

Abstract
An immunogenic aminopeptidase was purified from Brucella melitensis strain VTRM1. The purification procedure consisted of ammonium sulfate fractionation and three chromatographic steps. This procedure resulted in a yield of 29% and a 144-fold increase in specific activity. The aminopeptidase appeared to be a monomeric enzyme with a molecular mass of 96 kDa and an isoelectric point of 4.8. Its activity was optimal at pH 7.0 at 40°C. The enzyme was strongly inhibited by EDTA, 1,10-phenathroline, and divalent cations (Zn2+ and Hg2+), suggesting that this protein was a metalloaminopeptidase. The enzyme showed preference for alanine at the N termini of aminoacyl derivatives. The Km values for l-alanine-p-nitroanilide (Ala-pNA) and Lys-pNA were 0.35 and 0.18 mM, respectively. The N-terminal sequence of aminopeptidase was used for a homologous search in the genomes of B. melitensis 16M and Brucella suis 1330. The analysis revealed an exact match of the probe sequence (36 bp) with an open reading frame of 2,652 bp encoding a protein predicted to be alanyl aminopeptidase (aminopeptidase N). Collectively, these data suggest designation of the B. melitensis enzyme as an aminopeptidase N. The aminopeptidase was recognized by sera from patients with acute and chronic brucellosis, suggesting that the enzyme may have important diagnostic implications.
Brucellosis is a major problem in the Mediterranean region and parts of Asia, Africa, and Latin America, where it remains endemic, generating severe economic losses (4). Bovine and caprine brucellosis caused by Brucella abortus and Brucella melitensis, respectively, are characterized by late-term abortions in pregnant females (4). In Mexico, brucellosis, mainly produced by B. melitensis, is one of the most important zoonoses causing severe morbidity in humans. The infection in humans is commonly acquired by drinking unpasteurized milk or eating milk by-products derived from infected goats or cows (15). The disease is characterized by recurrent high fever, headache, cachexia, lethargy, arthritis, and splenomegaly. The genus Brucella consists of 6 recognized species classified as facultative intracellular pathogens. They are able to invade macrophages, adapt to the acidic environment, and multiply in the vacuolar compartments (27). Survival inside host cells allows the bacteria to evade the host's protective humoral immune mechanisms, such as those mediated by specific antibodies and complement. The macrophage subjects the bacteria to a harsh intracellular environment characterized by reactive oxygen intermediates, low pH, and decreased iron availability. Ironically, it has been shown that this environment, especially its acidity, activates Brucella genes coding for products essential for intracellular survival in this niche (28).
The induction of Brucella heat shock proteins has been involved in adaptive responses to adverse environmental conditions (28). Some of these proteins such as Brucella DnaK (11), HtrA (7), and Lon have been studied in detail (29). The Lon protein is an ATP-dependent protease (19), and in B. abortus, it functions as a stress response protease required during the initial stages of infection in the mouse model (29).
Intracellular proteolytic degradation is important in bacteria for the elimination of damaged proteins, modulation of protein levels, and maintenance of amino acid pools. Proteolytic enzymes represent one of the best-studied and understood classes of proteins. For many years it was accepted that proteolytic enzymes functioned primarily in the acquisition of nutrients for growth and proliferation through the degradation of host tissues (3). However, recent observations indicated that pathogen-derived proteolytic enzymes can also play important roles in the regulation of critical host processes in order for the invading microbes to survive in a hostile host environment (16).
Aminopeptidases (APEs) of many bacteria have been studied with some detail both structurally and enzymatically (2, 17, 31). On the other hand, it has been observed that interleukin-2 and gamma interferon are rapidly inactivated and degraded by proteinases from Pseudomonas aeruginosa and Legionella pneumophila (10, 24). Some specific proteinases have been shown to be related to severe periodontitis and cardiovascular disease (21). It has been suggested that extracellular enzymes of Aeromonas spp. play an important role in invasiveness and in establishment of the infection (14). Limited studies have been carried out with Brucella along these lines.
In this study, we describe the purification and characterization of an immunogenic APE obtained from B. melitensis. The properties of the purified enzyme are discussed in relation to those of previously characterized APEs. To our knowledge, this is the first report of the purification of an APE from B. melitensis.
MATERIALS AND METHODS
Bacterial strains and growth conditions.
B. melitensis 16M was kindly donated by Central Veterinary Laboratory (New Haw, Weybridge, United Kingdom), and the B. melitensis rough mutant VTRM1 was obtained in 1995 from the Brucella culture collection at Virginia Tech (S. M. Boyle). Strain VTRM1 was obtained by inserting a Tn5 element in the wboA gene of strain 16M (35). The strains were grown on Trypticase soy agar (Difco) supplemented with 0.5% (wt/vol) yeast extract (Difco) (TSA-Y) or Trypticase soy broth (Difco) supplemented with 0.5% (wt/vol) yeast extract (Difco) (TSA-B) at 37°C for 30 h.
Preparation of cell extract.
