

Abstract
Aims
To investigate prehepatic metabolism of verapamil and its inducibility by rifampicin in older subjects.
Methods
Eight older subjects (67.1±1.2 years mean±s.d.) received racemic, unlabelled verapamil orally for 16 days (120 mg twice daily). Rifampicin (600 mg daily) was coadministered from day 5 to 16. Using stable isotope technology (i.e. intravenous coadministration of 10 mg deuterated verapamil) during verapamil steady-state without (day 4) and with rifampicin (day 16) bioavailability, prehepatic and hepatic extraction of verapamil were determined. The effects of verapamil on AV-conduction were measured by the maximum PR interval prolongation (%).
Results
Bioavailability of the cardiovascularly more active S-verapamil decreased from 14.2±4.3% on day 4 to 0.6±0.5% on day 16 (P<0.001). As a consequence, effects of orally administered verapamil on the AV-conduction were nearly abolished (14.4±9.4%vs 2.7±2.6%, P<0.01). This could be attributed to a considerable increase of prehepatic extraction during treatment with rifampicin (41.7±22.1%vs 91.6±6.6%, P<0.01) and to a minor extent to induction of hepatic metabolism (73.7±9.4%vs 91.6±5.3%, P<0.01).
Conclusions
Prehepatic metabolism of verapamil occurred in the group of older people investigated. Induction of gut wall metabolism most likely was the major reason for the loss of verapamil effect during treatment with rifampicin in this group of older subjects.
Keywords: verapamil, rifampicin, prehepatic metabolism, older people, enzyme induction, pharmacokinetics, pharmacodynamics
Introduction
The fraction of people older than 65 years in the total population of developed countries is increasing rapidly [1]. Older people consume on average more drugs than the young. Drug action and disposition, however, can be altered in the aged patient. The β-adrenoceptor blocker propranolol, which undergoes substantial first pass metabolism in the young, shows a nearly twofold increased bioavailability in older subjects [2]. Thus, it is essential to identify the factors influencing drug effects in older people in order to recognize the need for dose adjustments [3], for example with cardiovascularly active medications, which are among the most commonly used drugs in the older people [4]. While the influence of age on hepatic drug metabolism [5] and on renal drug excretion [6] has been intensively studied, the relevance of intestinal drug metabolism in older people has not been investigated.
During recent years it has been realized that not only hepatic metabolism but also gut wall metabolism can contribute to first pass elimination after oral drug administration (e.g. with cyclosporin, verapamil, midazolam, [7–10]). Drug metabolizing enzymes have been localized in the gut wall mucosa (e.g. CYP3A4, [11–17]). Age-related alterations in the amount of drug metabolizing enzymes in the gut wall mucosa and in splanchnic blood flow could affect first pass metabolism and therefore alter bioavailability of orally administered drugs. As in the liver, gut wall metabolism is modified both by enzyme induction (e.g. rifampicin, [8, 9, 17]) and by enzyme inhibition (e.g. ketoconazole, [18]). We recently identified induction of prehepatic metabolism by rifampicin as the major reason for a pronounced decrease in bioavailability and hence drug effect of verapamil in young subjects [9]. To our knowledge, inducibility of prehepatic drug metabolism in older subjects has not been studied so far. By using stable isotope technology (i.e. simultaneous administration of deuterium labelled verapamil i.v. and unlabelled verapamil orally), gastrointestinal extraction and hepatic extraction of verapamil can be determined simultaneously [19].
The calcium channel blocker verapamil (S/R-verapamil), which is used for the treatment of hypertension, angina pectoris and supraventricular arrhythmias, was studied in this investigation, since it is one of the most commonly prescribed drugs in the older patients [4]. Its pharmacokinetics and pharmacodynamic effects have been determined in detail in young and older subjects [20–32]. Verapamil is subject to an extensive stereoselective first pass metabolism with the cardiovascularly more active S-enantiomer being cleared more rapidly than R-verapamil [21, 33]. The hepatic metabolism of verapamil is catalyzed by CYP3A4, CYP2C and CYP1A2 [34–36].
In this study, the verapamil-rifampicin interaction [9, 37] was used for evaluation of possible age-dependent changes in inducibility of drug metabolism [38, 39] and to characterize the importance of intestinal drug metabolism in older subjects.
Methods
Study population
Eight healthy, male, nonsmoking older volunteers were included in this study (age: 67.1±1.2 years, body weight: 83.5±9.9 kg, creatinine clearance: 118±39 ml min−1, mean±s.d.). The study protocol was approved by the ethics committee of the Robert Bosch Hospital (Stuttgart, Germany) according to the ethical guidelines of the 1975 Declaration of Helsinki. Prior to the study, volunteers were informed by a physician about the aim, course and possible risks of the study, and a written informed consent was obtained from all subjects. A physical examination and laboratory tests were carried out before and after completion of the study.
Study design
Subjects received orally 120 mg racemic (S/R), unlabelled verapamil HCl twice daily for 16 days (one dose in the morning, one dose in the evening; Isoptin KHK retard, Knoll AG, Ludwigshafen, Germany). A daily dose of 600 mg rifampicin (Rifa, Grünenthal GmbH, Stolberg, Germany) was coadministered in the evening from day 5 to day 16. Pharmacokinetics and pharmacodynamics were studied after an overnight fast during verapamil steady-state without comedication (day 4) and during verapamil steady-state plus rifampicin (day 16). On day 4 and day 16 the following identical procedure was carried out: deuterated, racemic verapamil was injected intravenously over 10 min (d7-S/R-verapamil; obtained from Knoll AG, Ludwigshafen, Germany) and 2 h later each volunteer received 120 mg unlabelled verapamil. On study days 4 and 16, no evening verapamil dose was administered. 10 ml of blood was taken before i.v. administration and after 5, 10, 20, 30, 40 min and 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 14, 24 and 26 h. In parallel, ECGs and blood pressure (automated Dinamap 1846SX, Critikon GmbH, Norderstedt, Germany) were recorded. The volunteers were instructed about refraining from ingestion of grapefruit juice, methylxanthine containing substances (e.g. coffee) and alcohol on the day before pharmacokinetics of verapamil were studied and during these study days. Additional blood samples were taken on day 8 and day 12 for determination of verapamil trough levels.
