

Abstract
Although anticancer agents are one of the most toxic classes of medication prescribed today, there is relatively little information available about clinically relevant drug–drug interactions. Pharmacokinetic drug interactions have been described, including alterations in absorption, catabolism, and excretion. For example, an increased bioavailability of 6-mercaptopurine has been observed when combined with either allopurinol or methotrexate, leading to increased toxicity in some patients. Induction of etoposide or teniposide clearance by anticonvulsants has also been described, resulting in a lower systemic exposure and risk for lower anticancer activity. Alterations in elimination of methotrexate has been observed with probenecid, presumably through competition for renal secretion. There are also several examples of pharmacodynamic interactions. The combination of 5-fluorouracil plus folinic acid results in more efficient inhibition of thymidylate synthase, a finding which is now utilized routinely in the treatment of colorectal cancer. Improvements in the in vitro and early clinical testing now allow a relatively high degree of prediction of potential clinical drug interactions, prior to observations of untoward drug effects. In conclusion, drug interactions among commonly used anticancer agents have been identified. Their clinical significance can have more impact than many other classes of medications due to the narrow therapeutic index of antineoplastic agents and the potential for lethal side-effects. It is only through prospective, preclinical and early clinical evaluation that the presence of clinically significant drug interactions can be identified and the information used to provide better therapy for this significant health problem.
Keywords: cancer chemotherapy, drug interactions, pharmacodynamics, pharmacokinetics
Introduction
Cancer is a major public health issue, with ≈1 in 3 individuals developing a malignancy, including nonmelanoma skin cancer, during their lifetime. Cancer is diagnosed in over 270 000 people each year in the United Kingdom alone and is the most common cause of death in adults. The diagnosis and treatment of cancer also represents a significant proportion of total health care expenditure throughout the world.
The current treatment of cancer requires administration of toxic poisons, many of which have a narrow therapeutic index [1]. For most anticancer drugs the dose required to have activity against the tumour also has a high level of haematopoietic toxicity, mucositis, or other systemic toxicities. A strong correlation between the degree of toxicity and the level of antitumour response has been found for several anticancer agents. Many anticancer agents undergo hepatic metabolism via common enzyme pathways such as cytochrome P450 3A (Table 1). In addition, several anticancer drugs are metabolized by polymorphic enzyme pathways such as thiopurine methyltransferase (6-mercaptopurine, azathioprine) and dihydropyrimidine dehydrogenase (5-fluorouracil). Renal elimination also has a significant influence on the disposition of anticancer drugs (Table 1). Carboplatin is primarily eliminated by glomerular filtration (≈75%) and many centres individualise the dose of this drug based on kidney function [2]. Methotrexate is also eliminated by the kidneys and has been known to influence its own elimination by inducing nephrotoxicity [3]. The narrow therapeutic index and the potential for lethal side-effects make drug interactions in oncology of particular concern, yet drug interactions are not widely recognized as a clinical problem in oncology [4]. Part of the reason for this is that most anticancer drugs are administered in combination regimens and therefore it is difficult to identify the specific agent(s) involved in the interaction. In addition, there is a high level of accepted (expected) systemic toxicity with anticancer agents, decreasing the concern over exaggerated toxicity in specific patients. There have only been a limited number of preclinical drug interaction studies or clinical blood level monitoring of anticancer therapies in order to detect significant pharmacokinetic interactions with commonly used agents.
Table 1.
Examples of anticancer agents which undergo hepatic metabolism and renal elimination.

Pharmacokinetic drug interactions
Pharmacokinetic drug interactions have been identified with several anticancer agents at the level of absorption, metabolism, and elimination. The thiopurine agent 6-mercaptopurine, which is commonly used in childhood leukaemia, is metabolized by both thiopurine methyltransferase and xanthine oxidase to inactive metabolites [5]. Administration of 6-mercaptopurine to patients receiving the xanthine oxidase inhibitor allopurinol leads to significantly higher 6-mercaptopurine plasma concentrations [6] (Figure 1). More extensive studies have found that it is the influence of allopurinol on gastrointestinal and hepatic xanthine oxidase that is most important, leading to a greater bioavailability of 6-mercaptopurine [6]. The increase in systemic exposure from this interaction has led to severe toxicity or death in a number of patients [7]. The anticancer agent methotrexate has also been reported to enhance the bioavailability of oral 6-mercaptopurine, probably through inhibition of xanthine oxidase [8, 9]. Pre-clinical and clinical studies have shown synergism between methotrexate and 6-mercaptopurine antineoplastic activity at both low and high dosages [8, 9]. This combination is commonly used in the treatment of lymphoblastic leukaemia, but the potential for interaction is not well recognized.
