

Abstract
Aims
Chemical inhibitors of cytochrome P450 (CYP) are a useful tool in defining the role of individual CYPs involved in drug metabolism. The aim of the present study was to evaluate the selectivity and rank the order of potency of a range of isoform-selective CYP inhibitors and to compare directly the effects of these inhibitors in human and rat hepatic microsomes.
Methods
Four chemical inhibitors of human cytochrome P450 isoforms, furafylline (CYP1A2), sulphaphenazole (CYP2C9), diethyldithiocarbamate (CYP2E1), and ketoconazole (CYP3A4) were screened for their inhibitory specificity towards CYP-mediated reactions in both human and rat liver microsomal preparations. Phenacetin O-deethylation, tolbutamide 4-hydroxylation, chlorzoxazone 6-hydroxylation and testosterone 6β-hydroxylation were monitored for enzyme activity.
Results
Furafylline was a potent, selective inhibitor of phenacetin O-deethylation (CYP1A2-mediated) in human liver microsomes (IC50 = 0.48 μm), but inhibited both phenacetin O-deethylation and tolbutamide 4-hydroxylation (CYP2C9-mediated) at equimolar concentrations in rat liver microsomes (IC50 = 20.8 and 24.0 μm respectively). Sulphaphenazole demonstrated selective inhibition of tolbutamide hydroxylation in human liver microsomes but failed to inhibit this reaction in rat liver microsomes. DDC demonstrated a low level of selectivity as an inhibitory probe for chlorzoxazone 6-hydroxylation (CYP2E1-mediated). DDC also inhibited testosterone 6β-hydroxylation (CYP3A-mediated) in man and rat, and tolbutamide 4-hydroxylase activity in rat. Ketoconazole was a very potent, selective inhibitor of CYP3A4 activity in human liver (IC50 = 0.04 μm). Although inhibiting CYP3A in rat liver it also inhibited all other reactions at concentrations ≤5 μm.
Conclusions
It is evident that CYP inhibitors do not exhibit the same selectivity in human and rat liver microsomes. This is due to differential selectivity of the inhibitors and/or differences in the CYP isoform responsible for metabolism in the different species.
Keywords: cytochrome P450, cytochrome P450 inhibition, differential selectivity
Introduction
The correct assignment of individual cytochrome P450 (CYP) isoforms to specific metabolic pathways is an area of considerable importance; in particular in the rational prediction of drug-drug interactions. Many different strategies are currently employed in the unambiguous identification of CYP isoforms responsible for the biotransformation of therapeutic agents. These include the use of selective chemical inhibitors of CYP isoforms, inhibitory CYP antibodies, studies with purified, reconstituted enzymes and the correlation of immunoquantified CYP levels and metabolic rates [1, 2]. Recent advances in the field of chemical inhibitors of CYP have greatly facilitated the characterization of the catalytic specificities of individual CYP isoforms involved in drug metabolism. The lack of availability of human liver samples for metabolism/inhibitor studies may mean that hepatic microsomal preparations from other species such as the rat are used and predictions made to man.
Currently gene families 1, 2 and 3 are thought to be involved in the biotransformation of xenobiotics in both humans and rodents. However, isoforms are not conserved between species and differences are known to occur in catalytic and regulatory specificities between human CYP isoforms and their rat orthologues, although the CYP1A and CYP2E subfamilies show remarkable conservation between human and rat [3]. The major human hepatic CYP subfamilies are CYP2C and CYP3A which account for 20% and 30% of total CYP respectively [4], with CYP1A2, CYP2E1, CYP2A6, CYP2D6 and CYP2B6 comprising 13%, 7%, 4%, 2% and <1% respectively. Levels of CYP isoforms in male rats are very different with CYP2C11 accounting for 54% of total CYP content, CYP3A2 being 17% abundant and CYP1A2 being expressed at much lower levels (2%) in untreated rat liver samples [5, 6].
Differences in the levels of individual CYP isoforms and indeed the expression of distinct isoforms may lead to differences in the metabolism of alleged probe substrates between species. In addition, these differences have also been shown to influence the selectivity of inhibitor probes. For example, the inhibition of tolbutamide 4-hydroxylation by sulphaphenazole (CYP2C9 inhibitor in human) differs markedly when this reaction is catalysed by human, rat or rabbit liver microsomes [7].
In the present study, four chemical inhibitors of human cytochrome P450 isoforms, namely, furafylline (CYP1A2, [1, 8–10]); sulphaphenazole (CYP2C9, [1, 2, 10]); diethyldithiocarbamate (DDC) (CYP2E1, [10, 11]) and ketoconazole (CYP3A4, [12–14]) were screened for their inhibitory specificity towards CYP-mediated reactions in both human and rat liver microsomal preparations. Phenacetin O-deethylation, tolbutamide 4-hydroxylation, chlorzoxazone 6-hydroxylation and testosterone 6β-hydroxylation were chosen as markers for human CYP1A2 [15], CYP2C9 [16], CYP2E1 [17] and CYP3A4 [18] activity respectively and incubations performed in human and rat liver microsomes.