Determination of labelled and unlabelled verapamil enantiomers
Labelled and unlabelled verapamil enantiomers were quantified in serum by means of chiral h.p.l.c. and GC/MS as previously described [9]. In brief, the S- and the R-enantiomers were separated by a modified h.p.l.c. method [40] using a Daicel AD column with a mean silica particle size of 10 μm (25×0.46 cm, purchased from Baker Chemikalien, Groß-Gerau, Germany), a mobile phase of hexane : 2-propanol (95:5, v/v; flow rate: 0.4 ml min−1) and a fluorescence detector (excitation: 273 nm, emission: 320 nm, Fluorescence Monitor 8400, Bischoff, Leonberg, Germany). After separation of the enantiomers, the d0- and d7-isotopes were quantified by GC/MS [41] using trideuterated verapamil as an internal standard (20 ng of each enantiomer; obtained from Knoll AG, Ludwigshafen, Germany). Quality control samples for S- and R-verapamil were routinely measured with intra-assay and inter-assay coefficients of variation of less than 5% and 10%, respectively. The limit of quantification was 0.25 ng ml−1 for both S- and R-verapamil.
Verapamil and other basic drugs bind in blood to α1-acid glycoprotein [42] and therefore alterations of α1-acid glycoprotein concentrations might influence drug effects. In order to determine the influence of rifampicin on α1-acid glycoprotein concentrations, serum samples were analyzed before drug administration and on day 16 using turbidimetry (Turbiquant, Behringwerke AG, Marburg, Germany).
Pharmacokinetic evaluation
The areas under the serum concentration-time curves (AUC) of intravenously administered verapamil enantiomers (AUCiv) and of orally administered verapamil enantiomers (AUCpo, 2, 14 h) were calculated by the trapezoidal rule with and without extrapolation to infinity, respectively. Systemic serum clearance (CL) of S- and R-verapamil was obtained from CL=(dose of enantiomer)/AUCiv and apparent oral clearance (CLO) was calculated by CLO=(dose of enantiomer)/AUCpo. Bioavailability (F) of the verapamil enantiomers was determined by F=(AUCpodoseiv)/(AUCivdosepo). Volume of distribution (VSS) was calculated by VSS=(doseAUMC)/AUC2 with AUMC being the area under the first-moment curve [43]. Elimination half-life (t1/2) was estimated by linear regression of the log transformed serum concentrations vs time relations in terminal elimination phase after i.v. administration.
Since verapamil is completely absorbed [44], its bioavailability depends on the fraction of verapamil, which is not metabolized in the gastrointestinal mucosa (1-EGI) and on the fraction of verapamil in the portal vein, which is not metabolized by the liver (1-EH) [43]:
Thus, prehepatic extraction of verapamil enantiomers can be estimated after rearrangement of the formula by EGI= 1-F/(1-EH). For example, a bioavailability of 40% (F=0.4) of a completely absorbed drug could be caused in the absence of gut wall metabolism (EGI=0) by an hepatic extraction of 60% (EH=0.6). The same bioavailability (F=0.4), however, could also be the result of a prehepatic extraction of 37% (EGI=0.37) and a subsequent hepatic extraction of 37% (EH=0.37). The hepatic extraction of verapamil enantiomers (EH) was calculated as previously described [9] by EH=CLblood/QH (assuming that no extrahepatic metabolism of verapamil occurs after i.v. administration) with CLblood being the blood clearance {obtained from CLblood=CL/(blood/serum verapamil concentration ratio), [9, 42, 45]} and with QH being the liver blood flow for each volunteer {QH=(19.3 ml min−1kg−1 body weight)body weight for the older subjects; modified from [46]}. The assumption of neglegible extrahepatic metabolism after intravenous administration is based on the following lines of evidence. First, the total mass of drug metabolizing enzymes (e.g. CYP3A) is much higher in the liver than in any other organ. Second, gut wall metabolism (e.g. by CYP3A) of an intravenously administered drugappears to play an insignificant role due to anatomical reasons. The drug has to be transported from the intestinal capillaries to the apical site of the enterocytes, where CYP3A is primarily located. Finally, there are experimental findings supporting these theoretical assumptions. It was shown that the intestinal extraction of the CYP3A substrate midazolam after intravenous administration contributed only 8% to overall clearance of the drug [10].
Statistical analysis
All data are presented as mean±s.d. The data before and during administration of rifampicin were compared by paired t-tests. Trough levels of both enantiomers on days 4, 8, 12 and 16 were analyzed by repeated measures ANOVA. The serum concentration-effect relationships were calculated for the first 2 h after i.v. administration of verapamil on day 4 using linear regression analysis. If a hysteresis effect was observed, the concentration-effect relationship was calculated from the offset of peak effect. The negative dromotropic activity of racemic verapamil resides mainly in the S-enantiomer [22, 23]. Thus, serum concentrations of unlabelled plus labelled S-verapamil were correlated (by linear regression) with PR interval prolongation (%, compared with baseline). Moreover, a linear regression analysis was calculated for the serum concentrations of labelled and unlabelled (R+S)-verapamil and the reduction of mean arterial pressure (%, compared with baseline).
