

Abstract
Aims
To study the pharmacokinetics and bioavailability of the prodrug bambuterol and its bronchodilator moiety terbutaline in healthy subjects.
Methods
Eight healthy subjects (four women) received intravenous doses of bambuterol and terbutaline. On a third occasion, they, plus another four subjects, ingested oral bambuterol as a single dose followed by repeated doses once daily for 7 days. Plasma concentrations and urinary excretion of bambuterol and terbutaline were measured.
Results
After intravenous administration, renal clearances of bambuterol and terbutaline were similar (about 140 ml min−1), but there was a five-fold difference in total clearance (bambuterol 1.25 l min−1, terbutaline 0.23 l min−1). Volume of distribution (Vss) was 1.6 l kg−1 b.w. for both substances. A similar renal clearance of bambuterol was found during oral administration but that of terbutaline decreased (to about 120 ml min−1). Mean terminal half-life of bambuterol was 2.6 h after intravenous and 12 h after oral administration, implying that uptake was rate-limiting. Mean residence time of terbutaline generated from oral bambuterol was 34 h compared with 8.0 h when terbutaline as such was infused. Generated terbutaline had a bioavailability of 36% (28–46) after intravenous and 10.2% (6.1–13.2) after oral administration of the prodrug. Bambuterol was well tolerated. The mean activity of plasma cholinesterase, an enzyme catalyzing bambuterol metabolism, was inhibited between 30–60% during repeated oral dosing. It virtually regained original activity within 48 h after the last dose.
Conclusions
The plasma concentration of terbutaline fluctuated little during repeated oral administration (mean peak:trough ratio 1.9), as a result of prolonged absorption of bambuterol and slow formation of terbutaline. Thus, the pharmacokinetic properties of bambuterol make it suitable for oral once-daily dosage.
Keywords: bambuterol, terbutaline, pharmacokinetics, bioavailability, plasma cholinesterase
Introduction
Terbutaline is a β2-sympathomimetic agent used to achieve bronchodilation in the management of asthma; bambuterol is its bisdimethylcarbamate prodrug (Figure 1). Bambuterol itself is devoid of sympathomimetic activity [1]. Terbutaline is formed from bambuterol by hydrolysis predominantly catalysed by plasma cholinesterase (pChE, EC 3.1.1.8) [2, 3] and probably by CYP-450 enzymes [4]. The pathways involve several intermediate metabolites [4, 5], none of which has pharmacological activity.
Figure 1.
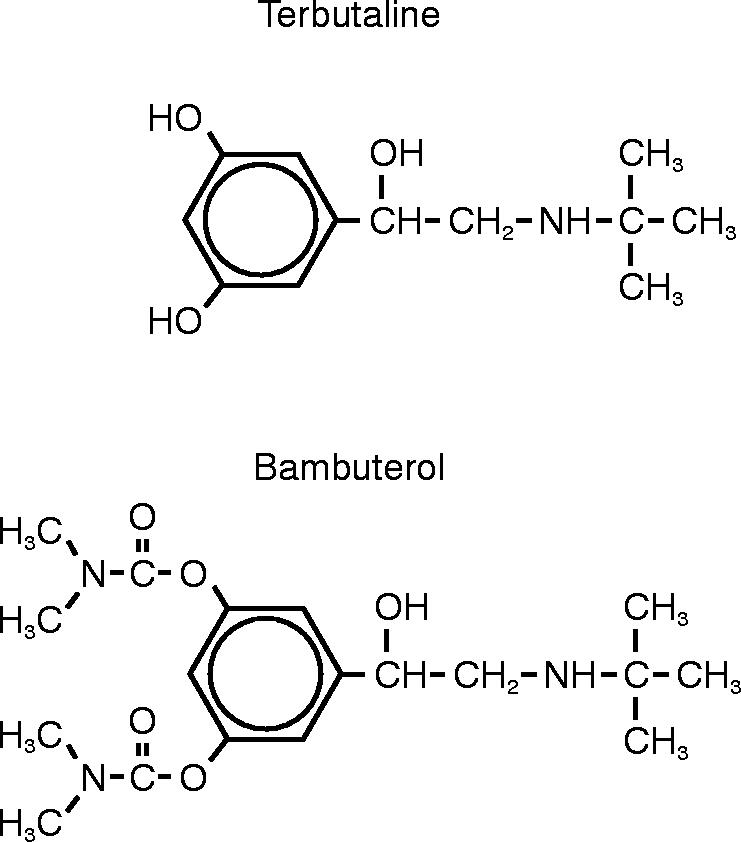
Structural formulae of terbutaline and bambuterol
Oral administration of bambuterol to patients with asthma prolongs the bronchodilating effect produced by terbutaline, thus making once-daily dosing sufficient [6, 7]. The clinical efficacy of once-daily bambuterol appears to be equivalent to that of twice-daily sustained-release terbutaline [8].
