

Abstract
Aims
To determine the effect of therapeutic loading doses of halofantrine and chloroquine on CYP2D6 activity in healthy black Zambians.
Methods
Twenty healthy black male Zambians were phenotyped for CYP2D6 activity by measuring the debrisoquine/4-hydroxydebrisoquine ratio in a 0–8 h urine sample after a 10 mg oral dose of debrisoquine hemi-sulphate. The subjects (all ‘extensive metabolizer’ phenotype with respect to CYP2D6) were randomized into two groups of 10, and 24 h later one group received 1500 mg halofantrine hydrochloride and the other group 1500 mg chloroquine phosphate both orally in divided doses. All subjects were given further 10 mg doses of debrisoquine at 2 h, 1 week and 2 weeks after the last dose of the antimalarial drug, and phenotyped as described above.
Results
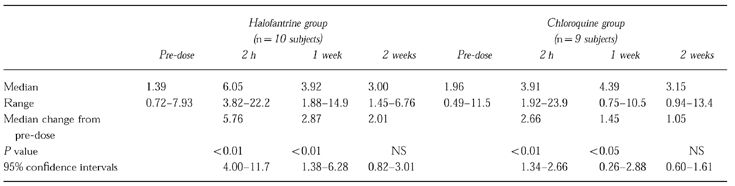
The median debrisoquine/4-hydroxydebrisoquine 0–8 h urinary ratio was increased by halofantrine (1.39 to 6.05; P<0.01; 95% confidence intervals 4.00–11.7) and chloroquine (1.96 to 3.91; P<0.01; 95% confidence intervals 1.34–2.66) when debrisoquine was given 2 h after treatment. The decrease in CYP2D6 activity remained statistically significant for 1 week after both drugs. Halofantrine was a significantly more potent inhibitor of CYP2D6 than chloroquine (P = 0.037). Phenocopying occurred in two subjects taking halofantrine and one taking chloroquine (i.e. the debrisoquine/4-hydroxydebrisoquine ratios became consistent with the poor metabolizer phenotype).
Conclusions
Given in therapeutic loading doses, both halofantrine and chloroquine caused significant inhibition of CYP2D6 activity in healthy black Zambians. With respect to halofantrine, this finding reinforces the recommendation that its combination with other drugs known to prolong the QT interval should be avoided, especially those that are metabolized significantly by CYP2D6.
Keywords: halofantrine, chloroquine, CYP2D6 inhibition, Zambians
Introduction
Cytochrome P450 2D6 (CYP2D6) exhibits a marked genetic polymorphism and is responsible for the metabolism of many drugs in widespread clinical use, including antidepressants, antipsychotics, antiarrhythmics, β-adrenoceptor antagonists and analgesics [1]. Individuals who are homozygous for an autosomal recessive trait affecting the CYP2D6 gene are termed poor metabolizers [2]. They constitute about 5–9% of Caucasians; the remainder are designated extensive metabolizers. The data on black Africans are inconsistent, but the prevalence of the poor metabolizer phenotype is probably less than 1% [3]. Thus, most black Africans should possess a catalytically active enzyme.
CYP2D6 activity is subject to inhibition by many drugs which, in some cases, leads to clinically significant interactions. For example, inhibition of CYP2D6 mediated metabolism of tricyclic antidepressants by coadministered selective serotonin reuptake inhibitor antidepressants results in severe cardiotoxicity [4].
Chloroquine, halofantrine and other agents that are used increasingly in the treatment of sulphadoxine-pyrimethamine resistant falciparum malaria, have been shown to be relatively potent inhibitors of CYP2D6 activity in human liver microsomes (Ki values: chloroquine 12 μm; halofantrine 4 μm) [5–7]. In rats, chloroquine was found to decrease substantially the clearance of metoprolol, a CYP2D6 substrate in humans [5]. However, Masimirembwa et al. [8] did not observe significant inhibition of CYP2D6 activity by chloroquine at a dose of 1500 mg in healthy black Zimbabwean subjects using debrisoquine as the substrate. The effect of halofantrine on CYP2D6 activity in vivo has not been studied.
The aim of the present work was to determine the effect of therapeutic loading doses of chloroquine and halofantrine on CYP2D6 activity in healthy black Zambian subjects using debrisoquine as the probe substrate.
Methods
Subjects
Twenty black male Zambians, aged 21 to 29 years, took part in the study. All were students or staff at the Copperbelt University, Kitwe, Zambia. Volunteers who had taken antimalarial drugs within the previous month, those with a positive Dill-Glasko urine test for chloroquine [9] and subjects taking any other drug 1 week prior to the study were excluded. None of the subjects had prolongation of the QT interval or were known to have any cardiac disorder associated with a prolonged QT interval. Although specific tests of liver and renal function were not performed, all subjects were considered to be healthy on the basis of a medical history. The study was approved by the Ethics Committee of the Tropical Diseases Centre, Ndola, Zambia and all subjects gave their written consent.