Results
Pharmacokinetics and pharmacodynamic effects of verapamil
The mean serum concentration-time curves of S- and R-verapamil after intravenous administration of S/R-verapamil on day 4 (before rifampicin) and on day 16 (during rifampicin) are shown in Figure 1. Systemic clearance of both enantiomers significantly increased during induction (S-verapamil: from 1149±146 ml min−1 to 1486±209 ml min−1, P<0.001; R-verapamil: from 599±74 ml min−1 to 1107±173 ml min−1, P<0.001; Figure 2). The AUC, volume of distribution and terminal half-life of both enantiomers are shown in Table 1. A considerable reduction of stereoselectivity was observed during induction (R/S-ratio day 4 vs day 16: 1.9±0.2 vs 1.4±0.1, P<0.001, Table 1).
Figure 1.

Mean serum concentration-time curves of a) intravenously (10 mg d7-S/R-verapamil) and b) orally (120 mg d0-S/R-verapamil) administered verapamil before (day 4; • S-verapamil, ▴ R-verapamil) and during (day 16, ○ S-verapamil; ▵ R-verapamil) treatment of eight older subjects with rifampicin. No evening dose of oral verapamil was administered on day 4 and day 16
Figure 2.

Systemic clearance (CL) and apparent oral clearance (CLO) of S- and R-verapamil after intravenous (10 mg d7-S/R-verapamil) and oral (120 mg d0-S/R-verapamil) administration of verapamil to the older subjects before and during treatment with rifampicin. Individual (•) and mean data (○,±s.d.) are shown (*P<0.01 before vs during rifampicin)
Table 1.
Pharmacokinetics (mean±s.d.) of intravenously (10 mg d7-S/R-verapamil) and orally (120 mg d0-S/R-verapamil) administered verapamil in older and young [9] subjects before (day 4) and during (day 16) treatment with rifampicin
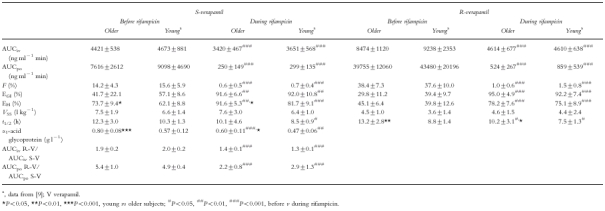
Major differences in pharmacokinetics were observed after oral administration of S/R-verapamil, when data before and during rifampicin treatment were compared. The mean serum concentration-time curves are shown in Figure 1. The respective pharmacokinetic data are given in Figures 2–4 and in Table 1. The apparent oral clearance of S- and R-verapamil increased from 8.9±3.5 l min−1 (day 4) to 299.4±134.9 l min−1 (day 16, P<0.001) and from 1.6±0.5 l min−1 to 149.6±86.2 l min−1 (P<0.01), respectively. As observed after intravenous administration, stereoselectivity of verapamil metabolism was reduced during treatment with rifampicin (R/S-ratio day 4 vs day 16: 5.4±1.0 vs 2.2±0.8, P<0.001, Table 1).
Figure 4.

Mean trough levels (±s.d.) of S- (○) and R-verapamil (▴; 120 mg d0-S/R-verapamil twice daily) before (day 4) and during (day 8, 12, 16) administration of rifampicin to eight older subjects (*P<0.001 day 4 vs days 8, 12, 16)
Bioavailability of S-verapamil decreased from 14.2% before rifampicin to 0.6% during rifampicin (P<0.001; R-verapamil: 38.4% to 1.0%, P<0.001; Figure 3, Table 1). The decrease in bioavailability was predominantly due to an increase in the prehepatic extraction of verapamil enantiomers (EGI, S-verapamil: from 41.7% to 91.6%, P<0.01; R-verapamil: from 29.8% to 95.0%, P<0.001, Table 1), whereas the hepatic extraction (EH) increased only moderately (S-verapamil: from 73.7% to 91.6%, P<0.01; R-verapamil: from 45.1% to 78.2%, P<0.001; Table 1).
Figure 3.

Bioavailability of S- and R-verapamil (10 mg d7-S/R-verapamil i.v., 120 mg d0-S/R-verapamil p.o.) and maximum PR interval prolongation (i.v.: 0–2 h, p.o.: 2–26 h) in the older subjects before and during treatment with rifampicin. Individual (•) and mean data (○,±s.d.) are shown. (*P<0.01 before vs during rifampicin)
The time course of S- and R-verapamil trough levels during the study is shown in Figure 4. After only 4 days of treatment with rifampicin trough levels of both verapamil enantiomers were grossly altered (S-verapamil day 4: 14.1±10.5 ng ml−1, day 8: 0.8±0.3 ng ml−1, P<0.001; R-verapamil day 4: 66.7±26.9 ng ml−1, day 8: 2.8±0.7 ng ml−1, P<0.001).
The PR interval prolongation was reduced after intravenous administration on day 16 when compared with day 4 (day 4 vs day 16: 17.0±5.8%vs 11.4±4.8%, P<0.01). On day 16, drug effect on AV conduction was nearly abolished after oral administration of verapamil (day 4 vs day 16: 14.4±9.4%vs 2.7±2.6%, P<0.01, Figure 3). A positive correlation was found between serum concentrations of S-verapamil (labelled+unlabelled) and PR interval prolongation (r=0.86, P<0.05). Maximum reduction of mean arterial pressure compared with pretreatment data was −16.1±5.6%. Serum concentrations of S/R-verapamil were negatively correlated with reduction of mean arterial pressure (r=−0.68, P<0.05).
Discussion
The results of the present investigation highlight the importance of gut wall metabolism after oral drug administration in this group of older people. In addition to drug metabolism in liver, prehepatic extraction by the gut wall contributed to first pass elimination of both verapamil enantiomers. Moreover, gut wall metabolism was increased by rifampicin in these older subjects, causing a major reduction in bioavailability of S- and R-verapamil and a loss of drug effect after oral administration of verapamil.