The growth of two 500-ml (TSA-B) batch cultures of B. melitensis 16M and B. melitensis VTRM1 was monitored every 2 h to establish a growth curve, and enzyme activity detection was performed at 6-h intervals. Cultures at different growth stages were subjected to the following fractionation procedure. The cells were harvested by centrifugation (10,000 × g, 30 min) at 4°C, washed twice with 10 mM Tris-HCl (pH 7.0), and resuspended in the same buffer. Glass beads (0.1-mm diameter) were added, and the suspension was stirred with a magnetic stirrer for 5 h at 4°C. Unbroken cells were removed by centrifugation (10,000 × g, 30 min), and the opalescent supernatant was subjected to ultracentrifugation (100,000 × g, 120 min), yielding a pellet (PE) containing insoluble bacterial components and a supernatant that was considered a membrane-free bacterial extract (MFBE). All fractions, cell-free culture medium (CM), PE, and MFBE, were assayed for APE activity with l-lysine-p-nitroanilide (Lys-pNA) as the substrate.
Enzyme purification.
As both strains exhibited APE activity, the purification was performed with the B. melitensis rough mutant VTRM1 strain to eliminate smooth lipopolysaccharide contamination. The strain was grown on TSA-Y plates at 37°C for 30 h. Bacterial cells were resuspended in Tris-HCl (pH 7.0), and the extract was obtained as described above. Solid ammonium sulfate was used to precipitate proteins between 40 and 70% saturation at 4°C; the insoluble proteins were collected by centrifugation (15,000 × g, 30 min), dissolved in 10 mM imidazole buffer (pH 7.0), and extensively dialyzed against the same buffer (overnight at 4°C). The dialyzed fraction was clarified by centrifugation (15,000 × g, 30 min) and then was filtered through a 0.22-μm-pore-size membrane. The sample was applied onto an XK50 column packed with high-performance Q Sepharose (60 ml) equilibrated with 10 mM imidazole buffer (pH 7.0). A linear gradient of 0 to 1 M NaCl was applied at a flow rate of 120 ml/h, and 15-ml fractions were collected and assayed for APE activity. Fractions with enzymatic activity were pooled and saturated with 1.5 M ammonium sulfate. The sample was then loaded onto a phenyl-Superose HR 5/5 column previously equilibrated with 100 mM sodium phosphate and 1.5 M ammonium sulfate (pH 7.0). The column was eluted with a descending gradient from 1.5 to 0 M ammonium sulfate at a flow rate of 30 ml/h. The fractions with enzymatic activity were pooled and concentrated in a Centricon 50 tube (Amicon). The sample was loaded onto a Superose 12 HR 10/30 column previously equilibrated with 50 mM phosphate buffer (pH 7.0) containing 150 mM NaCl and eluted at a flow rate of 12 ml/h. The fractions with APE activity obtained by gel filtration chromatography were used for characterization. All purification steps were performed at 4°C by using a fast protein liquid chromatography system (Amersham Pharmacia Biotech, Uppsala, Sweden).
Enzyme assays.
APE activity was assayed by measuring the hydrolysis of Lys-pNA (Sigma, St. Louis, Mo.). The incubation mixture consisted of 30 μl of substrate (10 mM), 250 μl of 100 mM Tris-HCl buffer (pH 7.0), 120 μl of water, and 100 μl of enzymatic samples. After incubation at 37°C for 20 min, the reaction was stopped by the addition of 500 μl of ZnSO4 (5%) and 100 μl of BaCl2 (7.5%). The mixture was centrifuged (10,000 × g for 10 min), and absorbance of the released p-nitroaniline was determined at 405 nm in the clear supernatant. One unit of APE activity was defined as the amount of enzyme that liberates 1 μmol of p-nitroaniline per minute at 37°C. The molar absorption coefficient for 4-nitroaniline at 405 nm is ɛ405 nm = 9,900 M−1 cm−1, and this value was used for the calculation of enzyme activity.
Protein determination.
Protein concentrations were determined with a Bradford reagent kit (Sigma) by following the manufacturer's protocol.
Gel electrophoresis.
The sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) system developed by Laemmli (13) was used to monitor enzyme purification and estimate the enzyme's molecular mass. Gels were stained with Coomasie blue G250. A wide molecular marker kit (200,000 to 14,000 kDa; Sigma) was used as a standard.
Amino acid sequencing.
For amino-terminal sequence analysis, the protein band resolved by SDS-PAGE was electrotransferred to a polyvinylidene difluoride membrane (18) and the N-terminal sequence was determined by automated Edman degradation on an Applied Biosystems Procise model 494 protein sequencer (Biomolecular Research Facility, University of Virginia Health System). The amino acid sequence obtained was compared with available sequences at the website of the National Center for Biotechnology Information (http://www.ncbi.nlm.nih.gov) by using the Psi-BLAST program (1). Hydrophobic regions and motif sequences were analyzed with the Prosite program (9).
Enzyme characterization. (i) Molecular mass measurement.
The relative molecular mass was estimated by fast protein liquid chromatography gel filtration and SDS-PAGE. Gel filtration (mean of four determinations) was executed on a Superose 12 HR 10/30 column (Amersham Pharmacia Biotech) calibrated with the following standard markers (molecular masses are given in parentheses): thyroglobulin (670 kDa), gamma globulin (158 kDa), ovalbumin (44 kDa), myoglobin (17 kDa), and vitamin B12 (1.3 kDa) (kit from Bio-Rad). Equilibrium and elution (0.2 ml/min) were performed with 50 mM phosphate buffer (pH 7.0) containing 150 mM NaCl.
(ii) Determination of pI.
The isoelectric point was determined by chromatofocusing on a MonoP column equilibrated with 25 mM imidazole (pH 7.4) by using a pH gradient from 7.0 to 4.0 (Polybuffer 74; Amersham Pharmacia Biotech).