Methods
Chemicals
Phenacetin, paracetamol, metacetamol, tolbutamide, chlorzoxazone, zoxazolamine, testosterone, 6β-OH testosterone, 11β-OH testosterone, sulphaphenazole, diethyldithiocarbamate and β-NADPH (reduced form) were purchased from the Sigma Chemical Company (Poole, Dorset, UK). Furafylline and 6-OH chlorzoxazone were obtained from Ultrafine Chemicals (Manchester, UK). Chlorpropamide and 4-OH tolbutamide were gifts from Hoescht AG (Frankfurt, Germany); ketoconazole was a gift from Janssen (Beerse, Belgium). H.p.l.c. grade acetonitrile (AcN), dichloromethane (DCM), methanol and ethyl acetate were purchased from Fisons Plc (Loughborough, UK). All other reagents were of the highest grade possible.
Human liver samples
Histologically normal human livers were obtained from kidney transplant donors. Liver samples were transferred on ice to the laboratory within 30 min where they were sectioned into 10–20 g portions and frozen in liquid nitrogen. These were then stored at −80° C until required. Washed microsomes (105, 000g pellets) were prepared from human liver samples by the differential centrifugation technique [19] and microsomal protein yield was determined by the method of Lowry et al. [20] using bovine serum albumin as standard.
Rat liver samples
Washed microsomes were prepared by the classical differential centrifugation technique from the livers of male Wistar rats (200–300 g) which were sacrificed by cervical dislocation.
Enzyme assays for CYP probes
a) Phenacetin O-deethylation
Initial linearity studies indicated that this reaction was linear up to 1.5 mg protein for human and 2.0 mg protein for rat liver microsomes and an incubation time of 30 min in both species. A 500 μl reaction mixture typically containing 0.5 mg microsomal protein (human or rat) was incubated with phenacetin in the presence of MgCl2 (10 mm) and NADPH (2.5 mm) in phosphate buffer (0.067 m; pH 7.4). Metacetamol was added as internal standard and the mixture was extracted with DCM (10 ml; 20 min) to remove unreacted phenacetin followed by ethylacetate (10 ml; 20 min). Samples were reconstituted in mobile phase (200 μl) prior to h.p.l.c. analysis. Paracetamol and metacetamol were separated using an isocratic mobile phase (flow rate 1 ml min−1) consisting of AcN: sodium phosphate buffer (10:90, v/v; 0.1 m; pH 4.3) and a Spherex 5 μC18 column (25 cm x 4.6 mm; Phenomenex, Macclesfield, UK) with u.v. detection at 245 nm. Formation of paracetamol was quantified by interpolating peak height ratios of paracetamol and metacetamol from a standard curve of known paracetamol concentrations. Inter- and intra- assay coefficients of variation were 8.7% and 6.5% (determined at 100 pmol paracetamol) respectively. The lower limit of determination was 25 pmol.
b) Tolbutamide 4-hydroxylation
Initial linearity studies indicated that this reaction was linear up to 4 mg and 2 mg microsomal protein for human and rat liver microsomes respectively and an incubation time of 16 min for human liver microsomes and 10 min for rat liver microsomal preparations. A 500 μl reaction mixture containing 0.5 mg microsomal protein (human or rat) was incubated with tolbutamide in the presence of MgCl2 (10 mm) and NADPH (1 mm) in phosphate buffer (0.067 m; pH 7.4) according to the method of Back et al. [21]. Termination, extraction and h.p.l.c. analysis of samples were as previously described. The inter- and intra- assay coefficients of variation (determined at 0.6 nmol hydroxytolbutamide) were 3% and 1.3% respectively, with a lower limit of determination of 20 pmol.
c) Chlorzoxazone 6-hydroxylation
Initial linearity studies revealed that this reaction was linear upto 2 mg microsomal protein in both species and an incubation time of 40 min. A 500 μl incubation volume containing 0.2 mg microsomal protein (human or rat) was incubated with chlorzoxazone (CLZ) in the presence of MgCl2 (10 mm) and NADPH (1 mm) in phosphate buffer (0.067 m; pH 7.4). The reaction was terminated by the addition of zoxazolamine as internal standard and the mixture was extracted with DCM (5 ml; 10 min). The organic phase was evaporated to dryness and reconstituted into mobile phase (200 μl) prior to h.p.l.c. analysis. Formation of 6-hydroxychlorzoxazone (6-OHCLZ) was measured by h.p.l.c. with u.v. detection at 295 nm and quantified by interpolating peak height ratios of 6-OHCLZ and zoxazolamine from a standard curve of known 6-OHCLZ concentrations. A 5 μC18 Spherex column (25 cm × 4.6 mm; Phenomenex, Macclesfield, UK) was employed to separate CLZ, 6OHCLZ and internal standard using a gradient mobile phase system. Initial chromatographic conditions were AcN:ammonium acetate buffer (28:22, v/v; 0.05; pH 3.3), followed by a linear increase of AcN to 33% between 10 and 15 min remaining so until 17 min then returning to the original run conditions at 20 min. This was followed by a 5 min re-equilibration period. The inter- and intra- assay coefficients of variation (determined at 3 and 10 nmol 6-OHCLZ) were 5.3% and 8.5% respectively. The lower limit of determination was 100 pmol.