Our observations of gut wall metabolism in these older subjects are in accordance with previous findings. In addition to a significant and inducible prehepatic metabolism of verapamil in young subjects [9], gut wall metabolism occurs with cyclosporin, midazolam and rifampicin [8, 10, 47, 48]. Similarly to observations in the liver, the gut wall metabolism of cyclosporin was altered both by enzyme induction (rifampicin, [8]) and by enzyme inhibition (ketoconazole, erythromycin; [18, 49]). Metabolism of cyclosporin was also inhibited by grapefruit juice after oral but not after intravenous administration of the immunosuppressant, indicating that the prehepatic metabolism of cyclosporin was affected [50]. The possible advantages of using this interaction clinically (e.g. reduction of costs for cyclosporin therapy after renal transplantation) have been dicussed elsewhere [50]. Similarly, the bioavailability of orally administered verapamil was increased during intake of grapefruit juice, most likely by inhibition of gut wall metabolism [51]. These pharmacokinetic data are in accordance with the presence of cytochrome P450 enzymes (e.g. CYP3A4) in the human small bowel [12–16]. CYP3A4, which is involved in the hepatic metabolism of verapamil and cyclosporin, is present in the duodenum and has been shown to be inducible by rifampicin in small bowel [12]. Thus, induction of CYP3A4-mediated metabolism of verapamil in small bowel is likely to be a major reason for the low bioavailability of S- and R-verapamil during treatment with rifampicin. However, other enzymes in the gut wall mucosa (e.g. CYP2C) might also contribute to the prehepatic metabolism of verapamil before and during induction. In this study gut wall metabolism was shown for a sustained release verapamil tablet. The extent of prehepatic metabolism of an immediate release tablet of verapamil might be different (especially without rifampicin) due to an altered concentration-time profile within the enterocytes and due to differences in the sites of absorption (with different expression of drug metabolizing enzymes). Moreover, blood flow alterations in gut wall or liver by rifampicin could affect pharmacokinetics of verapamil. We have, however, previously shown in young subjects that blood flow in the portal vein and in a hepatic vein is not altered by rifampicin [9]. We therefore assume that our observations in this study are not due to altered gut wall or hepatic blood flow. It should be noted that an error in the assumption of negligible extrahepatic metabolism after intravenous drug administration would lead to overestimation of hepatic extraction and, thus, underestimation of intestinal extraction, which would not alter the major findings of our study.
Prior to induction, the major fraction of verapamil was extracted in the liver, which is in agreement with previous investigations [9]. The low bioavailability of verapamil during treatment with rifampicin might also be caused by a decreased absorption of verapamil from gut lumen. Rifampicin is very unlikely, however, to alter absorption of verapamil, since cumulative excretion of parent compound and its metabolites was not altered during induction when compared with administration of verapamil alone [9].
The identical study design of this investigation in older people and our recently published study [9] in young, healthy volunteers (32.0±2.8 years, from day 1 to day 16) provides a good basis for evaluating age-related changes of prehepatic metabolism of verapamil and its inducibility by rifampicin for the specific age groups studied. It should, however, be noted that this inter-study comparison was not the primary aim of our investigation and further prospective studies are required to confirm these results. Clearance, apparent oral clearance, bioavailability and gastrointestinal extraction of S- and R-verapamil were not different between both age-groups (Table 1). Whereas gut wall metabolism in the older subjects has not been investigated so far, there have been studies on the influence of age on the hepatic metabolism of verapamil with somewhat contradictory results. After intravenous administration the systemic clearance is assumed to reflect hepatic drug metabolism. Abernethy et al. [25] found a significant reduction of systemic clearance of S- and R-verapamil (−25%; 20 mg racemic verapamil i.v. over 30 min) in healthy, older volunteers in comparison with younger subjects. In hypertensive, older patients, the same group reported a significant reduction of systemic clearance of racemic verapamil (−32%; 10 mg racemic verapamil i.v. over 10 min) when data were corrected for body weight [24]. Schwartz et al. [27, 28] detected no significant reduction in systemic clearance of R-verapamil after a single administration of the enantiomer (10–11 mg kg−1 over 15 min, CL in ml min−1 kg−1) and during multistage infusions of the racemate (CL in l h−1) in healthy, older subjects. In the same studies systemic clearance of S-verapamil was significantly decreased in the older people (−35%, [27, 28]). We did not observe any alteration of the systemic clearance of either S- or R-verapamil in the older subjects when data were compared with young subjects indicating that hepatic metabolism of verapamil was not affected between our age-groups.
Contradictory results have been also reported for the influence of age on the oral clearance of verapamil (which reflects age-related changes in the gut wall and hepatic metabolism). Abernethy et al. {chronic administration of a immediate-release preparation (120 mg three times daily) and a sustained-release formulation (240 mg daily), [25]} and Gupta et al. (chronic administration of 180 mg daily for 5 days, [32]) found a significant reduction of oral clearance of both enantiomers in healthy, older volunteers during steady-state conditions (−30% to −51%, CL in ml min−1), when compared with the respective data for the young. Similar to these studies, in a Japanese population, the apparent oral clearance of verapamil was also reduced in healthy, older people after administration of a single dose (−69%, 80 mg, CL in l h−1 kg−1, [31]). No differences, however, between apparent oral clearance of racemic verapamil in young and older people were observed by Ahmed et al. (240/360 mg daily for at least 1 week, [30]). Peak plasma concentration and bioavailability were not different between the young and the older patients after a single dose of 120 mg racemic verapamil [24]. The pharmacokinetics of orally administered verapamil during steady-state conditions were not altered in older subjects in our studies (CLO older people vs young[9]: S: 8.9±3.5 l min−1vs 8.6±4.9 l min−1, NS; R: 1.6±0.5 l min−1vs 1.7±0.9 l min−1, NS) confirming the results of the latter studies and indicating preserved gut wall metabolism (EGI older people vs young [9]: S: 41.7±22.1%vs 57.1±8.6%, NS; R: 29.8±11.2%vs 39.4±9.7%, NS) and hepatic metabolism in our group of older subjects. Due to the use of a very sensitive GC/MS system, we were able to quantify S- and R-verapamil after both intravenous and oral administration in all volunteers up to the last time point of blood sampling (26 h after the start of infusion). The tenfold lower limit of quantification of our analytical system in comparison with the frequently used h.p.l.c. methods provides a sound basis for studying possible age-related differences, which have not been detected in our study populations. In addition to the different analytical methods used, explanation of the discrepancies between our findings and some of the studies mentioned above has to take into account the differences in study populations, verapamil preparations and study design. It should be noted, however, that the average ages of the study populations of our investigation and of the other studies are in a very close range (67 to 71 years). Thus, age of these study populations is unlikely to contribute to the observed differences in pharmacokinetics of verapamil between some of these investigations.