(iii) Effect of temperature and pH dependence on APE.
The effect of pH on APE activity was examined by using McIlvaine buffers at pH values ranging from 3.0 to 7.0, 100 mM Tris-HCl at pH values from 7.0 to 9.0, and 100 mM carbonate-bicarbonate buffer at pH values from 9.0 to 11.0. The pH stability of the APE was tested by incubation of the purified enzyme for 12 h at 4°C in each buffer followed by the standard enzyme activity assay. The optimal temperature for enzyme activity was determined in 100 mM Tris-HCl buffer (pH 7.0) in the range of 4 to 70°C. The temperature stability was tested by incubation of the enzyme at 4, 10, 20, 30, 40, 50, 60, and 70°C for 30 min in 50 mM Tris-HCl buffer, pH 7.0, followed by the standard enzyme activity assay. The protein was subjected to several cycles of freezing and thawing, and the activity was measured every month. In every case, activity was expressed as a percentage of the activity obtained at either the optimal pH or the optimal temperature.
(iv) Determination of kinetic parameters.
Kinetics constants of the purified enzyme were estimated for Lys-pNa and Ala-pNA, with substrate concentrations ranging from 0.01 to 1.0 mM. Activity was measured under the same conditions as described above and expressed as the mean of three different determinations. The Km and Vmax were obtained from a Michaelis-Menten plot.
(v) Effect of protease inhibitors and metal cations.
The protease inhibitors bestatin (250 μM), pepstatin (250 μM), leupeptin (50 μM), pefabloc (5 μM), 1,10-phenanthroline (10 μM), EDTA (2.5 mM), phenylmethylsulfonyl fluoride (PMSF) (5 μM), and trans-epoxysuccinyl-l-leucylamido-(4-guanidino) butane (E64) (50 μM) and metal cations ZnCl2 (1 mM), CoCl2 (1 mM), CaCl2 (1 mM), HgCl2 (1 mM), and MgCl2 (1 mM) were added to the enzyme solution. The purified enzyme was incubated in presence of each inhibitor for 30 min followed by the standard enzyme assay with Lys-pNA as the substrate. Activity was expressed as a percentage of the activity obtained in the absence of the added inhibitor.
Substrate specificity.
The relative activities of the APE from B. melitensis against several aminoacyl-pNA (Glu-pNA, Ala-pNA, Leu-pNA, Gly-pNA, Met-pNA, Lys-pNA, and Pro-pNA) and dipeptidyl-pNA (Ala-Pro-pNA, Gly-Phe-pNA, Gly-Pro-pNA, Ala-Ala-pNA, and Ala-Phe-pNA) (Bachem) substrates were determined by the standard activity assay. Relative activity was expressed as a percentage of the rate of hydrolysis of Ala-pNA which was assigned a value of 100%.
Western blotting.
Immunoblotting was carried out by using the method of Towbin et al. (33). A prestained molecular marker kit (Gibco BRL) was used as a standard.
Human sera.
Sera from 20 patients with brucellosis at different stages of the disease belonging to our serum collection were used. The samples were selected according to the following criteria. Ten had acute brucellosis as determined by the clinical picture of brucellosis: an agglutination titer equal to or higher than 1:80 and positive 2-mercaptoethanol agglutination. Also, half of the patients had positive blood cultures. The other 10 patients were considered to have chronic brucellosis, as determined by a history of persisting symptoms or relapses and persisting agglutination titers for more than 1 year. Also, sera from 50 volunteers, without antecedents of brucellosis, and negative to standard and 2-mercaptoethanol agglutination tests were included.
Preparation of anti-APE serum.
Two mice were injected intraperitoneally with 20 μg of purified enzyme emulsified with complete Freund adjuvant and reinjected with 20 μg of mixed with incomplete Freund adjuvant 3 weeks later. The mice were bled before the first injection (preimmune serum) and 1 week after the booster (anti-APE serum) to obtain immune serum.
RESULTS
Purification of APE activity.
Preliminary experiments indicated the presence of an intracellular Lys-pNA activity in MFBE from both B. melitensis 16M and B. melitensis rough mutant VTRM1. Results of enzymatic activity detection in different fractions of the B. melitensis rough mutant VTRM1 indicated that the majority of this enzyme was found in the MFBE while some remaining activity was found associated with PE; no activity was found in CM alone (Fig. 1). Similar results were obtained with B. melitensis 16M strain (data not shown). Detection of free (MFBE) and cell-associated (PE) APE activity did not occur until the early stationary phase. A substantial increase in activity occurred after 30 h of incubation (Fig. 1). The ammonium sulfate precipitation (40 to 70%) and the subsequent chromatography steps (Fig. 2) resulted in the final isolation of a pure enzyme. The APE was purified 144-fold with a recovery of 29% of the enzyme activity (Table 1).
FIG. 1.

B. melitensis strain VTRM1 growth and activity curve. Culture turbidity was measured at 560 nm (♦). Determination of APE activity was measured every 6 h in CM (▪), MFBE (▴), and PE (•).
FIG. 2.

Chromatograms of the purification of B. melitensis APE. Proteins were detected by measuring the absorbance at 280 nm (optical density at 280 nm [OD280]) (dotted line); APE activity was assayed with Lys-pNA at pH 7.0 (solid circles). (A) High-performance Q Sepharose chromatography with NaCl gradient (solid line); (B) phenyl-Superose chromatography with (NH4)2SO4 gradient (solid line); (C) Superose chromatography.