d) Testosterone 6β-hydroxylation
Initial linearity studies were performed and revealed that this reaction was linear up to 0.2 mg microsomal protein for both species and an incubation time of 15 min in human and 30 min in rat microsomal incubations. A 500 μl incubation mixture containing 0.05 mg microsomal protein (human or rat) was incubated with testosterone in the presence of MgCl2 (10 mm) and NADPH (2.5 mm) in phosphate buffer (0.067 m; pH 7.4). The reaction was terminated by the addition of 11β-hydroxytestosterone as internal standard and immediate extraction with DCM (10 ml; 20 min). The DCM layer was then evaporated to dryness before reconstitution with mobile phase (200 μl). Testosterone 6β-hydroxylation was quantified by h.p.l.c. analysis. 6β-hydroxytestosterone was separated from internal standard, testosterone and other metabolites by a 5 μC18 Prodigy column (15 cm x 4.6 mm; Phenomenex, Macclesfield, UK) using a gradient mobile phase system comprising of solvent mixtures: mixture A 35:64:1 (v/v) methanol: distilled water: AcN and mixture B 80:18:2 (v/v) methanol: distilled water: AcN. Initial run conditions were 75% A:25% B. Between 9 and 28 min there was a linear increase of solvent B to 75% (25% A) remaining so until 30 min. Between 30 and 32 min there was a linear decrease back to the starting run conditions (25% A:75% B) with a 3 min re-equilibration period. U.v. detection was at 254 nm. The inter- and intra- assay coefficients of variation were 5.6% and 4.3% respectively (determined at 500 pmol 6β-hydroxytestosterone). The assay had a lower limit of determination of 50 pmol.
Km and Vmax determinations for CYP probes
Under predetermined linear conditions with respect to time and protein concentration, a range of substrate concentrations were incubated as outlined above to determine Km and Vmax values. The substrate concentrations were as follows: phenacetin 5–2000 μm, tolbutamide 25–400 μm, chlorzoxazone 10–1000 μm and testosterone 10–200 μm. For each substrate, metabolite formation was calculated (nmol mg−1 min−1) and Michaelis-Menten equations for a one or two enzyme model were fitted to the data using the iterative non-linear regression program GraFit (version 3).
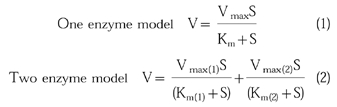 |
(1) |
| (2) |
where Km(1), Vmax(1) correspond to high affinity, low capacity site and Km(2), Vmax(2) correspond to low affinity, high capacity site.
Inhibition studies
The inhibitory potentials of furafylline, sulphaphenazole, diethyldithiocarbamate (DDC) and ketoconazole were investigated using the standard assay conditions listed above for each CYP probe substrate. A single concentration of each substrate was used: 20 μm for phenacetin, 100 μm for tolbutamide, 100 μm for chlorzoxazone and 100 μm for testosterone. Substrate concentrations for tolbutamide, chlorzoxazone and testosterone were chosen as approximating to the Km value for these substrates in human liver. A value of 20 μm was chosen for phenacetin as at this concentration greater than 95% of activity was catalysed by the high affinity component (CYP1A2). This value was calculated by substituting values for Km and Vmax into the Michaelis-Menten equation for a two enzyme system. Incubations were performed in the presence of a range of inhibitor concentrations. Inhibitors were prepared as methanolic stock solutions, with an appropriate amount dried down prior to reconstitution in the incubation volume containing protein and phosphate buffer. There was no visible evidence of precipitation of any of these compounds over the concentration range studied. In the case of the mechanism-based inactivators of CYP, namely furafylline and DDC, these compounds were preincubated with microsomes and NADPH for 15 min prior to the addition of substrate.
IC50 values (concentration of inhibitor to cause 50% inhibition of original enzyme activity) were determined by GraFit where appropriate using the following equation:
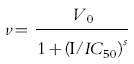 |
where V0 is uninhibited velocity, v is observed velocity, s is slope factor and I is inhibitor concentration.
Inhibition constants (Kis) were not calculated. Although we recognize that ultimately it is Ki values, and not IC50 values, which provide the most valid parameters for comparison, the purpose of this study was to obtain screening data which is important when a high throughput of samples is required.
Results
Km and Vmax determinations for CYP probe substrates
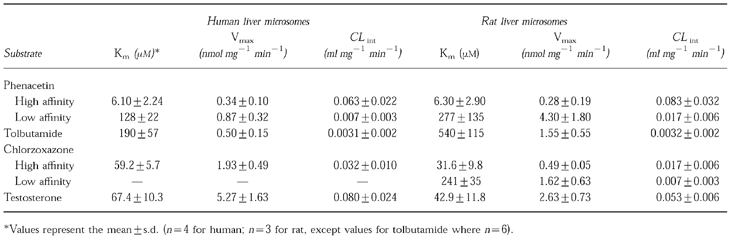
Apparent Km and Vmax values for all four substrates are presented in Table 1. Intrinsic clearance (CLint, Vmax/Km) was also calculated for each substrate in both human and rat liver microsomal incubations (Table 1). Km values in the two species were similar for the high affinity component of phenacetin O-deethylation and testosterone 6βhydroxylation. A greater difference in Km values for tolbutamide hydroxylation was observed between human and rat. A single enzyme model best fitted chlorzoxazone 6-hydroxylation by human liver microsomes. However, data from the rat was best fitted by a two enzyme model (curved Eadie-Hofstee plots indicating the involvement of at least two enzymes), which would indicate differences in the metabolism of chlorzoxazone in these two species.