The purpose of the present study was to study the fate of bambuterol given intravenously and orally in healthy subjects in order to evaluate the pharmacokinetics and bioavailability of bambuterol and its metabolite terbutaline.
Methods
Study design
This open study was approved by the local Ethics Committee and performed in accordance with the Declaration of Helsinki. Subjects were fully informed verbally and in writing about the nature and objectives of the procedures. Written informed consent was given before entry.
Eight healthy subjects (four women) received single doses of bambuterol and terbutaline intravenously in random order. On another occasion, these subjects (reference group), plus an additional four healthy subjects (two women), received single and multiple doses of bambuterol orally. Additional subjects were included to gain more information regarding the clinical use of bambuterol.
Subjects
Mean age of the 12 participants was 36 (23–62) years and mean weight 69 (55–83) kg. In the reference group, mean baseline pChE activity was normal: 15 (12–21) nmol butyrylthiocholine were hydrolysed per min per 5 μl blood using a modification of a published method [9]. The metabolic ratio for debrisoquine, used as a marker for CYP2D6 polymorphism, was <12.6 in all but one ‘poor metabolizer’, who had a ratio of 84 [10].
Treatments
The women were allowed to use contraceptive pills but other medicines were excluded. The study drugs were given in the evening after a meal, after that no food was allowed for at least 2 h. Isotonic aqueous solutions of bambuterol hydrochloride or terbutaline sulphate were given as 5 min infusions into an arm vein. Target doses were 30.0 and 7.00 μg kg−1 of bambuterol hydrochloride and terbutaline sulphate, respectively. Half the dose was given initially, the rest one hour later. The sequence of the treatments was randomized with a one-week washout period. Exact given doses were calculated after weighing: bambuterol hydrochloride 29.6 (28.4–30.0) μg kg−1 and terbutaline sulphate 6.91 (6.69–7.00) μg kg−1 (73.4 nmol kg−1 of bambuterol, 25.2 nmol kg−1 of terbutaline).
Orally, bambuterol hydrochloride was administered as an aqueous solution in a dose of 270 μg kg−1 (668 nmol kg−1). After the first dose there was an at least 7 day drug-free interval, then the same dose was taken once daily for a further 7 days.
Sampling schedule
Venous blood (10 ml) was drawn from an indwelling catheter and plasma obtained for the measurement of bambuterol and terbutaline up to 48 h after i.v. administration and the first oral dose and up to 72 h after the last oral dose. After i.v. dosing, sampling was frequent during 1.5 h (0, 5, 15, 30, 60, 75, 90 min) then successively more sparse (2, 2.5, 3, 4 h....). After the oral doses, samples were taken hourly up to 4 h, then less frequent. Blood was collected into sodium-heparinized Venoject® sampling tubes into which 100 μl of a 1 mm solution of an esterase inhibitor (D2456, a bambuterol analogue [11]) had been added. The tubes were immediately centrifuged and the plasma was stored at −20° C.
All urine was collected in consecutive pools up to 72 h after start of the infusions and after the single oral dose (0–4 h, 4–12 h, 12–18 h, 18–24 h, then less often). After the last oral dose, urine was sampled up to 96 h (0–24 h, 24–38 h, 38–43 h, then more sparse). Portions of the samples were frozen and saved at −20° C.
After the start of the first infusion of bambuterol, pChE activity in 1 ml blood samples was measured frequently up to 2 h, then less frequent. After the first oral dose, pChE activity was measured at 0, 1, and 24 h, after the last oral dose also at 48 h and at 2 weeks. The samples were immediately chilled on ice and assayed within 15 to 90 min.
Bioanalysis
Bambuterol was analysed by gas chromatography-mass spectrometry as described previously [11]. At 1 nmol l−1 in plasma and 8 nmol l−1 in urine, the within-day coefficient of variation (CV) was about 4% and 5.4%, respectively. The lower limit of quantification applied was 0.5 nmol l−1 in plasma and 4 nmol l−1 in urine. The between-day CV was 6.6% at 3 nmol l−1 for plasma samples and 10.6% at 12.8 nmol l−1 for urine samples.