Figure 1.
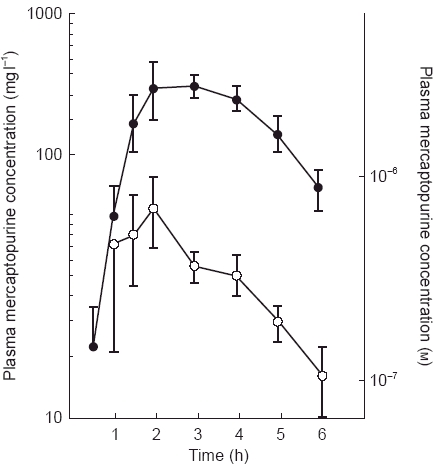
Plasma 6-mercaptopurine concentrations after oral administration (75 mg day−1) with (•) and without (○) allopurinol pretreatment (100 mg three times day−1 for 2 days). Reproduced from Reference 6, with permission.
The anticonvulsants phenobarbitone and phenytoin have long been recognized to induce hepatic enzymes and are involved in a number of clinically significant drug interactions [10]. Administration of etoposide and teniposide to patients receiving anticonvulsant therapy found a significant increase in systemic clearance of the anticancer agents with a lower achieved plasma area under the concentration-time curve (AUC) [11, 12]. A comparison of teniposide pharmacokinetics prior to and after the initiation of anticonvulsant therapy demonstrated a three-fold increase in systemic clearance (Figure 2) [11]. This drug interaction has a significant impact on the clinical use of teniposide and etoposide as the plasma concentrations of these agents are strongly correlated with both systemic toxicity and, in some cases, antitumour activity [1].
Figure 2.
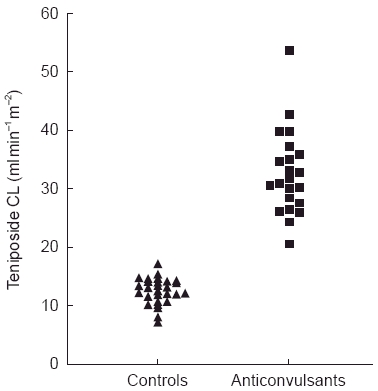
Teniposide systemic clearance values for all treatment courses administered to six children receiving anticonvulsant therapy (phenobarbitone or phenytoin) and six control children. All clearance values for the control patients were lower than all clearance values for the patients treated with anticonvulsants. Reproduced from reference 11, with permission.
A similar interaction has been demonstrated between 5-fluorouracil (5-FU) and interferon (Figure 3). Coadministration of these agents is common in the treatment of colorectal carcinomas and the mechanism behind their synergistic activity has been primarily evaluated at the cellular level. However, several pharmacokinetic studies have shown that interferon-α inhibits dihydropyrimidine dehydrogenase, the enzyme responsible for greater than 80% of 5-FU metabolism, leading to a significant increase in 5-FU plasma AUC [13]. This pharmacokinetic interaction has not been found with all studies [14] and the influence of 5-FU dose and schedule on this interaction is currently under evaluation.
Figure 3.
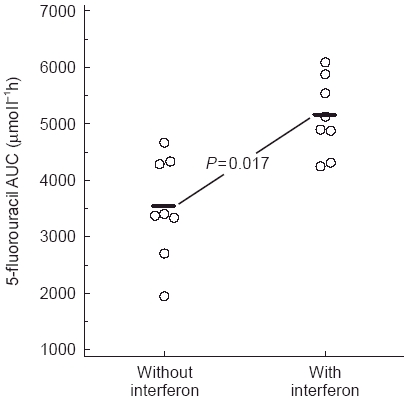
Area under the plasma concentration-time curve (AUC) of patients treated with 5-fluorouracil (375 mg m−2 day−1 for 7 days) with or without interferon α (5–10×106 units m−2 day−1 for 7 days). Reproduced from reference 13, with permission.