TABLE 1.
Purification of APE97 from B. melitensis
| Purification step | Amt of protein (mg) | Total activity (U) | Sp act (U/mg) | Yield (%) | Purification (fold) |
|---|---|---|---|---|---|
| Cell extract | 901.0 | 998.6 | 1.1 | 100 | 1.0 |
| Ammonium sulfate (40-70%) | 542.1 | 773.3 | 1.4 | 77.0 | 1.3 |
| High-performance Q Sepharose | 40.0 | 502.0 | 12.5 | 50.0 | 11.3 |
| Phenyl Sepharose | 4.2 | 411.2 | 98.0 | 41.1 | 89.0 |
| Superose 12HR | 1.8 | 286.8 | 159.0 | 29.0 | 144.5 |
Molecular mass.
SDS-PAGE analysis of the purified enzyme revealed the presence of a single protein band with an apparent molecular mass of 96,000 Da (designated APE97) (Fig. 3). The molecular mass of the native enzyme obtained by gel filtration was 97,000 Da. Similarities in both results suggested that the purified enzyme was a monomer.
FIG. 3.
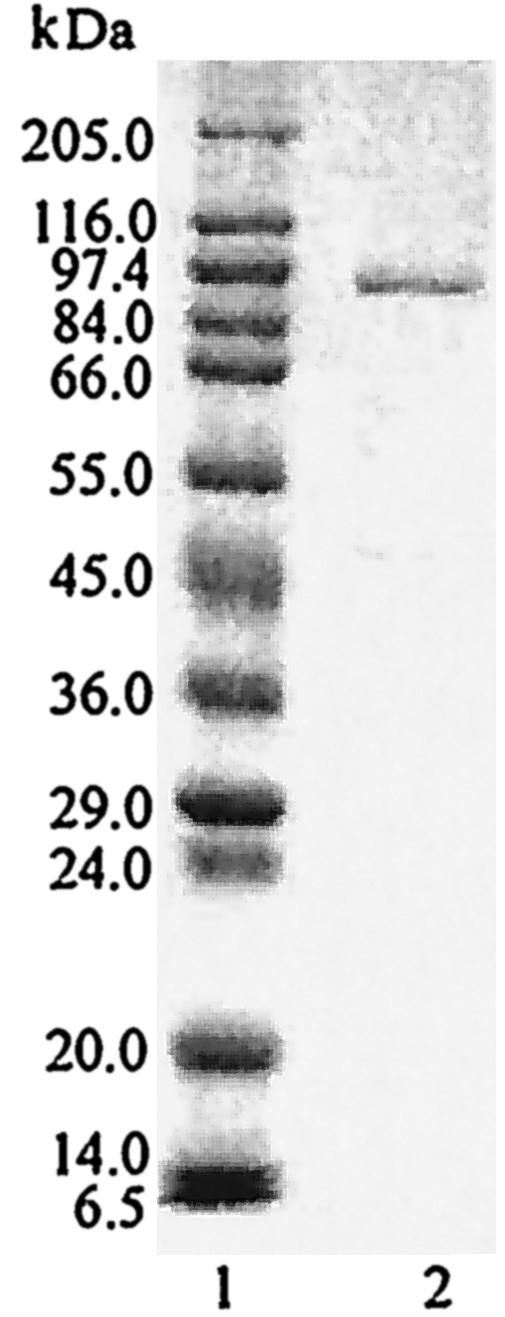
SDS-PAGE of purified APE. Electrophoresis was performed on an SDS-polyacrylamide gel (10%). Lane 1, molecular mass markers; lane 2, purified APE (5 μg). The molecular mass markers were as follows: myosin, 205 kDa; beta-galactosidase, 116 kDa; phosphorylase b, 97 kDa; fructose-6-phosphate kinase, 84 kDa; albumin, 66 kDa; glutamic dehydrogenase, 55 kDa; ovalbumin, 45 kDa; glyceraldehyde-3-phosphate dehydrogenase, 36 kDa; carbonic anhydrase, 30 kDa; trypsinogen, 24 kDa; trypsin inhibitor, 20 kDa; alfa-lactalbumin, 14 kDa; aprotinin, 6.5 kDa.
Isoelectric point determination.
The isoelectric point was estimated to be 4.8 by isoelectric chromatofocusing.
Effects of pH and temperature.
Using the APE activity assay with Lys-pNA as a substrate, it was found that the purified enzyme showed an optimal activity at pH 7.0. The enzyme was stable in the pH range of 6.0 to 8.5. APE97 had no activity at a pH below 6.0 or over 9.0. The activity of the purified enzyme was optimal at 40°C. Enzyme activity decreased significantly at temperatures over 50°C, although around 15% of the activity was observed at 60°C. The thermal stability results showed that the enzyme was inactivated >95% after incubation for 30 min at 50°C. We also determined that APE97 was resistant to several cycles of freezing and thawing. The enzyme showed no appreciable loss of activity when kept frozen at −70°C for at least 6 months, whereas it retained approximately 50% of its original activity when kept at −20°C over the same period of time.
Effect of inhibitors on enzyme activity.