Table 1.
Apparent Km and Vmax values for phenacetin O-deethylation, tolbutamide 4-hydroxylation, chlorzoxazone 6-hydroxylation and testosterone 6β-hydroxylation in human and rat liver microsomal preparations.
Inhibition studies
To assess the specificity of the cytochrome P450 inhibitors in both rat and human liver microsomal preparations their inhibitory potential against the activities of probe substrates was evaluated (Figures 1 and 2).
Figure 1.
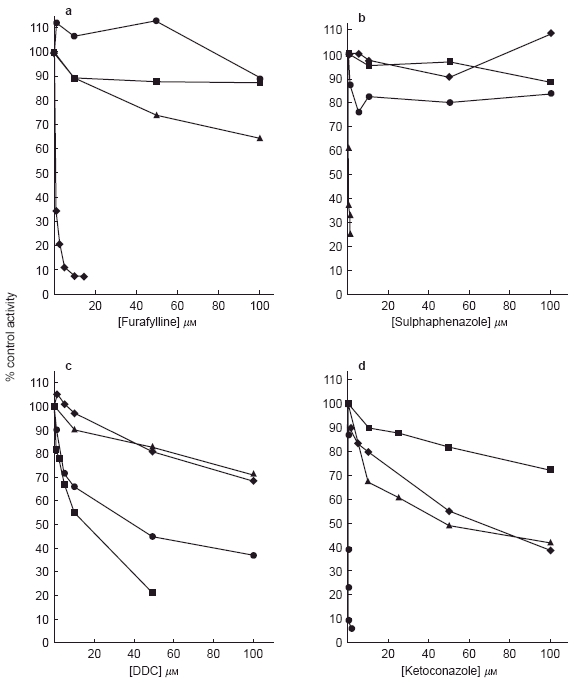
The effects of a) furafylline, b) sulphaphenazole, c) DDC and d) ketoconazole on CYP-mediated reactions in human liver microsomes. Values represent the mean of three determinations. ✦ phenacetin O-deethylation; ▴ tolbutamide 4-hydroxylation; ▪ chlorzoxazone 6-hydroxylation; • testosterone 6β-hydroxylation.
Figure 2.
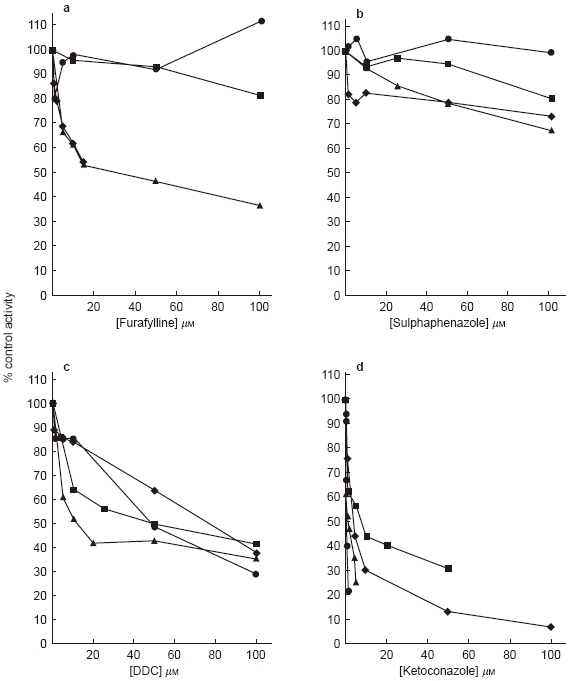
The effects of a) furafylline, b) sulphaphenazole, c) DDC and d) ketoconazole on CYP-mediated reactions in rat liver microsomes. Values represent the mean of three determinations. ✦ phenacetin O-deethylation; ▴ tolbutamide 4-hydroxylation; ▪ chlorzoxazone 6-hydroxylation; • testosterone 6β-hydroxylation.
The IC50 data (Table 2) and Figures 1a and 2a, indicate furafylline to be a potent inhibitor of phenacetin O-deethylase in human liver microsomes (IC50 = 0.48 μm) but a far less potent inhibitor in rat liver microsomes (IC50 = 20.8 μm). In rat liver microsomes furafylline also inhibited tolbutamide 4-hydroxylation with equal potency (IC50 = 24.0 μm).
Table 2.
The effect of selective inhibitors on the metabolism of CYP probe substrates. IC50 values are the mean±s.d. (n = 3 individual livers).
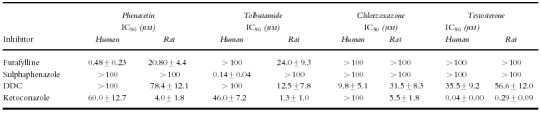
Sulphaphenazole was a highly selective inhibitor of CYP2C9-catalysed tolbutamide metabolism in human liver microsomes and had little effect on any other CYP-dependent reactions over the concentration ranges studied (Table 2; Figures 1b and 2b). However, sulphaphenazole did not inhibit tolbutamide 4-hydroxylation in rat liver microsomes.
