994;72:233–238. [PMC free article] [PubMed] [Google Scholar]
- 7.Batty KT, Davis TME, Thu LTA, Binh TQ, Anh TK, Ilett KF. Selective high-performance liquid chromatographic determination of artesunate and α- and β-dihydroartemisinin in patients with falciparum malaria. J Chromatogr B. 1996;677:345–350. doi: 10.1016/0378-4347(95)00428-9. [DOI] [PubMed] [Google Scholar]
- 8.Yang SD, Ma JM, Sun JH, Chen DX, Song ZY. Clinical pharmacokinetics of a new effective antimalarial artesunate, a qinghaosu derivative. Chin J Clin Pharmacol. 1985;1:106–109. [Google Scholar]
- 9.Zhao KC, Chen ZX, Lin BL, Guo XB, Li GQ, Song ZY. Studies on the phase 1 clinical pharmacokinetics of artesunate and artemether. Chin J Clin Pharmacol. 1988;4:76–81. [Google Scholar]
- 10.Bethell DB, Teja-Isavadharm P, Phuong CXT, et al. Pharmacokinetics of oral artesunate in children with moderately severe Plasmodium falciparum malaria. Trans R Soc Trop Med Hyg. 1997;91:195–198. doi: 10.1016/s0035-9203(97)90222-4. [DOI] [PubMed] [Google Scholar]
- 11.Hien TT, Arnold K, Vinh H, et al. Comparison of artemisinin suppositories with intravenous artesunate and intravenous quinine in the treatment of cerebral malaria. Trans R Soc Trop Med Hyg. 1992;86:582–583. doi: 10.1016/0035-9203(92)90137-2. [DOI] [PubMed] [Google Scholar]
- 12.Looareesuwan S, Viravan C, Vanijanonta S, et al. Randomised trial of artesunate and mefloquine alone and in sequence for acute uncomplicated falciparum malaria. Lancet. 1992;339:821–824. doi: 10.1016/0140-6736(92)90276-9. [DOI] [PubMed] [Google Scholar]
- 13.Hien TT, Phu NH, Mai NTH, et al. An open randomized comparison of intravenous and intramuscular artesunate in severe falciparum malaria. Trans R Soc Trop Med Hyg. 1992;86:584–585. doi: 10.1016/0035-9203(92)90138-3. [DOI] [PubMed] [Google Scholar]
- 14.Looareesuwan S, Viravan C, Vanijanonta S, Wilairatana P, Pitisuttithum P, Andrial M. Comparative clinical trial of artesunate followed by mefloquine in the treatment of acute uncomplicated falciparum malaria: two- and three-day regimens. Am J Trop Med Hyg. 1996;54:210–213. doi: 10.4269/ajtmh.1996.54.210. [DOI] [PubMed] [Google Scholar]
- 15.Karbwang J, Na-Bangchang K, Thanavibul A, Ditta-in M, Bunnag D, Harinasuta T. Comparative clinical trial of artesunate and the combination of artesunate-mefloquine in multidrug-resistant falciparum malaria. Clin Drug Invest. 1996;11:84–89. [Google Scholar]
- 16.Bunnag D, Kanda T, Karbwang J, Thimasarn K, Pungpak S, Harinasuta T. Artemether or artesunate followed by mefloquine as a possible treatment for multidrug resistant falciparum malaria. Trans R Soc Trop Med Hyg. 1996;90:415–417. doi: 10.1016/s0035-9203(96)90529-5. [DOI] [PubMed] [Google Scholar]
- 17.Luxemburger C, ter Kuile FO, Nosten F, et al. Single day mefloquine-artesunate combination in the treatment of multi-drug resistant falciparum malaria. Trans R Soc Trop Med Hyg. 1994;88:213–217. doi: 10.1016/0035-9203(94)90303-4. [DOI] [PubMed] [Google Scholar]
- 18.Brewer TG, Grate SJ, Peggins JO, et al. Fatal neurotoxicity of arteether and artemether. Am J Trop Med Hyg. 1994;51:251–259. doi: 10.4269/ajtmh.1994.51.251. [DOI] [PubMed] [Google Scholar]
- 19.Smith SL, Fishwick J, McLean WG, Edwards G, Ward SA. Enhanced in vitro neurotoxicity of artemisinin derivatives in the presence of haemin. Biochem Pharmacol. 1997;53:5–10. doi: 10.1016/s0006-2952(96)00591-6. [DOI] [PubMed] [Google Scholar]
- 20.Kamchonwongpaisan S, McKeever P, Hossler P, Ziffer H, Meshnick SR. Artemisinin neurotoxicity: neuropathology in rats and mechanistic studies in vitro. Am J Trop Med Hyg. 1997;56:7–12. doi: 10.4269/ajtmh.1997.56.7. [DOI] [PubMed] [Google Scholar]
- 21.Batty KT, Ilett KF, Davis TME. Chemical stability of artesunate injection and proposal for its administration by intravenous infusion. J Pharm Pharmacol. 1996;48:22–26. doi: 10.1111/j.2042-7158.1996.tb05870.x. [DOI] [PubMed] [Google Scholar]
- 22.Thomann P. Non-compartmental analysis methods. In: Heinzel G, Woloszczak R, Thomann P, editors. Topfit Version 2.0; Pharmacokinetic and Pharmacodynamic Data Analysis System for the PC. Stuttgart: Gustav Fischer; 1993. [Google Scholar]