The influences of several agents on the activity of APE97 are summarized in Table 2. The activity of APE97 was almost completely inhibited in the presence of the chelating agents 1,10-phenanthroline (10 μM) and EDTA (2.5 mM). Bestatine, a typical inhibitor of exopeptidases, inhibited more than 50% the activity. APE97 was insensitive to serine-protease inhibitors such as leupeptin, pefabloc, and PMSF. E64, an inhibitor of cysteine proteinases and the aspartate-specific inhibitor pepstatin, had no effect on the activity of APE97. The presence of Zn2+ and Hg2+ caused complete inhibition at 1.0 mM while slight effects (10 to 15% inhibition) were observed in the presence of Ca2+, Mg2+, and Co2+ (Table 2). Concentrations of ammonium sulfate, used in the purification procedure, had no inhibitory effect on APE97 activity.
TABLE 2.
Effect of proteinase inhibitors and divalent cations on the activity of B. melitensis APE97
| Inhibitor | Concn | Relative activity (%) |
|---|---|---|
| None | None | 100 |
| Zn2+ | 1 mM | 0 |
| Co2+ | 1 mM | 93 |
| Ca2+ | 1 mM | 86 |
| Hg2+ | 1 mM | 0 |
| Mg2+ | 1 mM | 96 |
| EDTA | 2.5 mM | 0 |
| Pefabloc | 5 μM | 60 |
| Bestatin | 250 μM | 45 |
| Pepstatin | 250 μM | 90 |
| Leupeptin | 50 μM | 100 |
| PMSF | 5 μM | 94 |
| E-64 | 50 μM | 90 |
| 1,10-Phenanthroline | 10 μM | 20 |
Substrate specificity.
The specificity of the enzyme towards the N-terminal residue was tested with several aminoacyl-pNA and dipeptidyl-pNA substrates. APE97 was able to release the hydrophobic amino acids Ala (100%), Leu (25.5%), and Met (18.5%), the uncharged amino acid Gly (13.5%), and the hydrophilic amino acid Lys (50%). Ala-pNA was most actively hydrolyzed, whereas Lys-pNA, Leu-pNA, Met-pNA, and Gly-pNA were hydrolyzed at lower rates. The acid derivative Glu-pNA showed no detectable reactivity with APE97. Weak activity was detected against Ala-Pro-pNA (1.5%), and the other dipeptidyl substrates were not hydrolyzed.
Kinetics parameters.
The Km and Vmax were 0.35 mM and 45 μmol min−1 mg of protein−1, respectively, for Ala-pNA and 0.18 mM and 6.1 μmol min−1 mg of protein−1, respectively, for Lys-pNA.
N-terminal amino acid sequencing.
The N-terminal sequence was determined to be MRTETGHTFRLE. This sequence was then used as a probe to search for the complete sequence of the APE gene in the genome bank of B. melitensis 16M (5). The results revealed an exact match of this sequence with a potential open reading frame of 2,652 bp (GenBank accession number NP_540241). This gene codes for a single protein consisting of 884 amino acid residues and predicted to be an alanyl APE (PepN). The apparent molecular mass determined for APE97, based on its migration position in the SDS gel, was close to the theoretical value of 98,048 Da calculated for the pepN gene product based on its amino acid composition.
Sequence comparison.
The comparison obtained by using the Psi-BLAST program showed that the deduced primary structure of the purified APE from B. melitensis was extremely similar (99%) to the putative APE N from B. suis 1330 (GenBank accession number NP_697631). Lower identities were observed with the putative APE N from Mesorhizobium loti (63%; GenBank accession number NP_107963), a putative APE N from Sinorhizobium meliloti (59%; GenBank accession number NP_385132), and a putative APE N from Agrobacterium tumefaciens (58%; GenBank accession number NP_354008).
Reactivity of human sera.
To establish the ability of this protein to induce a specific antibody response in humans, Western blot analysis was performed. Using human sera from patients with acute and chronic brucellosis and sera from healthy individuals, it was demonstrated that patients with brucellosis responded to the protein while healthy individuals were seronegative (Fig. 4). We used a mouse anti-APE serum as a positive control.
FIG. 4.

Immunoreaction of serum to purified APE. Lane 1, molecular mass markers; lanes 2 and 3, sera from patients with acute brucellosis; lanes 4 and 5, sera from patients with chronic brucellosis; lanes 6 and 7, sera from healthy people; lane 8, APE antisera. The molecular mass markers were as follows: phosphorylase b, 97 kDa; bovine serum, 68 kDa; ovalbumin, 43 kDa; carbonic anhydrase, 30 kDa; beta-lactoglobulin, 18.4 kDa; lysozyme, 14.3 kDa.
DISCUSSION
The role of proteases from several bacteria has been the focus of many studies in bacterial pathogenesis (10, 21, 24). APEs belong to the M1 family, which forms part of the MA clan of metalloproteases (3). APEs occur in a wide variety of microbial species, including bacteria and fungi, act on a free N-terminal amino acid of a polypeptide chain, and release a single amino acid residue (3). The APEs of Escherichia coli as well as those of lactic acid bacteria have previously been purified and characterized by several workers (8, 36). There appear to be no reports on the purification of Brucella APEs.