Terbutaline was analysed by use of coupled-column high performance liquid chromatography with electrochemical detection [12]. At 8 nmol l−1 in plasma, the CV was 2.1% within a day and 3.6% between days. The lower limit of quantification was 4 nmol l−1 in plasma. The same method was applied for terbutaline in urine. The assay calibration curve was then extended up to 5000 nmol l−1 to allow for measurement of the higher concentrations in urine. At 320 nmol l−1, the between-day CV was 4.6% and at 120 nmol l−1 7.2%. Concentrations of terbutaline in urine down to 80 nmol l−1 were accepted in the calculations (within-day variation 3.7%).
Clinical assessments
All adverse events reported by the subjects or observed by the staff were documented. Blood pressure and radial pulse were monitored up to 2 h after the start of each infusion and up to 24 h after oral doses. Subjects rested supine for 5 min and refrained from smoking for 30 min before assessment. Tremor was assessed up to 24 h after the first and last oral doses of bambuterol. Small vertical movements of the sitting subject's right midfinger were measured using an optoelectronic position sensor [13]. The distance travelled by the finger during a 10 s interval was recorded. Baseline measurements were made during a drug-free interval.
Symbols and calculations
Terbutaline-T=terbutaline given as such
Terbutaline-B=terbutaline derived from administration of bambuterol
Cmax, Cmin=maximum and minimum plasma concentrations, respectively
tmax=the time when Cmaxoccurred
AUC=the area under the curve of plasma concentration vs. time, calculated by the linear trapezoidal rule up to time t, i.e. AUC(0,t), where t after the last oral dose is 24 h. After a single dose, extrapolation to infinite time was estimated as last measurable concentration divided by λz, where λz is the terminal rate constant calculated from plasma or urinary excretion data
Css=AUC divided by dosing interval (24h)
AUMC=area under the first moment of the plasma concentration-time curve, calculated after single doses by the linear trapezoidal rule with extrapolation to infinite time, the latter portion being estimated as the last measurable plasma concentration, at time tlast, multiplied by {1/λz× (1/λz+tlast)}
CL=total plasma clearance, calculated as intravenous dose divided by AUC
Ae=amount excreted in urine
CLR=renal clearance, in a defined time (0,t) divided by AUC(0,t)
MRT=mean residence time, calculated as AUMC/AUC after intravenous dosage minus t piv, where t piv (pivotal infusion time) is t1+(t2−t1)×Dose 2/(Dose 1+Dose 2), t1 and t2 being the infusion time midpoints
MAT=mean absorption time of bambuterol
MFT=mean formation time of terbutaline
AUMC/AUC=apparent mean residence time of terbutaline-B:
(MFT+MRTTT) after intravenous bambuterol
(MAT+MFT+MRTTT) after oral bambuterol
where subscript TT stands for terbutaline-T
Vss=steady-state volume of distribution, defined by CL× MRT
t1/2=terminal half-life, ln 2/λz
F=bioavailability, AUC of orally given bambuterol or generated terbutaline divided by the intravenous AUC of bambuterol or terbutaline, respectively. The AUCs were corrected for dose tCss=the time during a steady-state dosing interval when plasma concentration equals Css. tCss was calculated by interpolation
MIT=mean inhibition time for pChE activity calculated analogously to MRT, with plasma concentration being replaced by degree of enzyme inhibition, i.e. {100–100×pChE/pChE(baseline)}
When plasma concentrations fell below acceptable values after intravenous administration, urinary excretion rates were converted to plasma concentrations situated in the middle of the collection interval by dividing excretion rate in the interval (dAe/dt) by CLR(0,4h).
Statistical considerations and methods
Data are given as arithmetic means and ranges. By paired t-tests, parameters after the first oral dose were compared with those after the intravenous doses regarding t1/2 for bambuterol plus CLR(0–4h) for terbutaline and with those after the last oral dose regarding AUCs of bambuterol and terbutaline. The model was multiplicative for AUC and CLR and additive for t1/2. The level of significance was 0.05.
Results
Pharmacokinetics after intravenous bambuterol and terbutaline (Table 1)
Table 1.