Protocol
After emptying the bladder each subject took an oral dose of 10 mg debrisoquine hemisulphate. All urine was collected for the following 8 h and a 20 ml aliquot was stored at −20° C. The subjects were randomized into two groups of 10, and 24 h later one group received 1500 mg (500 mg at 6 hourly intervals) of halofantrine hydrochloride orally and the other 1500 mg (600 mg followed by 600 mg at 6 h and 300 mg at 24 h) of chloroquine phosphate orally. Both drugs were taken 2 h after a low fat meal. All subjects were again phenotyped with further doses of debrisoquine given at 2 h, 1 week and 2 weeks after the last dose of the antimalarial drug. One subject in the chloroquine group did not comply with the protocol and was excluded from the analysis.
Drug analysis
The frozen urine samples were transported by air to the Department of Medicine and Pharmacology, University of Sheffield, UK, where debrisoquine and 4-hydroxydebrisoquine were assayed by g.l.c. [10]. The coefficient of variation of the assay was less than 5% at the lower limit of determination (0.05 μg ml−1).
Data analysis
The debrisoquine/4-hydroxydebrisoquine (D/HD) urine ratios in each group were analysed initially by a two way analysis of variance followed by Dunnett's test [11]. The latter was used because repeated measurements of the D/HD ratio were made on each subject. The difference in the baseline corrected area under the D/HD ratio–time curve (estimated to the last time point that significant differences were found) between halofantrine and chloroquine was compared using the Mann-Whitney U test.
The study was designed with an 80% power to detect a change in the D/HD ratio of 1.95 at a significance level of 5%.
Results
The effects of halofantrine and chloroquine on the D/HD 0–8 h urine ratios are shown in Figure 1. The pre-dose values indicated that none of the subjects was a poor metabolizer as defined by the antimode (12.4) obtained from studies in Caucasian populations [12]. Both halofantrine (P<0.0001) and chloroquine (P = 0.001) increased the mean D/HD ratio significantly. The greatest changes occurred when debrisoquine was given two hours compared with 1 and 2 weeks after their administration (Figure 1 and Table 1). In two of the subjects taking halofantrine and one taking chloroquine the magnitude of the change was sufficient to convert their extensive metabolizer phenotype to that of an apparent poor metabolizer. The median D/HD ratio remained significantly higher than the pre-dose ratio at 1 week but not at 2 weeks after administration of both halofantrine and chloroquine (Table 1). The median area under the baseline-corrected D/HD ratio–time curve (0–1 week) was significantly (P = 0.037) higher after halofantrine (635 units) than after chloroquine admninistration (326 units).
Figure 1.

Effects of 1500 mg loading doses of a) halofantrine and b) chloroquine on the debrisoquine/4-hydroxydebrisoquine 0–8 h urinary ratio in healthy black Zambians measured from 2 h, 1 week and 2 weeks after dosage of the antimalarials
Table 1.
A comparison of the effects of 1500 mg loading doses of halofantrine and chloroquine on the debrisoquine/4-hydroxydebrisoquine 0–8 h urinary ratio in healthy black Zambians
Discussion
All of the subjects had detectable amounts of 4-hydroxydebrisoquine in the urine and, therefore, exhibited significant CYP2D6 activity. Both chloroquine and halofantrine given as therapeutic loading doses significantly impaired the 4-hydroxylation of debrisoquine. Inhibition of CYP2D6 activity was greatest when debrisoquine was given 2 h compared with 1 and 2 weeks after antimalarial drug therapy. However, enzyme inhibition was still significant 1 week after dosing, consistent with both drugs and their major metabolites having very long elimination half-lives [13, 14]. Halofantrine inhibited CYP2D6 activity to a greater extent than chloroquine. Neither drug was as potent an inhibitor as quinidine (50 mg), which in most cases causes phenocopying [2], the apparent conversion of an extensive into a poor metabolizer. Based on criteria obtained in Caucasian populations [12], two subjects taking halofantrine and one taking chloroquine would be classified as poor metabolizers.
The data for halofantrine confirm in vitro findings [6] that it is a moderately potent inhibitor of CYP2D6. The extent of inhibition differed markedly amongst the subjects, which might be due in part to the highly variable absorption of halofantrine from the gastrointestinal tract [13]. Halofantrine is avidly bound to human liver and converted mainly to N-desbutylhalofantrine, which is also an inhibitor of CYP2D6 [6]. Halliday et al. [6] have reported that the major form of cytochrome P450 responsible for catalysing this reaction is CYP3A, although a contribution from CYP2D6 could not be excluded. The effect of halofantrine on CYP3A activity or of CYP3A inhibitors on halofantrine metabolism has not been studied in vitro or in vivo.