One possible factor influencing the hepatic drug metabolism in older people could be a decrease in the protein content or function of the drug metabolizing enzymes in the liver. Data from several studies indicate, however, that the content of CYP3A-, CYP1A2- and CYP2C-proteins in the liver is not affected by age [52 (for histologically normal livers), 53, 54]. Moreover, CYP3A function assessed by the erythromycin N-demethylation in human liver microsomes [55] and by the erythromycin breath test [56] was not altered in the older subjects. Other determinants of hepatic drug metabolism are liver blood flow and liver weight (total enzyme content=enzyme content g−1 liverliver weight), which are reduced in older people in comparison with young subjects. In our study, however, systemic clearance of verapamil was not impaired in the older volunteers, indicating that these parameters did not have major impact on the pharmacokinetics of verapamil. To our knowledge there are until now no data on age-dependent expression and function of drug metabolizing enzymes in the gut wall mucosa.
Whereas the pharmacokinetics of verapamil enantiomers were not affected by age, there might be an altered response to verapamil in the older people. The maximum PR interval prolongation after intravenous and oral administration, however, was not different between the young and the older subjects at similar concentrations of the more active S-enantiomer (Figure 3 and [9]). As previously reported [23] a correlation was observed between the concentrations of S-verapamil and PR interval prolongation. The concentration-effect relationship, however, was not altered in the older people, which is in contrast to previous studies [25, 26]. In accordance with data published by Gupta et al. [32], the decrease of mean arterial pressure was negatively correlated with the total verapamil concentration with no differences in the response between the older subjects and the young ([9], data not shown).
In addition to the determination of the influence of age on the response to verapamil and on its pharmacokinetics, we investigated the influence of age on the inducibility of drug metabolism, which has been a field of intensive research in the past (for review [38, 39]). Depending on the inducing agent investigated and on the substrate evaluated, inducibility of drug metabolism was impaired [57] or remained preserved in the older people [58]. According to our data, inducibility of the prehepatic and the hepatic metabolism of verapamil by rifampicin was not different between the young[9] and our group of older people (CLO in the older subjects vs the young during rifampicin: S: 299±135 l min−1vs 237±98 l min−1, NS; R: 150± 86 l min−1vs 92±46 l min−1, NS; CL: S: 1486± 209 ml min−1vs 1398±203 ml min−1, NS; R: 1107±173 ml min−1vs 1049±174 ml min−1, NS), indicating that reduced drug effects are to be expected in both groups due to the rifampicin induced drug metabolism.
With racemic drugs, effects do not necessarily depend on total [i.e. (R+S)-enantiomer] serum concentrations. The aspect of stereoselective drug metabolism has to be taken into account if the enantiomers differ in their pharmacodynamic properties (e.g. with verapamil, propafenone, [59]). The induction caused a significant decrease in the R/S-ratio of verapamil in serum after intravenous and even more pronounced after oral administration. In vitro experiments with human liver microsomes showed only moderate stereoselectivity for the CYP3A4-mediated verapamil metabolism, but a considerable stereoselectivity for the CYP2C-mediated reactions with preferential metabolism of the S-enantiomer [34–36]. We therefore assume that the considerable loss of stereoselectivity after oral administration of verapamil is primarily due to induction of CYP3A4 rather than CYP2C in the gut wall mucosa.
We found a major induction within the first 4 days of administration of rifampicin. A rapid loss of drug effect occurred in the older subjects when verapamil and rifampicin are coadministered. In order to guarantee optimal pharmacotherapy, a change of the medications might be indicated.
In summary, we found preserved pharmacokinetics and effects of verapamil in our group of older individuals compared with young subjects. Moreover, prehepatic metabolism of verapamil occurred most likely in the gut wall mucosa of these older people and was inducible by rifampicin leading to a nearly complete loss of verapamil effects after oral drug administration.
Acknowledgments
We are grateful to Mrs E. Schneider and Mr F. Schönberger for their excellent technical assistance. We thank Dr D. Ratge (Department of Clinical Pathology, Robert Bosch Hospital, Stuttgart, Germany) for determination of α1-acid glycoprotein. Deuterated verapamil was a generous gift from the Knoll AG (Ludwigshafen, Germany). This work was supported by the Robert Bosch Foundation (Stuttgart, Germany) and by a grant of the Bundesministerium für Bildung und Forschung (grant number 01 EC 9405).