The present study describes the purification and characterization of an APE from B. melitensis not previously reported. The peak enzyme production occurred at the early stationary growth phase (30 h). The APE activity was found predominantly in a bacterial cell extract, although some residual activity was found associated with Brucella membranes (PE) but was considered a product of contamination. Immunogold labeling of Lactococcus lactis cells with antibodies against APE N strongly supported an intracellular location (34); this seems to be the case for APE97 from Brucella. Purification of B. melitensis APE from strain VTRM1 was achieved by ammonium sulfate fractionation and three consecutive chromatographic steps. The enzymatic activity remained stable during the purification process and was not inactivated by ammonium sulfate. The native enzyme obtained by gel filtration chromatography showed a peak activity with an apparent molecular mass of 97,000 Da. Results from the SDS-PAGE gel revealed a single protein band with an apparent molecular mass of 96,000 Da and implied that the purified enzyme was a monomer.
APE97 shared several features in common with APEs purified from other bacteria. The molecular mass of the purified enzyme was similar to those described for Streptococcus salivarius (98 kDa) (22), L. lactis (95 kDa) (8), and E. coli K-12 (87 kDa) (20). APE97 exhibited a pI of 4.8, which is similar to the pI of 4.6 reported for the APE N of E. coli K-12 (20) and to the pI of 4.5 reported for the APE N of L. lactis (8). In order to further characterize the enzymatic activity, various inhibitors were tested. The activity of APE97 was almost completely inhibited in the presence of chelators, 1,10-phenanthroline, and EDTA. APE97 was insensitive to serine-protease inhibitors such as leupeptin, pefabloc, and PMSF. E64, an inhibitor of cysteine proteinases and the aspartate-specific inhibitor pepstatin, had no effect on the activity of APE97. Zn2+ and Hg2+ caused complete inhibition of APE97. Inhibition by the metal cation Zn2+ has been reported for S. salivarius (22) and L. lactis (8). Altogether, these results strongly suggest that APE97 is a metallo-APE.
APE97 showed optimal activity at 40°C, a temperature similar to that found for other bacterial APE N′s (PepN) (8, 22, 36). APE97 showed narrow pH requirements, as the enzyme was stable at a pH range of 6.0 to 8.0, with maximal activity at pH 7.0. This is consistent with bacterial PepN, which in general has a pH optimum between 6.5 and 7.5 (3). Bacterial PepN is capable of hydrolyzing a broad range of peptides, removing the N-terminal amino acid. APE97 released a single amino acid and was able to cleave a variety of N-terminal amino acids from pNA derivates. In decreasing order of preference, they were Ala, Lys, Leu, and Met; the enzyme was not able to liberate either Glu or dipeptidyl substrates. The profile of these relative activities within a range of pNA derivates was therefore similar to those reported for the APE N from E. coli K-12, S. salivarius, and L. lactis (8, 22, 36). In general, APE97 showed a strong preference for bonds involving the hydrophobic amino acid Ala and hydrophilic Lys N-terminal aminocyl residues, a relatively high activity toward hydrophobic amino acids Leu and Met, and very little activity toward glutamyl residues.
The N-terminal sequence obtained from the APE served as a probe to search for the complete DNA sequence of the APE gene in the genome bank of B. melitensis 16M (5). The results revealed an exact match of this sequence with a potential open reading frame of 2,652 bp. The identified gene codifies a single protein consisting of 884 amino acid residues, which is predicted to be an alanyl APE. This homology, in addition to our experimental results on molecular mass and biochemical and enzymatic properties, appears sufficient to warrant designation of APE97 as an APE N.
A high similarity between the APE97 amino acid sequence and the putative PepN APE from B. suis 1330 was found. Lower similarities were observed with the putative PepN APE from M. loti, S. meliloti, and A. tumefaciens, all of which are phylogenetically related to Brucella (25).
The databanks currently assign the sequences of bacterial alanyl APE to EC 3.4.11.2, for which the recommended name is membrane alanine APE. However, this appears inappropriate, since the bacterial sequences contain no transmembrane or membrane-associated helices or any hydrophobic segments likely to be part of a signal peptide. In contrast, many of the mammalian APE N′s, which are thought to play important roles in the hydrolysis of peptides, are membrane-associated glycoproteins (3). We did not identify hydrophobic segments in the sequence of the putative APE N from B. melitensis M16; on the other hand, it contains the characteristic catalytic site of metallopeptidases in which the zinc ligands are the two histidines in the His-Glu-Xaa-Xaa-His (HEXXH) motif (3).
Bacterial PepN plays an essential role in bacterial metabolism and catabolism, since it may be involved in nitrogen supply and in the degradation of intracellular peptides generated by protein breakdown during normal growth as well as in response to nutrient starvation (3). The functions of five intracellular peptidases, PepN, PepC, PepX, PepT, and PepO, involved in the degradation of oligopeptides in L. lactis have been investigated. Mutations in all of these genes led to slower growth rates in milk relative to the wild-type strain (23). Furthermore, a single mutation of PepN leads to a significant decrease in the growth rate, thus PepN seems to play a more prominent role than do the other proteases (23). Some studies have revealed that environmental stress (e.g., heat, oxidative, and acid shock) rapidly modified gene expression and the corresponding synthesis of proteins in Brucella; these proteins may be related to resistance to live intramacrophagic brucellae (28, 32). In prokaryotes, most ATP-dependent ClpP proteases are involved in protein catabolism under both optimal and stress conditions (26). As heat shock leads to severe down-regulation of ClpP expression in B. melitensis, it is possible that alternative proteases may compensate for its absence. In this context, the role of APE97 and other intracellular enzymes may be involved in the degradation (turnover) and the de novo synthesis of proteins under stress conditions in brucellae. A phenotypic evaluation of an isogenic B. melitensis pepN mutant could shed some light on the mouse infection process and its possible participation in eliciting protective immunity.