DDC, an alleged selective inhibitor of CYP2E1 inhibited the metabolism of chlorzoxazone in human liver microsomes (IC50 = 9.8 μm) but also inhibited the 6β-hydroxylation of testosterone with an IC50 value of 35.5 μm (Table 2, Figures 1c and 2c). In addition, DDC inhibited all CYP-mediated reactions in rat liver microsomes in increasing order of inhibitory potency phenacetin<testosterone<chlorzoxazone<tolbutamide (Table 2).
Ketoconazole, a known selective inhibitor of CYP3A4-mediated metabolism at low concentrations in human liver studies, demonstrated a degree of inhibitory selectivity against testosterone 6β-hydroxylation in human liver microsomes (IC50 = 0.04 μm) and inhibited the metabolism of phenacetin, tolbutamide and chlorzoxazone only at much higher concentrations, with IC50 values of 60.0, 46.0 and >100 μm respectively (Table 2, Figure 1d). In rat liver microsomal incubations, ketoconazole inhibited testosterone 6β-hydroxylation with an IC50 value of 0.29 μm and inhibited all other reactions with a roughly equal degree of potency (Table 2, Figure 2d).
Discussion
It is clear from the present study that there are differences in the kinetic parameters generated with the same substrates in human and rat liver microsomes. This is highlighted with chlorzoxazone metabolism. Although only one isoform appeared to be involved in this biotransformation in human liver microsomal preparations, CYP1A1 has been previously implicated at high substrate concentration; [22]. In addition, other studies on chlorzoxazone metabolism by human liver microsomes and heterologously expressed enzymes have indicated that CYP3A4, is involved but with an identical Km value [23]. At least two isoforms are involved in rat microsomes (as evidenced by curved Eadie-Hofstee plots). Jayyosi et al. [24] have previously reported that CYP2E1 is not the sole catalyst for chlorzoxazone hydroxylation in the rat as demonstrated by enzyme induction, kinetic and immunoinhibition studies. These authors determined that CYP3A was the high affinity component of this reaction with CYP2E1 involvement at higher concentrations.
When screening for specific CYP isoform involvement in the metabolism of a drug, inhibition studies are often conducted with alleged isoform selective inhibitor probes. As highlighted in this study, the effects of inhibitors are not the same in rat and human. Inhibition of the metabolism of a drug may arise as a consequence of a number of mechanisms which include competition for the active site of the enzyme, non-competitive interaction with the enzyme or suicideinactivation of the enzyme. Boobis et al. [25] have proposed three possible reasons for species differences in the effects of chemical inhibitors of drug metabolism in vitro:
the active site is different amongst different species,
metabolism is catalysed by different isoforms in different species,
the inhibition is not via direct competition at the active site, and the inhibitory site is different between the species.
High affinity phenacetin O-deethylation is known to be catalysed by CYP1A2 in both man and rat [15, 26] and therefore the 40-fold difference in the inhibitory potency of furafylline against phenacetin O-deethylation in human and rat liver microsomes observed here must be due to differences in the structure of CYP1A2 in these two species. The two orthologues are 75% homologous [27] which may indicate differences in the active site geometry between human and rat orthologues of CYP1A2. Furafylline is a non-competitive inhibitor of CYP1A2 mediated phenacetin O-deethylation [9], this is consistent with its mechanism based mode of inhibition [8]. Lewis & Lake [26] have modeled both rat and human CYP1A2 on the structure of CYP102 and identified amino acid residues in the CYP1A active site which may be involved in substrate binding. Differences were observed between the rat and human orthologues which may affect the binding of substrates and inhibitors, including furafylline. Our results in human liver are in agreement with Sesardic et al. [9] who demonstrated that furafylline selectively inhibited CYP1A2 and had little effect on CYP2D, CYP2C or CYP3A mediated metabolism.
Sulphaphenazole demonstrated potent, selective inhibition of CYP2C9-mediated tolbutamide hydroxylation in human liver microsomes; this selectivity has been demonstrated previously [28]. However, in rat liver microsomes, sulphaphenazole was much less potent at inhibiting tolbutamide metabolism (IC50 around 100 μm compared to 0.14 μm in human liver microsomes). Veronese et al. [7] have also demonstrated differential inhibition characteristics of sulphaphenazole against tolbutamide metabolism in animal species, with IC50 values being 10-fold higher in rat liver microsomes compared with human microsomes and 3–4 orders of magnitude greater in rabbit liver microsomes. Following pretreatment of rats with phenobarbitone the authors suggested a role for CYP2B1 and/or CYP2B2 in the hydroxylation of tolbutamide. By site directed mutagenesis and cDNA expression studies, differences in the kinetics of tolbutamide hydroxylation by seven CYP2C proteins (including 2C8, 2C9 and variants and 2C10) have been highlighted [28]. Sulphaphenazole was shown to inhibit CYP2C9/10 but had no inhibitory effect on CYP2C8. Results from these authors indicated that subtle differences in the amino acid sequence of the CYP2C9/10 protein can affect the functional specificity of the enzyme towards tolbutamide. An active site template for CYP2C9 has been proposed based on a hydrogen donor/acceptor model and it was shown that sulphaphenazole would fit this model along with alleged CYP2C9 substrates including tolbutamide and phenytoin [29]. It is apparent that sulphaphenazole potently inhibits CYP2C9-mediated tolbutamide metabolism by binding to the active site and that differences in the structure of the isoform responsible for tolbutamide hydroxylation in rat liver alters its inhibitory propensity (with >50% activity remaining at 100 μm concentrations of sulphaphenazole). Even though sulphaphenazole is a highly selective inhibitor of CYP2C9-mediated tolbutamide metabolism in man, it is apparent from the studies of Veronese et al. [7] that tolbutamide is metabolised by other CYP isoforms in the rat against which sulphaphenazole has altered potency.