References
- 1.Vestal RE, Montamat SC, Nielson CP. Drugs in special patient groups: the elderly. In. In: Melmon KL, editor. Clinical Pharmacology. New York: McGraw-Hill Inc.; 1992. pp. 851–874. [Google Scholar]
- 2.Castleden CM, George CF. The effect of ageing on the hepatic clearance of propranolol. Br J Clin Pharmacol. 1979;7:49–54. doi: 10.1111/j.1365-2125.1979.tb00896.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Thomson AH, Tucker GT. Gerontokinetics—a reappraisal. Br J Clin Pharmacol. 1992;33:1–2. doi: 10.1111/j.1365-2125.1992.tb03992.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meyer BR, Reidenberg MM. Clinical pharmacology and ageing. In: Evans TF, editor. Oxford Textbook of Geriatric Medicine. Oxford: Oxford University Press; 1992. pp. 107–116. [Google Scholar]
- 5.Klotz U, Avant GR, Hoyumpa A, Schenker S, Wilkinson GR. The effects of age and liver disease on the disposition and elimination of diazepam in adult man. J Clin Invest. 1975;55:347–359. doi: 10.1172/JCI107938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fromm MF, Botsch S, Heinkele G, Evers J, Kroemer HK. Influence of renal function on the steady-state pharmacokinetics of the antiarrhythmic propafenone and its phase I and phase II metabolites. Eur J Clin Pharmacol. 1995;48:279–283. doi: 10.1007/BF00198312. [DOI] [PubMed] [Google Scholar]
- 7.Kolars JC, Awni WM, Merion RM, Watkins PB. First-pass metabolism of cyclosporin by the gut. Lancet. 1991;338:1488–1490. doi: 10.1016/0140-6736(91)92302-i. [DOI] [PubMed] [Google Scholar]
- 8.Hebert MF, Roberts JP, Prueksaritanont T, Benet LZ. Bioavailability of cyclosporine with concomitant rifampin administration is markedly less than predicted by hepatic enzyme induction. Clin Pharmacol Ther. 1992;52:453–457. doi: 10.1038/clpt.1992.171. [DOI] [PubMed] [Google Scholar]
- 9.Fromm MF, Busse D, Kroemer HK, Eichelbaum M. Differential induction of prehepatic and hepatic metabolism of verapamil by rifampin. Hepatology. 1996;24:796–801. doi: 10.1002/hep.510240407. [DOI] [PubMed] [Google Scholar]
- 10.Paine MF, Shen DD, Kunze KL, et al. First-pass metabolism of midazolam by the human intestine. Clin Pharmacol Ther. 1996;60:14–24. doi: 10.1016/S0009-9236(96)90162-9. [DOI] [PubMed] [Google Scholar]
- 11.Watkins PB, Wrighton SA, Schuetz EG, Molowa DT, Guzelian PS. Identification of glucocorticoid-inducible cytochromes P-450 in the intestinal mucosa of rats and man. J Clin Invest. 1987;80:1029–1036. doi: 10.1172/JCI113156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kolars JC, Schmiedlin-Ren P, Schuetz JD, Fang C, Watkins PB. Identification of rifampin-inducible P450IIIA4 (CYP3A4) in human small bowel enterocytes. J Clin Invest. 1992;90:1871–1878. doi: 10.1172/JCI116064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kolars JC, Lown KS, Schmiedlin-Ren P, et al. CYP3A gene expression in human gut epithelium. Pharmacogenetics. 1994;4:247–259. doi: 10.1097/00008571-199410000-00003. [DOI] [PubMed] [Google Scholar]
- 14.De Waziers I, Cugnenc PH, Yang CS, Leroux J-P, Beaune PH. Cytochrome P 450 isoenzymes, epoxide hydrolase and glutathione transferases in rat and human hepatic and extrahepatic tissues. J Pharmacol Exp Ther. 1990;253:387–394. [PubMed] [Google Scholar]
- 15.Kivistö KT, Bookjans G, Fromm MF, Griese E-U, Münzel P, Kroemer HK. Expression of CYP3A4, CYP3A5 and CYP3A7 in human duodenal tissue. Br J Clin Pharmacol. 1996;42:387–389. doi: 10.1046/j.1365-2125.1996.42615.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Krishna DR, Klotz U. Extrahepatic metabolism of drugs in humans. Clin Pharmacokinet. 1994;26:144–160. doi: 10.2165/00003088-199426020-00007. [DOI] [PubMed] [Google Scholar]
- 17.McDonnell WM, Scheiman JM, Traber PG. Induction of cytochrome P450IA genes (CYP1A) by omeprazole in the human alimentary tract. Gastroenterology. 1992;103:1509–1516. doi: 10.1016/0016-5085(92)91171-y. [DOI] [PubMed] [Google Scholar]
- 18.Gomez DY, Wacher VJ, Tomlanovich SJ, Hebert MF, Benet LZ. The effects of ketoconazole on the intestinal metabolism and bioavailability of cyclosporine. Clin Pharmacol Ther. 1995;58:15–19. doi: 10.1016/0009-9236(95)90067-5. [DOI] [PubMed] [Google Scholar]
- 19.Eichelbaum M, Dengler HJ, Somogyi A, von Unruh GE. Superiority of stable isotope techniques in the assessment of the bioavailability of drugs undergoing extensive first pass elimination. Eur J Clin Pharmacol. 1981;19:127–131. doi: 10.1007/BF00568399. [DOI] [PubMed] [Google Scholar]