- 23.Quinton A, Lebedev G, Youtz P, Tuerke T, Sinkler R, Kuo J. SigmaStat Version 2.0. Vol. 1995. San Rafael: Jandel Scientific; [Google Scholar]
- 24.Baird JD, Truswell AS. Nutritional factors in disease. In: Macleod J, Edwards C, Bouchier I, editors. Davidson's Principles and Practice of Medicine. 15. Edinburgh: Churchill Livingstone; 1987. pp. 49–83. [Google Scholar]
- 25.Skinner TS, Manning LS, Johnston WA, Davis TME. In vitro stage-specific sensitivity of Plasmodium falciparum to quinine and artemisinin drugs. Int J Parasitol. 1996;26:519–525. doi: 10.1016/0020-7519(96)89380-5. [DOI] [PubMed] [Google Scholar]
- 26.Hassan Alin M, Bjorkman A, Landberg-Lindgren A, Ashton M. The effect of artemisinin, its derivatives and mefloquine against chloroquine-resistant strains of Plasmodium falciparum in vitro. Trans R Soc Trop Med Hyg. 1992;86:365–367. doi: 10.1016/0035-9203(92)90220-7. [DOI] [PubMed] [Google Scholar]
- 27.Lee IS, Hufford CD. Metabolism of antimalarial sesquiterpene lactones. Pharmacol Ther. 1990;48:345–355. doi: 10.1016/0163-7258(90)90053-5. [DOI] [PubMed] [Google Scholar]
- 28.Benakis A, Paris M, Plessas C, Hien TT, Waller D, White NJ. Pharmacokinetics of sodium artesunate after im and iv administration. Am J Trop Med Hyg. 1993;49(Suppl):293. [Google Scholar]
- 29.Benakis A, Paris M, Loutan L, Plessas CT, Plessas S. Pharmacokinetics of artemisinin and artesunate after oral administration in healthy volunteers. Am J Trop Med Hyg. 1997;56:17–23. doi: 10.4269/ajtmh.1997.56.17. [DOI] [PubMed] [Google Scholar]
- 30.Gibaldi M. Biopharmaceutics and Clinical Pharmacokinetics. 4. Philadelphia: Lea & Febiger; 1991. Gastrointestinal Absorption-Biologic Considerations; pp. 24–39. [Google Scholar]
- 31.Warrell DA, Molyneux ME, Beales PF. Severe and complicated malaria (2nd edition) Trans R Soc Trop Med Hyg. 1990;84(Suppl 2):1–65. [PubMed] [Google Scholar]
- 32.Hassan Alin M, Ashton M, Kihamia CM, Mtey GJ, Bjorkman A. Multiple dose pharmacokinetics of oral artemisinin and comparison of its efficacy with that of oral artesunate in falciparum malaria patients. Trans R Soc Trop Med Hyg. 1996;90:61–65. doi: 10.1016/s0035-9203(96)90480-0. [DOI] [PubMed] [Google Scholar]
- 33.Miller LG, Panosian CB. Ataxia and slurred speech after artesunate treatment for falciparum malaria. N Engl J Med. 1997;336:1328. doi: 10.1056/NEJM199705013361818. [DOI] [PubMed] [Google Scholar]
- 34.Senanayake N, de Silva HJ. Delayed cerebellar ataxia complicating falciparum malaria: a clinical study of 74 patients. J Neurol. 1994;241:456–459. doi: 10.1007/BF00900965. [DOI] [PubMed] [Google Scholar]
- 35.Mai NTH, Day NP, Ly VC, et al. Post-malaria neurological syndrome. Lancet. 1996;348:917–921. doi: 10.1016/s0140-6736(96)01409-2. [DOI] [PubMed] [Google Scholar]
- 36.Luxemburger C, Nosten F, Raimond SD, Chongsuphajaisiddhi T, White NJ. Oral artesunate in the treatment of uncomplicated hyperparasitemic falciparum malaria. Am J Trop Med Hyg. 1995;53:522–525. doi: 10.4269/ajtmh.1995.53.522. [DOI] [PubMed] [Google Scholar]
- 37.Hassan Alin M, Kihamia CM, Bjorkman A, et al. Efficacy of oral and intravenous artesunate in male Tanzanian adults with Plasmodium falciparum malaria and in vitro susceptibility to artemisinin, chloroquine, and mefloquine. Am J Trop Med Hyg. 1995;53:639–645. doi: 10.4269/ajtmh.1995.53.639. [DOI] [PubMed] [Google Scholar]
- 38.Duarte EC, Fontes CJ, Gyorkos TW, Abrahamowicz M. Randomized controlled trial of artesunate plus tetracycline versus standard treatment (quinine plus tetracycline) for uncomplicated Plasmodium falciparum malaria in Brazil. Am J Trop Med Hyg. 1996;54:197–202. doi: 10.4269/ajtmh.1996.54.197. [DOI] [PubMed] [Google Scholar]
- 39.Price RN, Nosten F, Luxemburger C, et al. Artesunate versus artemether in combination with mefloquine for the treatment of multidrug-resistant falciparum malaria. Trans R Soc Trop Med Hyg. 1995;89:523–527. doi: 10.1016/0035-9203(95)90094-2. [DOI] [PubMed] [Google Scholar]