Pharmacokinetics of bambuterol and generated terbutaline (B) plus terbutaline (T) after intravenous infusions of bambuterol and terbutaline
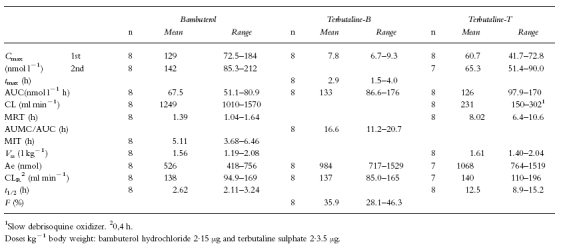
The decline in plasma concentrations of bambuterol after intravenous dosing appeared to be biphasic (Figure 2a). The terminal phase predominated from approximately 4 h after the start of the first infusion. CL was rapid (1250 ml min−1) with an about 10% renal contribution. Vss was 1.56 l kg−1, greater in the women (1.50–2.08 l kg−1) than in the men (1.19–1.43 l kg−1). MRT was 1.39 h, which was approximately 50% of the mean t1/2 (2.62 h).
Figure 2.

a) Log plasma concentration vs time curves of bambuterol (triangles, dashed line) and terbutaline-B (circles, full line) after intravenous infusion of bambuterol (mean 73.4 nmol kg−1). Filled symbols are measured concentrations, open symbols are concentrations calculated from urinary excretion rate and renal clearance. b) Log plasma concentration vs time curves of terbutaline-T after intravenous infusion of terbutaline (mean 25.2 nmol kg−1). Filled symbols are measured concentrations, open symbols are concentrations calculated from urinary excretion rate and renal clearance
There was no evident relation between the rate of generation of terbutaline-B and the CYP2D6 phenotype or hydrolytic capacity of the subjects. The decline in plasma terbutaline-B and -T (Figures 2a and b) appeared to be biphasic or possibly triphasic. The terminal phase was not entered until at least 12 h after the start of the first infusion.
CL of terbutaline-T was 231 ml min−1 after intravenous terbutaline (60% renal contribution). Vss of terbutaline-T (1.61 l kg−1) was similar to that seen for bambuterol, but men and women did not differ. The mean MRT of terbutaline-T (8.02 h) was approximately 65% of its mean t1/2 (12.5 h).
Approximately one-third (36%) of intravenously administered bambuterol was converted to terbutaline. The average AUMC/AUC for terbutaline-B (16.6 h) was twice as long as MRT of terbutaline-T, but terminal half-lives were similar (Figures 2a and b).
Pharmacokinetics after oral bambuterol (Tables 2 and 3)
Table 2.
Pharmacokinetic parameters of bambuterol and generated terbutaline after a single oral dose of 270 μg bambuterol hydrochloride kg−1 body weight. Italic numbers are values in the subgroup also given intravenous reference doses (eight subjects with documented normal pChE activity and seven being fast debrisoquine oxidizers), in the cases when their values differed from the full group
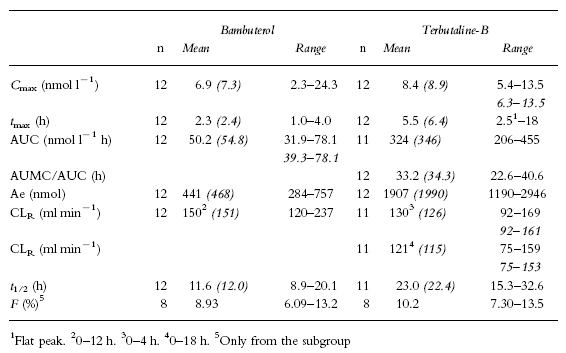
Table 3.