- 20.Eichelbaum M, Mikus G, Vogelgesang B. Pharmacokinetics of (+)-, (−)- and (+/−)-verapamil after intravenous administration. Br J Clin Pharmacol. 1984;17:453–458. doi: 10.1111/j.1365-2125.1984.tb02371.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vogelgesang B, Echizen H, Schmidt E, Eichelbaum M. Stereoselective first-pass metabolism of highly cleared drugs: studies of the bioavailability of l- and d-verapamil examined with a stable isotope technique. Br J Clin Pharmacol. 1984;18:733–740. doi: 10.1111/j.1365-2125.1984.tb02536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Echizen H, Brecht T, Niedergesäss S, Vogelgesang B, Eichelbaum M. The effect of dextro-, levo-, and racemic verapamil on atrioventricular conduction in humans. Am Heart J. 1985;109:210–217. doi: 10.1016/0002-8703(85)90585-x. [DOI] [PubMed] [Google Scholar]
- 23.Echizen H, Vogelgesang B, Eichelbaum M. Effects of d,l-verapamil on atrioventricular conduction in relation to its stereoselective first-pass metabolism. Clin Pharmacol Ther. 1985;38:71–76. doi: 10.1038/clpt.1985.137. [DOI] [PubMed] [Google Scholar]
- 24.Abernethy DR, Schwartz JB, Todd EL, Luchi R, Snow E. Verapamil pharmacodynamics and disposition in young and elderly hypertensive patients. Altered electrocardiographic and hypotensive responses. Ann Intern Med. 1986;105:329–336. doi: 10.7326/0003-4819-105-3-329. [DOI] [PubMed] [Google Scholar]
- 25.Abernethy DR, Wainer IW, Longstreth JA, Andrawis NS. Stereoselective verapamil disposition and dynamics in aging during racemic verapamil administration. J Pharmacol Exp Ther. 1993;266:904–911. [PubMed] [Google Scholar]
- 26.Schwartz JB. Aging alters verapamil elimination and dynamics: single dose and steady-state responses. J Pharmacol Exp Ther. 1990;255:373. [PubMed] [Google Scholar]
- 27.Schwartz JB, Troconiz IF, Verotta D, Liu S, Capili H. Aging effects on stereoselective pharmacokinetics and pharmacodynamics of verapamil. J Pharmacol Exp Ther. 1993;265:690–698. [PubMed] [Google Scholar]
- 28.Schwartz JB, Capili H, Wainer IW. Verapamil stereoisomers during racemic verapamil administration: effects of aging and comparisons to administration of individual stereoisomers. Clin Pharmacol Ther. 1994;56:368–376. doi: 10.1038/clpt.1994.151. [DOI] [PubMed] [Google Scholar]
- 29.Schwartz JB, Capili H, Daugherty J. Aging of women alters S-verapamil pharmacokinetics and pharmacodynamics. Clin Pharmacol Ther. 1994;55:509–517. doi: 10.1038/clpt.1994.64. [DOI] [PubMed] [Google Scholar]
- 30.Ahmed JH, Meredith PA, Elliott HL. The influence of age on the pharmacokinetics of verapamil. Pharmacol Res. 1991;24:227–233. doi: 10.1016/1043-6618(91)90085-c. [DOI] [PubMed] [Google Scholar]
- 31.Sasaki M, Tateishi T, Ebihara A. The effects of age and gender on the stereoselective pharmacokinetics of verapamil. Clin Pharmacol Ther. 1993;54:278–285. doi: 10.1038/clpt.1993.148. [DOI] [PubMed] [Google Scholar]
- 32.Gupta SK, Atkinson L, Tu T, Longstreth JA. Age and gender related changes in stereoselective pharmacokinetics and pharmacodynamics of verapamil and norverapamil. Br J Clin Pharmacol. 1995;40:325–331. doi: 10.1111/j.1365-2125.1995.tb04554.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Echizen H, Manz M, Eichelbaum M. Electrophysiologic effects of dextro- and levo-verapamil on sinus node and AV node function in humans. J Cardiovasc Pharmacol. 1988;12:543–546. doi: 10.1097/00005344-198811000-00007. [DOI] [PubMed] [Google Scholar]
- 34.Kroemer HK, Echizen H, Heidemann H, Eichelbaum M. Predictability of the in vivo metabolism of verapamil from in vitro data: contribution of individual metabolic pathways and stereoselective aspects. J Pharmacol Exp Ther. 1992;260:1052–1057. [PubMed] [Google Scholar]
- 35.Kroemer HK, Gautier J-C, Beaune P, Henderson C, Wolf CR, Eichelbaum M. Identification of P450 enzymes involved in metabolism of verapamil in humans. Naunyn-Schmiedeberg's Arch Pharmacol. 1993;348:332–337. doi: 10.1007/BF00169164. [DOI] [PubMed] [Google Scholar]
- 36.Busse D, Cosme J, Beaune P, Kroemer HK, Eichelbaum M. Cytochromes of the P450 2C subfamily are the major enzymes involved in the O-demethylation of verapamil in humans. Naunyn-Schmiedeberg's Arch Pharmacol. 1995;353:116–121. doi: 10.1007/BF00168924. [DOI] [PubMed] [Google Scholar]
- 37.Barbarash RA, Bauman JL, Fischer JH, Kondos GT, Batenhorst RL. Near-total reduction in verapamil bioavailability by rifampin. Chest. 1988;94:954–959. doi: 10.1378/chest.94.5.954. [DOI] [PubMed] [Google Scholar]
- 38.Durnas C, Loi C-M, Cusack BJ. Hepatic drug metabolism and aging. Clin Pharmacokinet. 1990;19:359–389. doi: 10.2165/00003088-199019050-00002. [DOI] [PubMed] [Google Scholar]
- 39.Woodhouse K, Wynne HA. Age-related changes in hepatic function. Implications for drug therapy. Drugs Aging. 1992;2:243–255. doi: 10.2165/00002512-199202030-00007. [DOI] [PubMed] [Google Scholar]
- 40.Fischer C, Schönberger F, Mück W, Heuck K, Eichelbaum M. Simultaneous assessment of the intravenous and oral disposition of the enantiomers of racemic nimodipine by chiral stationary-phase high-performance liquid chromatography and gas chromatography/mass spectroscopy combined with a stable isotope technique. J Pharm Sci. 1993;82:244–250. doi: 10.1002/jps.2600820305. [DOI] [PubMed] [Google Scholar]