DDC inhibited all four CYP mediated reactions in rat liver microsomes (IC50 30–80 μm) and actually inhibited tolbutamide metabolism to a greater extent than chlorzoxazone metabolism. In human liver microsomes, DDC inhibited testosterone 6β-hydroxylation in addition to chlorzoxazone metabolism with only a three fold higher concentration of DDC required to reach IC50. It has been suggested previously that DDC is a selective, mechanism based inhibitor of CYP2E1-mediated metabolism in human liver microsomes [11, 32]. However, Chang et al. [33] screened DDC as an inhibitor probe against a panel of 10 individually cDNA expressed CYP isoforms, and at IC50 concentrations against chlorzoxazone metabolism, found that DDC inhibited CYPs 1A1, 1A2, 2A6, 2B6, 2C8, 3A3 and 3A4. Mechanism based inhibitors of CYP usually exhibit a high degree of selectivity requiring metabolism by the target enzyme to intermediates or products which then inactivate the enzyme [34]. From the present study and evidence from the literature it would appear that there are doubts as to the selectivity of DDC as an inhibitor of CYP2E1. DDC is not a suitable inhibitor probe for CYP2E1 in rat liver microsomal studies. A lack of selectivity in the rat could, in part, be due to the concentration of chlorzoxazone used in this inhibitor study, which is lower than the observed Km for CYP2E1, the low affinity component in this reaction (100 μm cf >200 μm) [24]. At this concentration high affinity enzyme (CYP3A) would also have been involved in the metabolism of chlorzoxazone. As mentioned previously Gorski et al (1997) have proposed the involvement of CYP3A4 in the hydroxylation of chlorzoxazone by human liver microsomes and that heterologously expressed CYP3A4 is capable of mediating this biotransformation which may also indicate chlorzoxazone metabolism is a poor marker substrate for CYP2E1 activity in human liver.
Ketoconazole potently inhibited the 6β-hydroxylation of testosterone in human liver microsomes (IC50 = 0.04 μm). Other CYP activities were inhibited at much higher concentrations. Maurice et al. [14] have also demonstrated similar selective inhibition of CYP3A-mediated metabolism of erythromycin and cyclosporin A by ketoconazole. In other studies ketoconazole has inhibited CYP3A4-mediated metabolism with an IC50 at least an order of magnitude lower than those observed for CYP1A2, CYP2B6, 2C9/8, CYP2C19 and CYP2D6 [29]. In rat liver microsomes ketoconazole exhibits a far lesser degree of selectivity, inhibiting all reactions with IC50 values <6 μm. The eight fold difference in the potency of ketoconazole against testosterone 6β-hydroxylation in human and rat liver microsomes could be due to the involvement of other CYP isoforms in the rat. The constitutive 6ß-hydroxylase in male rat liver is CYP3A2 [35] against which ketoconazole may have altered inhibitory potency. Although other isoforms have been shown to possess 6β-hydroxylase activity in the rat, including CYP1A1, CYP1A2 [36] and CYP2C11 [37], this biotransformation fitted best to a one enzyme model.
In conclusion, furafylline, sulphaphenazole and ketoconazole are potent selective inhibitors of human CYP1A2, CYP2C9 and CYP3A4. DDC preferentially inhibits CYP2E1-mediated metabolism in human liver microsomes although with a lower degree of selectivity. It is clear that these inhibitors do not exhibit the same selectivity in rat microsomal studies and this may lead to the incorrect assignment of CYP involvement if rat liver microsomes are used as a model for human drug metabolism. This may be due to differential selectivity of the inhibitors between the two species, when the same isoform is implicated in both (for example phenacetin O-deethylation) or due to the involvement of completely different CYP isoforms in the two species. Caution must be exercised when extrapolating the effects of inhibitors from rat to human and making predictions concerning the involvement of CYP isoforms.
Acknowledgments
We are grateful to the Medical Research Council for financial support.