Pharmacokinetic parameters of bambuterol and generated terbutaline after repeated oral dosing of 270 μg bambuterol hydrochloride kg−1 body weight. Italic numbers are values in the subgroup also given intravenous reference doses (eight subjects with documented normal pChE activity and seven being fast debrisoquine oxidizers), in the cases when their values differed from the full group
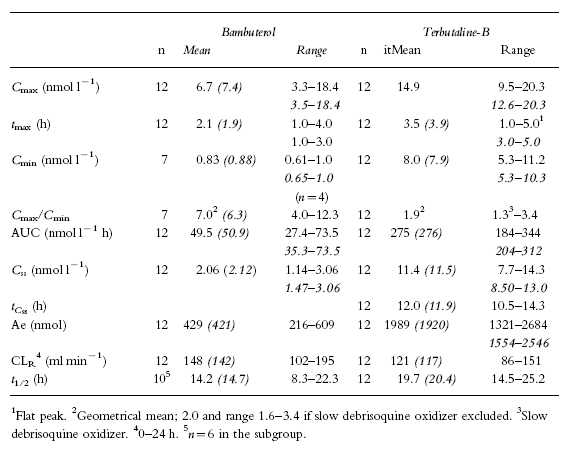
Plasma concentrations after the first and last dose of bambuterol were similar (Figures 3a and b). Mean AUC after the single dose and AUC(0,24 h) after the last dose of bambuterol were not significantly different (P=0.837). The initial decline in plasma concentration resembled that observed after intravenous bambuterol, but the the mean t1/2 of the single dose in the reference group was 12.0 h, that is 4–5 times longer (P=0.002) than after intravenous bambuterol. After repeated doses (Figure 4) it was 14.7 h. The mean bioavailability of a single oral dose of bambuterol was 8.9%.
Figure 3.

a) Plasma concentration vs time curves of bambuterol and terbutaline-B after the first oral dose of bambuterol (0.668 μmol kg−1). Symbols and lines as in Figure 2a. b) Plasma concentration vs time curves of bambuterol and terbutaline-B after the last oral dose of bambuterol (0.668 μmol kg−1 day−1). Symbols and lines as in Figure 2a
Figure 4.

Log urinary excretion rate vs time curves of bambuterol (▵) and terbutaline-B (○) after the last oral dose of bambuterol (0.668 μmol kg−1 day−1). The rate is plotted in the middle of the dosing interval
The plasma concentrations of terbutaline-B were higher during repeated dosing than after the first dose of bambuterol, but the mean AUC(0,24 h) of terbutaline-B after the last dose of bambuterol did not significantly differ from the mean AUC (extrapolated to infinite time) after the first dose (P=0.108). The bioavailability of terbutaline-B evaluated from the single dose was approximately 10%. CLR after the first oral dose (0–4 h) was not significantly different from that after the intravenous dose of bambuterol (P=0.316). Calculations of CLR up to infinite time were not reliable because of a large extrapolated part of the AUC; however, CLR up to 18 h was similar to that during the first 4 h. t1/2 of terbutaline-B (about 20 h) was longer after oral administration than after intravenous infusion of terbutaline-T.
Mean AUMC/AUC for terbutaline-B after oral administration of bambuterol was 33–34 h. This was calculated by use of t1/2 after repeated administration because the estimates of terbutaline t1/2 were less reliable after a single dose of bambuterol. The calculated parameter covers all the processes from absorption of bambuterol to the elimination of terbutaline. It was about twice as long as AUMC/AUC for terbutaline-B after infusion, implying that the absorption process gives a great contribution to the sustaining of the plasma concentrations of terbutaline. Indeed, they were sustained to such an extent that only in three subjects did the peak-to-trough concentration ratio of terbutaline exceed 2 (mean value 1.9) during repeated dosing with bambuterol.
Plasma cholinesterase activity
There was an immediate, almost complete inhibition of the activity of pChE after each of the two intravenous doses of bambuterol (Figure 5). However, 24 h after the start of infusion, the activity on average had returned to 99.1% of baseline (range 88.4–109.8%). The degree of inhibition was related to plasma bambuterol levels, but mean MIT following intravenous bambuterol (3.7–6.5 h, Table 1) was longer than the MRT for bambuterol (1.0–1.6 h).
Figure 5.

Plasma cholinesterase activity after intravenous administration of bambuterol (73.4 nmol kg−1)
Inhibition was less pronounced but more prolonged after oral bambuterol. One hour after the first oral dose, mean pChE activity was 59.7 (12.7–92.4)% of baseline, whereas after 24 h it was 71.1 (55.4–84.3)%. During the last dosing interval, mean pChE activity at dosing was 67.3 (54.7–77.2)% of baseline, i.e. similar to that found 24 h after the first dose, but at 1 h it was more reduced: 40.1 (16.8–59.6)%. At 48 h after the last oral dose, the activity was 95.2 (85.2–123.9)%. Measurements 2 weeks after the last dose showed complete restoration of pChE activity.