Compared with halofantrine, chloroquine is a less potent inhibitor of human CYP2D6 in vitro [5–7]. However, total tissue concentrations of chloroquine as high as 100 μm have been found in human liver after normal doses [14]. Thus, it might be anticipated that chloroquine could decrease CYP2D6 activity in vivo. The present findings confirm this suggestion and show that the degree of inhibition by chloroquine is similar to that caused by halofantrine given in the same dose. Why Masimirembwa et al. [8] did not observe inhibition of CYP2D6 by chloroquine in black Zimbabweans is unclear. Although the dosing regimen was similar to that of the present study, debrisoquine was administered 6 h after the last dose of chloroquine compared to 2 h in the present study.
In summary, we have shown that both halofantrine and chloroquine inhibit CYP2D6 activity in healthy black Zambians and that this effect is sustained for one week after therapeutic doses of the drugs. This finding for halofantrine reinforces the current recommendation that, unless there are compelling clinical reasons, it should not be used in combination with other drugs known to prolong the QT interval (manufacturer's data sheet), especially those metabolized by CYP2D6 (e.g. tricyclic antidepressants, antipsychotics and some antiarrhythmics).
Acknowledgments
We are grateful to Sister C. Mawila for nursing assistance and to Drs P. Ghaharamini and A. Rostami-Hodjegan for statistical advice.
References
- 1.Tucker GT. Clinical implications of genetic polymorphism in drug metabolism. J Pharm Pharmacol. 1994;46(Suppl. 1):417–424. [PubMed] [Google Scholar]
- 2.Lennard MS. Genetic polymorphism of sparteine/debrisoquine oxidation: a reappraisal. Pharmacol Toxicol. 1990;67:273–283. doi: 10.1111/j.1600-0773.1990.tb00830.x. [DOI] [PubMed] [Google Scholar]
- 3.Masimirembwa CM, Johansson I, Hasler JA, Ingelman-Sundberg M. Genetic polymorphism of cytochrome P450 CYP2D6 in a Zimbabwean population. Pharmacogenetics. 1993;3:275–280. doi: 10.1097/00008571-199312000-00001. [DOI] [PubMed] [Google Scholar]
- 4.Bertilsson L, Dahl M-L. Polymorphic drug oxidation: relevance to the treatment of psychiatric disease. CNS Drugs. 1996;5:200–223. [Google Scholar]
- 5.Lancaster DL, Adio RA, Tai KK, et al. Inhibition of metoprolol metabolism by chloroquine and other antimalarial drugs. J Pharm Pharmacol. 1990;42:267–271. doi: 10.1111/j.2042-7158.1990.tb05405.x. [DOI] [PubMed] [Google Scholar]
- 6.Halliday RC, Jones BC, Smith DA, Kitteringham NR, Park BK. An investigation of the interaction between halofantrine and CYP2D6 and CYP3A4: studies with human liver microsomes and heterologous enzyme expression systems. Br J Clin Pharmacol. 1995;40:369–378. doi: 10.1111/j.1365-2125.1995.tb04559.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masimirembwa CM, Hasler JA, Johansson I. Inhibitory effects of antiparasitic drugs on cytochrome P450 2D6. Eur J Clin Pharmacol. 1995;48:35–38. doi: 10.1007/BF00202169. [DOI] [PubMed] [Google Scholar]
- 8.Masimirembwa CM, Gustafsson LL, Dahl M-L, Abdi YA, Hasler JA. Lack of effect of chloroquine on the debrisoquine (CYP2D6) and S-mephenytoin (CYP2C19) hydroxylation phenotypes. Br J Clin Pharmacol. 1996;41:344–346. doi: 10.1046/j.1365-2125.1996.30713.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lelijveld J, Kortmann H. The eosin colour test of Dill and Glasko: a simple field test to detect chloroquine in urine. Bull WHO. 1970;42:477–479. [PMC free article] [PubMed] [Google Scholar]
- 10.Lennard MS, Silas JH, Smith AJ, Tucker GT. Determination of debrisoquine and its 4-hydroxymetabolite in biological fluids by gas chromatography with flame ionisation and nitrogen-selective detection. J Chromatogr. 1977;133:161–166. doi: 10.1016/s0021-9673(00)89216-x. [DOI] [PubMed] [Google Scholar]
- 11.Dunnett CW. A multiple comparison procedure for comparing several treatments with a control. J Am Stat Assoc. 1955;50:1096–1121. [Google Scholar]
- 12.Alvan G, Bechtel P, Iselius L, Gundert-Remy U. Hydroxylation polymorphisms of debrisoquine and mephenytoin in European populations. Eur J Clin Pharmacol. 1990;39:533–537. doi: 10.1007/BF00316090. [DOI] [PubMed] [Google Scholar]
- 13.Karbwang J, Bangchang KN. Clinical pharmacokinetics of halofantrine. Clin Pharmacokin. 1994;27:104–119. doi: 10.2165/00003088-199427020-00003. [DOI] [PubMed] [Google Scholar]
- 14.Ducharme J, Farinotti R. Clinical pharmacokinetics and metabolism of chloroquine. Clin Pharmacokin. 1996;31:257–274. doi: 10.2165/00003088-199631040-00003. [DOI] [PubMed] [Google Scholar]