The use of metalloproteases in the diagnosis of infectious diseases has been reported, as in the case for systemic candidiasis (6). In the last few years, several immunoreactive Brucella proteins have been identified (12, 30). Nevertheless, there are no reports regarding the antigenic properties of any APE in Brucella. In order to assess whether the APE97 of B. melitensis is immunoreactive, we determined whether patient serum reacted with the protein. Sera from patients with acute and chronic brucellosis recognized the protein while sera from healthy people did not. These results suggest that the described Brucella APE could be used diagnostically to identify Brucella-infected humans. Further studies should be undertaken with sera from humans with brucellosis and from people with other infections produced by gram-negative bacteria to discriminate cross-reactions by enzyme-linked immunosorbent assay.
In summary, this study describes for the first time the presence of an APE in B. melitensis and suggests that such enzyme can be used for diagnostic purposes. At present, the cloning of this 97-kDa APE gene is under way and will permit large-scale production and purification of this protein. Such preparations will enable us to further analyze the peptidase's characteristics as well as to better define the value of this protein in Brucella immunity and diagnosis.
Acknowledgments
This work was partially granted by CONACYT 1631PM and CGPI-IPN 32.1. A.C.-R. and B. R.-Z. were supported by fellowships from CONACYT and PIFI.
We thank S. M. Boyle for critically reading the manuscript and C. H. Hernández-Rodríguez for helpful discussions.
Editor: D. L. Burns
REFERENCES
- 1.Altschul, S. F., T. L. Madden, A. A. Schäffer, J. Zhang, Z. Zhang, W. Miller, and D. J. Lipman. 1997. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25:3389-3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Banbula, A., P. Mak, M. Bugno, J. Silberring, A. Dubin, D. Nelson, J. Travis, and J. Potempa. 1999. Prolyl trypeptidyl peptidase from Porphyromonas gingivalis. J. Biol. Chem. 274:9246-9252. [DOI] [PubMed] [Google Scholar]
- 3.Barret, A. J., N. D. Rawlings, and J. F. Woessner. 1998. Handbook of proteolytic enzymes. Academic Press, San Diego, Calif.
- 4.Corbel, M. J. 1997. Brucellosis: an overview. Emerg. Infect. Dis. 3:213-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.DelVecchio, V., V. Kapatral, R. J. Redkar, G. Patra, C. Mujer, T. Los, N. Ivanova, I. Anderson, A. Bhattacharyya, A. Lykidis, G. Reznik, L. Jablonski, N. Larsen, M. D′Souza, A. Bernal, M. Mazur, E. Goltsman, E. Selkiv, P. H. Elzer, S. Hagius, D. O′Callaghan, J. J. Letesson, R. Haselkon, N. Kyrpides, and R. Overbeek. 2002. The genome sequence of the facultative intracellular pathogen Brucella melitensis. Proc. Natl. Acad. Sci. USA 99:443-448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.El Moundi, B., M. H. Rodier, G. Daniault, and J. L. Jacquemin. 1998. Improved immunodiagnostis of human candidiasis by an enzyme-linked immunosorbent assay using a Candida albicans 52-kilodalton metallopeptidase. Clin. Diagn. Lab. Immunol. 5:823-825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elzer, P. H., R. W. Phillips, G. T. Robertson, and R. M. Roop II. 1996. The HtrA stress response protease contributes to resistance of Brucella abortus to killing by murine phagocytes. Infect. Immun. 64:4838-4841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Exterkate, F. A., M. De Jong, G. J. C. M. De Veer, and R. Baankreis. 1992. Localization and characterization of aminopeptidase N in Lactococcus lactis subsp. cremoris HP. Appl. Microbiol. Biotechnol. 37:46-54. [Google Scholar]
- 9.Falquet, L., M. Pagni, P. Bucher, N. Hulo, C. J. Sigrist, K. Hofmann, and A. Bairoch. 2002. The PROSITE database, its status in 2002. Nucleic Acids Res. 30:235-238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Horvat, R. T., and M. J. Parmely. 1988. Pseudomonas aeruginosa alkaline protease degrades human gamma interferon and inhibits its bioactivity. Infect. Immun. 56:2925-2932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kohler, S., J. Teyssier, A. Cloeckaert, B. Rouot, and J. P. Liautard. 1996. Participation of the molecular chaperone DnaK in intracellular growth of Brucella suis within U937-derived phagocytes. Mol. Microbiol. 20:701-712. [DOI] [PubMed] [Google Scholar]
- 12.Kovach, M. E., P. H. Elzer, G. T. Robertson, R. L. Chirhart-Gilleland, M. A. Christensen, K. M. Peterson, and R. M. Roop II. 1997. Cloning and nucleotide sequence analysis of a Brucella abortus gene encoding an 18 kDa immunoreactive protein. Microb. Pathog. 22:241-246. [DOI] [PubMed] [Google Scholar]
- 13.Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature 227:680-685. [DOI] [PubMed] [Google Scholar]
- 14.Leung, K. Y., and R. M. Stevenson. 1988. Characteristics and distribution of extracellular proteases from Aeromonas hydrophila. J. Gen. Microbiol. 134:151-160. [Google Scholar]
- 15.López-Merino, A. 1989. Brucellosis in Latin America, p. 151-161. In E. J. Young and M. Corbel (ed.), Brucellosis: clinical and laboratory aspects. CRC Press, Inc., Boca Raton, Fla.