β2-mediated systemic adverse events
Terbutaline-T increased pulse by a mean of 25 (8–52) beats min−1 and reduced diastolic blood pressure by a mean of 18 (0–40) mm Hg. These cardiovascular effects, normally associated with β2-adrenoceptor stimulation [14], were much less pronounced after bambuterol. For example, the mean peak increase in pulse was 3.1 (0–6) beats min−1 after intravenous bambuterol and 4.8 (0–12) beats min−1 after the last oral dose. Baseline tremor was rather stable over the day: the finger oscillations corresponded to 6.3–7.4 mm/10 s as a mean during drug-free measurement. Skeletal muscle tremor is increased by β2-adrenoceptor agonists [15], but the increase after oral bambuterol was modest and similar after the first single dose and in steady state, except at 1 h where mean tremor was increased by 26% after the single dose and by 47% at steady state. In most subjects, peak tremor occurred 4 h after dose representing a mean increase of 80–90% over baseline. Fifteen hours after dose, mean tremor was only increased by 30–40%. Mean peak tremor correlated with mean peak plasma concentrations of terbutaline, but on an individual basis there was not a good correlation.
Discussion
Basal pharmacokinetics of terbutaline-T and bambuterol
The plasma concentration of intravenously given terbutaline has previously been shown to decrease in a multi-exponential manner, indicating that terbutaline is to a great extent distributed outside the plasma compartment. Volume of distribution was found to exceed body water (Vss>0.6 l kg−1) and the terminal phase of elimination was entered approximately 12 h post-dose [16]. The present study confirms these previous findings.
Also bambuterol, a more lipophilic substance than terbutaline [4], would be expected to distribute considerably outside the plasma compartment. The biphasic shape of the decline of bambuterol in plasma after intravenous administration and a Vss as great as 1.56 l kg−1 confirm this prediction. Volume of distribution of bambuterol normalized to body weight was greater in the women, possibly related to a higher percentage of body fat. On the other hand, no gender difference was seen in the distribution volume of the very hydrophilic terbutaline.
The mean residence time after a single dose of a drug will be longer than the half-life only if the drug is solely distributed within the sampling compartment [17]. If a significant fraction is distributed into deeper compartments, and if redistribution from those compartments limits the overall rate of elimination, MRT will be smaller than seen with a single-compartmental behaviour [18]. It may even be shorter than the terminal half-life which was found with bambuterol and terbutaline in this study and which appears logical since the two drugs distribute considerably outside total body water.
The renal clearance of bambuterol (0.14 l min−1) is only about 1/10 of total plasma clearance (1.25 l min−1), most of which should therefore be the result of elimination in liver (CYP 450) and in plasma (pChE).
Generation and pharmacokinetics of terbutaline-B after intravenous bambuterol
About one-third of the intravenous bambuterol dose became bioavailable as terbutaline. As residence times are additive [18], the apparent mean residence time of terbutaline-B (AUMC/AUC) is the sum of the MRT of terbutaline itself (terbutaline-T) plus the mean formation time (MFT) of terbutaline from bambuterol. The MRT of bambuterol is in turn included in the MFT of terbutaline-B. Subtracting MRT of terbutaline-T (8.0 h, Table 1) from AUMC/AUC of terbutaline-B (16.6 h) gives a MFT of terbutaline-B of 8–9 h, i.e. about six times longer than MRT of (intravenous) bambuterol. It must therefore be a large contribution to the MFT of terbutaline-B from the residence times of intermediate metabolites formed in the conversion of bambuterol. The clearance values show that approximately 90% of the infused dose of bambuterol was eliminated by nonrenal routes; still only one-third of the dose was available as terbutaline-B. Thus, a large proportion of the generated intermediary metabolites was excreted before being transformed into terbutaline-B. The influence of these intermediates is also reflected by the fact that mean inhibition time of pChE activity (MIT) was almost four times longer than MRT of bambuterol.
Oral bioavailability and pharmacokinetics of bambuterol and terbutaline-B
The time for a drug to reach steady state is governed by its longest half-life. The terminal half-life of bambuterol after a single oral dose ranged from 9 to 20 h, implying that all subjects should have attained steady-state levels of plasma bambuterol during the 7th dosing interval. Provided linear pharmacokinetics, total AUC after the single dose should be the same as AUC(0,24 h) during a steady-state dosing interval. This was found with bambuterol to the extent that its plasma concentration-time profiles were similar after the first and last dose, thus implying that virtually all of the parent drug disappeared already during the dosing interval.