References
- 1.Birkett DJ, Mackenzie PI, Veronese ME, Miners JO. In vitro approaches can predict human drug metabolism. Trends Pharmacol Sci. 1993;4:292–294. doi: 10.1016/0165-6147(93)90043-j. [DOI] [PubMed] [Google Scholar]
- 2.Wrighton SA, Vandenbranden M, Stevens JC, Shipley LA, Ring BR. In vitro methods for assessing human hepatic drug metabolism: Their use in drug development. Drug Metab Rev. 1993;25:453–483. doi: 10.3109/03602539308993982. [DOI] [PubMed] [Google Scholar]
- 3.Paine AJ. Heterogeneity of cytochrome P450 and its toxicological significance. Human Exp Toxicol. 1995;14:1–7. doi: 10.1177/096032719501400101. [DOI] [PubMed] [Google Scholar]
- 4.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich Guengerich, FP FP. Interindividual variations in human liver cytochrome P450 enzymes involved in the oxidation of drugs, carcinogenes and toxic chemicals. Studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 5.Nedelcheva D, Gut I. P450 in the rat and man: methods of investigation, substrate specificities and relevance to cancer. Xenobiotica. 1994;24:1151–1175. doi: 10.3109/00498259409038673. [DOI] [PubMed] [Google Scholar]
- 6.Guengerich FP, Dannan GA, Wright ST, Martin MV, Kaminski LS. Purification and characterization of liver microsomal cytochromes P450: Electrophoretic, spectral, catalytic and immunochemical properties and inducibility of eight isozymes isolated from rats treated with phenobarbital and alpha-naphthoflavone. Biochem. 1982;21:6019–6031. doi: 10.1021/bi00266a045. [DOI] [PubMed] [Google Scholar]
- 7.Veronese ME, McManus ME, Laupattarakasem P, Miners JO, Birkett DJ. Tolbutamide hydroxylation by human, rabbit and rat liver microsomes and by purified forms of cytochrome P450. Drug Metab Dispos. 1990;18:356–361. [PubMed] [Google Scholar]
- 8.Kunze KL, Trager WF. Isoform-selective mechanism-based inhibition of human cytochrome P450 1A2 by furafylline. Chem Res Toxicol. 1993;6:649–656. doi: 10.1021/tx00035a009. [DOI] [PubMed] [Google Scholar]
- 9.Sesardic D, Boobis AR, Murray BP, et al. Furafylline is a potent and selective inhibitor of cytochrome P4501A2 in man. Br J Clin Pharmacol. 1990;29:651–663. doi: 10.1111/j.1365-2125.1990.tb03686.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Halpert JR, Guengerich FP, Bend JR, Correia MA. Selective inhibitors of cytochromes P450. Toxicol Appl Pharmacol. 1994;125:163–175. doi: 10.1006/taap.1994.1061. [DOI] [PubMed] [Google Scholar]
- 11.Guengerich FP, Kim DH, Iwasaki M. Role of human cytochrome P450 IIE1 in the oxidation of many low molecular weight cancer suspects. Chem Res Toxicol. 1991;4:168–179. doi: 10.1021/tx00020a008. [DOI] [PubMed] [Google Scholar]
- 12.Sheets JJ, Mason JI. Ketoconazole: a potent inhibitor of cytochrome P-450-dependent drug metabolism in rat liver. Drug Metab Dispos. 1984;12:603–606. [PubMed] [Google Scholar]
- 13.Meredith CG, Maldonado AL, Speeg KV. The effect of ketoconazole on hepatic oxidative drug metabolism in the rat in vivo and in vitro. Drug Metab Dispos. 1985;13:156–162. [PubMed] [Google Scholar]
- 14.Maurice M, Pichard L, Daujat M, et al. Effects of imidazole derivatives on cytochromes P-450 from human hepatocytes in primary culture. FASEB. 1992;6:752–758. doi: 10.1096/fasebj.6.2.1371482. [DOI] [PubMed] [Google Scholar]
- 15.Tassaneeyakul W, Birkett DJ, Veronese ME, et al. Specificity of substrate and inhibitor probes for human cytochromes 1A1 and 1A2. J Pharmacol Exp Ther. 1993;265:401–407. [PubMed] [Google Scholar]
- 16.Relling MV, Aoyama T, Gonzalez FJ, Meyer UA. Tolbutamide and mephenytion hydroxylation by human cytochrome P450s in the CYP2C subfamily. J Pharmacol Exp Ther. 1990;252:442–447. [PubMed] [Google Scholar]
- 17.Peter R, Bocker R, Beaune PH, Iwasaki M, Guengerich FP, Yang CS. Hydroxylation of chlorzoxazone as a specific probe for human liver cytochrome P-450IIE1. Chem Res Toxicol. 1990;3:566–573. doi: 10.1021/tx00018a012. [DOI] [PubMed] [Google Scholar]
- 18.Waxman DJ, Attisano C, Guengerich FP, Lapenson DP. Human liver microsomal steroid metabolism. Identification of the major microsomal steroid 6β hydroxylase cytochrome P450 enzyme. Arch Biochem Biophys. 1998;290:160–166. doi: 10.1016/0003-9861(88)90655-8. [DOI] [PubMed] [Google Scholar]