- 41.Mikus G, Eichelbaum M, Fischer C, Gumulka S, Klotz U, Kroemer HK. Interaction of verapamil and cimetidine: stereochemical aspects of drug metabolism, drug disposition and drug action. J Pharmacol Exp Ther. 1990;253:1042–1048. [PubMed] [Google Scholar]
- 42.Gross AS, Heuer B, Eichelbaum M. Stereoselective protein binding of verapamil enantiomers. Biochem Pharmacol. 1988;37:4623–4627. doi: 10.1016/0006-2952(88)90330-9. [DOI] [PubMed] [Google Scholar]
- 43.Rowland M, Tozer TN. Clinical Pharmacokinetics. Concepts and applications. Philadelphia: Lea & Febiger; 1995. [Google Scholar]
- 44.Schomerus M, Spiegelhalder B, Stieren B, Eichelbaum M. Physiological disposition of verapamil in man. Cardiovasc Res. 1976;10:605–612. doi: 10.1093/cvr/10.5.605. [DOI] [PubMed] [Google Scholar]
- 45.Somogyi A, Albrecht M, Kliems G, Schäfer K, Eichelbaum M. Pharmacokinetics, bioavailability and ECG response of verapamil in patients with liver cirrhosis. Br J Clin Pharmacol. 1981;12:51–60. doi: 10.1111/j.1365-2125.1981.tb01854.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wynne HA, Cope LH, Mutch E, Rawlins MD, Woodhouse KW, James OFW. The effect of age upon liver volume and apparent liver blood flow in healthy man. Hepatology. 1989;9:297–301. doi: 10.1002/hep.1840090222. [DOI] [PubMed] [Google Scholar]
- 47.Thummel KE, O'Shea D, Paine MF, et al. Oral first-pass elimination of midazolam involves both gastrointestinal and hepatic CYP3A-mediated metabolism. Clin Pharmacol Ther. 1996;59:491–502. doi: 10.1016/S0009-9236(96)90177-0. [DOI] [PubMed] [Google Scholar]
- 48.Loos U, Musch E, Jensen JC, Mikus G, Schwabe HK, Eichelbaum M. Pharmacokinetics of oral and intravenous rifampin during chronic administration. Klin Wochenschr. 1985;63:1205–1211. doi: 10.1007/BF01733779. [DOI] [PubMed] [Google Scholar]
- 49.Gupta SK, Bakran A, Johnson RWG, Rowland M. Erythromycin enhances the absorption of cyclosporin. Br J Clin Pharmacol. 1988;25:401–402. doi: 10.1111/j.1365-2125.1988.tb03320.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ducharme MP, Warbasse LH, Edwards DJ. Disposition of intravenous and oral cyclosporine after administration with grapefruit juice. Clin Pharmacol Ther. 1995;57:485–491. doi: 10.1016/0009-9236(95)90032-2. [DOI] [PubMed] [Google Scholar]
- 51.Fuhr U, Harder S, Lopez-Rojas P, Müller-Peltzer H, Kern R, Staib AH. Increase of verapamil concentrations in steady state by coadministration of grapefruit juice. Naunyn-Schmiedeberg's Arch Pharmacol. 349(suppl):R134. doi: 10.1007/s00228-002-0436-7. (abstract) [DOI] [PubMed] [Google Scholar]
- 52.George J, Byth K, Farrell GC. Age but not gender selectively affects expression of individual cytochrome P450 proteins in human liver. Biochem Pharmacol. 1995;50:730. doi: 10.1016/0006-2952(95)00192-3. [DOI] [PubMed] [Google Scholar]
- 53.Shimada T, Yamazaki H, Mimura M, Inui Y, Guengerich FP. Interindividual variations in human liver cytochrome P-450 enzymes involved in the oxidation of drugs, carcinogens and toxic chemicals: studies with liver microsomes of 30 Japanese and 30 Caucasians. J Pharmacol Exp Ther. 1994;270:414–423. [PubMed] [Google Scholar]
- 54.Schmucker DL, Woodhouse KW, Wang RK, et al. Effects of age and gender on in vitro properties of human liver microsomal monooxygenases. Clin Pharmacol Ther. 1990;48:365–374. doi: 10.1038/clpt.1990.164. [DOI] [PubMed] [Google Scholar]
- 55.Hunt CM, Westerkam WR, Stave GM. Effect of age and gender on the activity of human hepatic CYP3A. Biochem Pharmacol. 1992;44:275–283. doi: 10.1016/0006-2952(92)90010-g. [DOI] [PubMed] [Google Scholar]
- 56.Watkins PB, Murray SA, Winkelman LG, Heuman DM, Wrighton SA, Guzelian PS. Erythromycin breath test as an assay of glucocorticoid-inducible liver cytochromes P-450. J Clin Invest. 1989;83:688–697. doi: 10.1172/JCI113933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vestal RE, Wood AJJ, Branch RA, Shand DG, Wilkinson GR. Effects of age and cigarette smoking on propranolol disposition. Clin Pharmacol Ther. 1979;26:8–15. doi: 10.1002/cpt19792618. [DOI] [PubMed] [Google Scholar]
- 58.Crowley JJ, Cusack BJ, Jue SG, Koup JR, Park BK, Vestal RE. Aging and drug interactions. II. Effect of phenytoin and smoking on the oxidation of theophylline and cortisol in healthy men. J Pharmacol Exp Ther. 1988;245:513–523. [PubMed] [Google Scholar]
- 59.Kroemer HK, Fromm MF, Bühl K, Terefe H, Blaschke G, Eichelbaum M. An enantiomer-enantiomer interaction of (S)-and (R)-propafenone modifies the effect of racemic drug therapy. Circulation. 1994;89:2396–2400. doi: 10.1161/01.cir.89.5.2396. [DOI] [PubMed] [Google Scholar]