The terminal half-life of bambuterol was 4–5 times longer after the oral dosage than after intravenous administration. Slow absorption of a drug can mask the rate of elimination so that the terminal half-life reflects absorption rather than elimination [19], suggesting that uptake from the gut is a major determinant for the terminal decline of plasma bambuterol after oral administration. This was also reflected by the twofold greater AUMC/AUC-value of terbutaline-B after oral as compared with intravenous bambuterol. It appears, however, that the elimination rate after oral bambuterol was not entirely limited by its uptake rate, since the initial decline in plasma levels (Figures 3a and b) resembled that observed after intravenous administration (Figure 2a).
The terminal half-life of terbutaline-T when administered as plain tablets has been estimated to 17 h [16]. Apparently, the decline in terbutaline-B after oral bambuterol was slower, possibly as a result of sustained formation of terbutaline-B. However, as seen in Table 3 individual terminal half-lives were ≤1 day and, therefore, also terbutaline should be in steady state during the 7th dosing interval of bambuterol. This conclusion was corroborated by the fact that similar plasma concentrations of terbutaline-B were found at the end of the last two dosing intervals (0 and 24 h in Figure 3b).
The bioavailability of terbutaline-B after the first oral dose of bambuterol was approximately 10%, which is identical to the oral bioavailability of terbutaline given as such during nonfasting conditions [16]. The AUC(0,24 h) of terbutaline-B after the last dose did not significantly differ from the AUC after the first dose. It was the same as has been found in asthmatic patients during repeated treatment with bambuterol [7]. The addition of four subjects when bambuterol was given orally did not change the found parameters to any appreciable extent (Tables 2 and 3). Furthermore, despite the finding that one subject was a poor metabolizer of debrisoquine, he was still well capable of forming terbutaline but did it in a more sustained way (very flat steady-state plasma concentration profile).
It has been proposed [20] that if one aims at a peak-to-trough concentration ratio during the dosing interval of 2, the length of the interval should be ln2MRT (∼0.7MRT). In the case of terbutaline formed from oral bambuterol, the apparent mean residence time (AUMC/AUC) was 33.2 h (Table 2). This would then predict that dosing every 23 h would give a Cmax/Cmin-ratio of 2 which is in good agreement with the present finding (1.9) and supports the clinically recommended once-daily regimen with bambuterol.
Tremor
The maximum increase of tremor over baseline in this study was 80–90%. In a study where terbutaline was given repeatedly 3 times daily in a dose which would produce a similar AUC of terbutaline as in the present study, the maximum increase of tremor was 250% after the first dose and at steady state nearly 200% [13]. A doubling of baseline tremor is usually needed before a subject becomes aware of the side-effect [21].
Conclusion
The bronchodilator terbutaline is generated slowly after administration of its prodrug bambuterol. The bioavailability of terbutaline is approximately 1/3 of the actual intravenous dose of bambuterol. After oral administration of bambuterol, the bioavailability of bambuterol and terbutaline is about the same, i.e. circa 10% of the administered dose. The plasma profile of terbutaline after bambuterol administration shows little fluctuation within dosing intervals, seemingly a consequence of prolonged absorption of bambuterol and slow generation of terbutaline. The pharmacokinetic properties of bambuterol make it suitable for an oral once-daily dosage regimen.