- 19.Purba HS, Maggs JL, Orme ML’E, Back DJ, Park BK. The metabolism of 17α-ethinyloestradiol by human liver microsomes: formation of catechol and chemically reactive metabolites . Br J Clin Pharmacol. 1987;23:447–453. doi: 10.1111/j.1365-2125.1987.tb03074.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein determination with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 21.Back DJ, Tjia JF, Karbwang J, Colbert J. In vitro inhibition studies of tolbutamide hydroxylase activity of human liver microsomes by azoles, sulphonamides and quinolines. Br J Clin Pharamacol. 1998;26:23–29. doi: 10.1111/j.1365-2125.1988.tb03359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carriere V, Goasduff T, Ratanasavanh D, Morel F. Both cytochromes P4502E1 and 1A1 are involved in the metabolism of chlorzoxazone. Chem Res Toxicol. 1993;6:852–857. doi: 10.1021/tx00036a015. [DOI] [PubMed] [Google Scholar]
- 23.Gorski JC, Jones DR, Wrighton SA, Hall SD. Contribution of human CYP3A subfamily members to the 6-hydroxylation of chlorzoxazone. Xenobiotica. 1997;27:243–256. doi: 10.1080/004982597240578. [DOI] [PubMed] [Google Scholar]
- 24.Jayyosi Z, Knoble D, Muc M, Eric J, Thomas PE, Kelley M. Cytochrome P450 2E1 is not the sole catalyst of chlorzoxazone hydroxylation in rat liver microsomes. J Pharmacol Exp Ther. 1995;273:1156–1161. [PubMed] [Google Scholar]
- 25.Boobis AR, Sesardic D, Murray BP, et al. Species variation in the response of the cytochrome P450-dependent monooxygenase system to inducers and inhibitors. Xenobiotica. 1990;20:1139–1161. doi: 10.3109/00498259009046835. [DOI] [PubMed] [Google Scholar]
- 26.Sesardic D, Edwards RJ, Davies DS, et al. High affinity O-deethylase is catalysed specifically by cytochrome P450d (P450IA2) in the liver of the rat. Biochem Pharmacol. 1990;39:489–498. doi: 10.1016/0006-2952(90)90055-p. [DOI] [PubMed] [Google Scholar]
- 27.Soucek P, Gut I. Review. Cytochromes P-450 in rats: Structures, functions, properties and relevant human forms. Xenobiotica. 1992;22:83–103. doi: 10.3109/00498259209053106. [DOI] [PubMed] [Google Scholar]
- 28.Lewis DFV, Lake BG. Molecular modelling of CYP1A subfamily members based on sequence alignment with CYP102: rationalisation of CYP1A substrate specificity in terms of active site amino acid residues. Xenobiotica. 1996;26:723–753. doi: 10.3109/00498259609046745. [DOI] [PubMed] [Google Scholar]
- 29.Baldwin JJ, Bloomer JC, Smith GJ, Ayrton AD, Clarke SE, Chenery RJ. Ketoconazole and sulphaphenazole as the respective selective inhibitors of P4503A and 2C9. Xenobiotica. 1995;25:261–270. doi: 10.3109/00498259509061850. [DOI] [PubMed] [Google Scholar]
- 30.Veronese ME, Doecke CJ, Mackenzie PI, et al. Site directed mutagenesis of human liver cytochrome P450 isozymes in the CYP2C subfamily. Biochem J. 1993;289:533–538. doi: 10.1042/bj2890533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jones BC, Hawksworth G, Newlands A, Morsman J, Tute MS, Smith DA. Putative active site template model for Cytochrome P4502C9 (tolbutamide hydroxylase) Drug Metab Dispos. 1996;24:260–266. [PubMed] [Google Scholar]
- 32.Newton DJ, Wang RW, Lu AYH. Cytochrome P450 inhibitors. Evaluation of specificities in the in vitro metabolism of therapeutic agents by human liver microsomes. Drug Metab Dispos. 1995;23:154–158. [PubMed] [Google Scholar]
- 33.Chang TKH, Gonzalez FJ, Waxman DJ. Evaluation of triacetyloleandomycin, α-naphthoflavone and diethyldithiocarbamate as selective chemical probes for inhibition of human cytochrome P450. Arch Biochem Biophys. 1994;311:437–442. doi: 10.1006/abbi.1994.1259. [DOI] [PubMed] [Google Scholar]
- 34.Halpert JR. Structural basis of selective cytochrome P450 inhibition. Ann Rev Pharmacol Toxicol. 1995;35:29–53. doi: 10.1146/annurev.pa.35.040195.000333. [DOI] [PubMed] [Google Scholar]
- 35.Imaoka S, Terano Y, Funae Y. Constitutive 6β-hydroxylase in rat liver. J Biochem. 1988;104:481–487. doi: 10.1093/oxfordjournals.jbchem.a122494. [DOI] [PubMed] [Google Scholar]
- 36.Wood AW, Ryan DE, Thomas PE, Levin W. Regio- and stereoselective metabolism of two C19 steroids by five highly purified and reconstituted rat hepatic cytochrome P-450 isozymes. J Biol Chem. 1983;14:8839–8847. [PubMed] [Google Scholar]
- 37.Sonderfan AJ, Arlotto MP, Dutton DR, McMillen SK, Parkinson A. Regulation of testosterone hydroxylation by rat liver microsomal cytochrome P-450. Arch Biochem Biophys. 1987;255:41. doi: 10.1016/0003-9861(87)90291-8. [DOI] [PubMed] [Google Scholar]