- 16.Maeda, H., and T. Yamamoto. 1996. Pathogenic mechanism induced by microbial proteases in microbial infections. Biol. Chem. 377:217-226. [DOI] [PubMed] [Google Scholar]
- 17.Mathew, Z., T. M. Knox, and C. Miller. 2000. Salmonella enterica serovar Typhimurium peptidase B is a leucyl aminopeptidase with specificity for acidic amino acids. J. Bacteriol. 182:3383-3393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsudaira, P. 1987. Sequence from picomole quantities of proteins electroblotted onto polyvinylidene difluoride membranes. J. Biol. Chem. 262:10035-10038. [PubMed] [Google Scholar]
- 19.Maurizi, M. R. 1992. Proteases and protein degradation in Escherichia coli. Experientia 48:178-200. [DOI] [PubMed] [Google Scholar]
- 20.McCaman, M. T., and M. R. Villarejo. 1982. Structural and catalytic properties of peptidase N from Escherichia coli K-12. Arch. Biochem. Biophys. 213:384-394. [DOI] [PubMed] [Google Scholar]
- 21.Meyer, D. H., and P. M. Fives-Taylor. 1998. Oral pathogens: from dental plaque to cardiac disease. Curr. Opin. Microbiol. 1:88-95. [DOI] [PubMed] [Google Scholar]
- 22.Midwinter, R. G., and G. G. Pritchard. 1994. Aminopeptidase N from Streptococcus salivarius subsp. thermophilus NCDO 573: purification and properties. J. Appl. Bacteriol. 77:288-295. [DOI] [PubMed] [Google Scholar]
- 23.Mierau, I., E. R. S. Kunji, K. J. Leenhouts, M. A. Hellendoorn, A. J. Haandrikman, B. Poolman, W. N. Konings, G. Venema, and J. Kok. 1996. Multiple-peptidase mutants of Lactococcus lactis are severely impaired in their ability to grow in milk. J. Bacteriol. 178:2794-2803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mintz, C. S., R. D. Miller, N. S. Gutgsell, and T. Malek. 1993. Legionella pneumophila protease inactivates interleukin-2 and cleaves CD4 on human T cells. Infect. Immun. 61:3416-3421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moreno, E., E. Stackebrandt, M. Dorsch, J. Wolters, M. Busch, and H. Mayer. 1990. Brucella abortus 16S rRNA and lipid A reveal a phylogenetic relationship with members of the alpha-2 subdivision of the class Proteobacteria. J. Bacteriol. 172:3569-3576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Porankiewicz, J., J. Wang, and A. K. Clarke. 1999. New insights into the ATP-dependent Clp protease: Escherichia coli and beyond. Mol. Microbiol. 32:449-458. [DOI] [PubMed] [Google Scholar]
- 27.Porte, F., J. P. Liautard, and S. Kohler. 1999. Early acidification of phagosomes containing Brucella suis is essential for intracellular survival in murine macrophages. Infect. Immun. 67:4041-4047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rafie-Kolpin, M., R. C. Essenberg, and J. H. Wyckoff III. 1996. Identification and comparison of macrophage-induced proteins and proteins induced under various stress conditions in Brucella abortus. Infect. Immun. 64:5274-5283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson, T. G., M. E. Kovach, C. A. Allen, T. A. Ficht, and R. M. Roop II. 2000. The Brucella abortus Lon functions as a generalized stress response protease and is required for wild-type virulence in BALB/c mice. Mol. Microbiol. 35:577-588. [DOI] [PubMed] [Google Scholar]
- 30.Roop, R. M., T. W. Fletcher, N. Sriranganathan, S. M. Boyle, and G. G. Schurig. 1994. Identification of an immunoreactive Brucella abortus HtrA stress response protein homolog. Infect. Immun. 62:1000-1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sanz, Y., and F. Toldrá. 2002. Purification and characterization of an arginine aminopeptidase from Lactobacillus sakei. Appl. Environ. Microbiol. 64:1980-1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Texeira-Gomes, A. P., A. Cloeckaert, and M. S. Zygmunt. 2000. Characterization of heat, oxidative, and acid stress responses in Brucella melitensis. Infect. Immun. 68:2954-2961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Towbin, H., T. Staehelin, and J. Gordon. 1979. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc. Natl. Acad. Sci. USA 76:4350-4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.van Alen-Boerrigter, I. J., R. Bankreis, and W. M de Vos. 1991. Characterization and overexpression of the Lactococcus lactis pepN gene and localization of its product, aminopeptidase N. Appl. Environ. Microbiol. 57:2555-2561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winter, A. J., G. G. Schurig, S. M. Boyle, N. Siranganathan, J. S. Bevins, F. M. Enright, P. H. Elzer, and J. Kopec. 1996. Protection of BALB/c mice against homologous and heterologous species of Brucella by rough strain vaccines derived from Brucella melitensis and Brucella suis biovar 4. Am. J. Vet. Res. 57:677-683. [PubMed] [Google Scholar]
- 36.Yoshimoto, T., Y. Tamesa, K. Gushi, N. Murayama, and D. Tsuru. 1988. An aminopeptidase N from Escherichia coli HB1010: purification and demonstration that the enzyme possesses arylamidase and peptidase activities. Agric. Biol. Chem. 52:217-225. [Google Scholar]