References
- 1.Olsson OAT, Svensson L. New lipophilic terbutaline ester prodrugs with long effect duration. Pharm Res. 1984;3:19–23. doi: 10.1023/A:1016322524471. [DOI] [PubMed] [Google Scholar]
- 2.Tunek A, Levin E, Svensson L. Hydrolysis of 3H-bambuterol, a carbamate prodrug of terbutaline, in blood from humans and laboratory animals in vitro. Biochem Pharmacol. 1988;37:3867–3876. doi: 10.1016/0006-2952(88)90068-8. [DOI] [PubMed] [Google Scholar]
- 3.Tunek A, Svensson L. Bambuterol, a carbamate ester prodrug of terbutaline, as inhibitor of cholinesterase in human blood. Drug Metab Dispos. 1988;16:759–764. [PubMed] [Google Scholar]
- 4.Svensson L, Tunek A. The design and bioactivation of presystemically stable prodrugs. Drug Metab Rev. 1988;19:165–194. doi: 10.3109/03602538809049622. [DOI] [PubMed] [Google Scholar]
- 5.Lindberg C, Roos C, Tunek A, Svensson LÅ. Metabolism of bambuterol in rat liver microsomes: identification of hydroxylated and demethylated products by liquid chromatography mass spectrometry. Drug Metab Dispos. 1989;17:311–322. [PubMed] [Google Scholar]
- 6.Petrie GR, Chookang JY, Hassan WU, et al. Bambuterol: effective in nocturnal asthma. Respir. 1993;87:581–585. doi: 10.1016/s0954-6111(05)80260-4. [DOI] [PubMed] [Google Scholar]
- 7.D’Alonzo G, Smolensky M, Feldman S, et al. Bambuterol in the treatment of asthma. A placebo-controlled comparison of once-daily morning vs evening administration. Chest. 1995;107:406–412. doi: 10.1378/chest.107.2.406. [DOI] [PubMed] [Google Scholar]
- 8.Fugleholm AM, Ibsen TB, Laxmyr L, Svendsen UG. Therapeutic equivalence between bambuterol, 10 mg once daily, and terbutaline controlled release, 5 mg twice daily, in mild to moderate asthma. Eur Respir J. 1993;6:1474–1478. [PubMed] [Google Scholar]
- 9.Augustinsson K-B, Eriksson H, Faijersson Y. A new approach to determining cholinesterase activities in samples of whole blood. Clin Chim Acta. 1978;89:239–252. doi: 10.1016/0009-8981(78)90322-4. [DOI] [PubMed] [Google Scholar]
- 10.Steiner E, Iselius L, Alvàn G, et al. A family study of genetic and environmental factors determining polymorphic hydroxylation of debrisoquine. Clin Pharmacol Ther. 1985;38:394–401. doi: 10.1038/clpt.1985.193. [DOI] [PubMed] [Google Scholar]
- 11.Lindberg C, Jönsson S, Paulson J. Determination of bambuterol, a prodrug of terbutaline, in plasma and urine by gas chromatography mass spectrometry. Biomed Environ Mass Spectrom. 1990;19:218–224. doi: 10.1002/bms.1200190403. [DOI] [PubMed] [Google Scholar]
- 12.Kennedy B-M, Blomgren A, Edholm L-E, Roos C. Quantitative determination of terbutaline in human plasma after administration of bambuterol using coupled columns and electrochemical detection. Chromatographia. 1987;24:895–899. [Google Scholar]
- 13.Bengtsson B, Fagerström P-O. Extrapulmonary effects of terbutaline during prolonged administration. Clin Pharmacol Ther. 1982;31:726–732. doi: 10.1038/clpt.1982.102. [DOI] [PubMed] [Google Scholar]
- 14.Weiner N. Norepinephrine, epinephrine, and the sympathomimetic amines. In: Goodman Gilman A, Goodman LS, Rall TW, Murad F., editors. The Pharmacological Basis of Therapeutics. Seventh. New York: Macmillan Publishing Company; 1985. pp. 145–180. [Google Scholar]
- 15.Ahrens RC. Skeletal muscle tremor and the influence of adrenergic drugs. J Asthma. 1990;27:11–20. doi: 10.3109/02770909009073289. [DOI] [PubMed] [Google Scholar]
- 16.Nyberg L. Pharmacokinetic parameters of terbutaline in healthy man. An overview. Eur J Resp Dis. 1984;65(suppl.134):149–160. [PubMed] [Google Scholar]
- 17.Rowland M, Tozer T. Clinical Pharmacokinetics, Concepts and Applications. Second. London: Lea and Febiger; 1989. Mean residence time (Appendix F) pp. 479–485. [Google Scholar]
- 18.Riegelman S, Collier P. The application of statistical moment theory to the evaluation of in vivo dissolution time and absorption time. J Pharmacokinet Biopharm. 1980;8:509–534. doi: 10.1007/BF01059549. [DOI] [PubMed] [Google Scholar]
- 19.Rowland M, Tozer T. Clinical Pharmacokinetics, Concepts and Applications. Second. London: Lea & Febiger; 1989. Extravascular dose; pp. 33–48. [Google Scholar]
- 20.Benet LZ. Mean residence time in the body versus mean residence time in the central compartment. J Pharmacokinet Biopharm. 1985;13:555–558. doi: 10.1007/BF01059337. [DOI] [PubMed] [Google Scholar]
- 21.Hörnblad Y, Ripe E, Magnusson P-O, Tegnér K. The metabolism and clinical activity of terbutaline and its prodrug ibuterol. Eur J Clin Pharmacol. 1976;10:9–18. [Google Scholar]