While the role of dihydropyrimidine dehydrogenase in the degradation of 5-FU has been well recognized for greater than 30 years, there have been recent failures in prospectively identifying potential drug interactions with this anticancer agent. In 1993, 16 Japanese patients died from drug interactions of oral 5-FU pro-drugs with the new oral antiviral drug sorivudine, within 40 days after its approval by the Japanese Government for the clinical treatment of herpes zoster [15, 16]. All the patients were receiving 5-FU pro-drugs daily for long-term anticancer chemotherapy when sorivudine was administered by daily treatment for 7 days. Within several days after the sorivudine administration patients showed symptoms of severe toxicity including bloody diarrhoea, and marked decreases in white blood cell and platelet counts. It is thought that a sorivudine metabolite which is generated by gut flora, (E)-5- (2-bromovinyl)uracil, suppressed DPD catabolism of 5-FU in these patients, resulting in an increased incorporation of FU into cytotoxic nucleotides [16]. (E)-5- (2-bromovinyl)uracil is a potent inhibitor of rat liver dihydropyrimidine dehydrogenase and enhances the plasma concentration of 5-FU when 5-FU and (E)-5- (2-bromovinyl)uracil are coadministered [16]. Although studies from 1986 identified the inhibition of DPD by related compounds, this information did not appear to assist in the clinical development of sorivudine by identifying patients in which significant drug interactions could occur [15, 16].
Interactions between 5-FU and the anticoagulant warfarin have also been reported in the literature [17]. While some studies have suggested a pharmacokinetic (metabolic) interaction between these two agents, the clinical relevance of this mechanism is not clear. However, 5-FU has been shown to increase bleeding time and PTT in patients on warfarin therapy and has been responsible for severe morbidity and prolonged hospitalization in several cases [17].
Few drug interactions involving elimination have been reported, primarily due to multiple mechanisms of elimination (biliary, renal) found with most drugs. As mentioned above, methotrexate is heavily reliant on renal elimination. Coadministration of probenecid, which influences drug secretion in the kidney, led to prolonged methotrexate elimination, increased elimination half-life, and an elevated AUC [18] (Figure 4). This interaction has led to increased systemic toxicity, including severe neutropenia, in patients taking low-dose methotrexate [19]. Altered methotrexate elimination has also been found in patients receiving amphotericin and similar drugs with renal toxicity including patients receiving the anticancer drug cisplatin [3].
Figure 4.
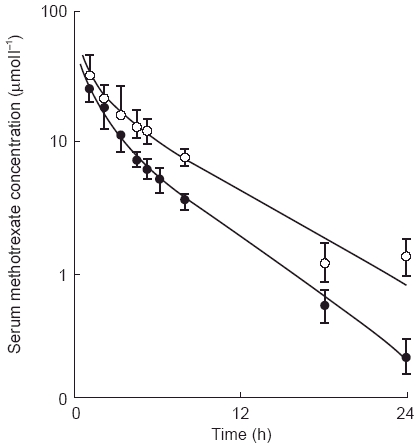
Serum concentrations of methotrexate after intravenous bolus injection (200 mg m−2) in patients receiving methotrexate alone (•) and with probenecid (○) (500 mg 1 h prior and 5 h after methotrexate administration). Reproduced from reference 18, with permission.
Pharmacodynamic drug interactions
Pharmacodynamic drug interactions with anticancer agents have not been extensively evaluated. One positive pharmacodynamic drug interaction has been found between 5-FU and folinic acid (leucovorin) [20]. 5-FU induces its anticancer activity in part, by inhibiting thymidylate synthase (TS) through formation of a ternary complex between 5-FU-TS-and reduced folates. Many tumour cells have been found to have relatively low levels of reduced folates, thereby influencing the efficiency of thymidylate synthase inhibition by 5-FU. Administration of folinic acid prior to 5-FU therapy is now commonly used as a mechanism to optimize the use of this anticancer therapy for the treatment of colorectal cancer, breast cancer and head/neck carcinomas. Pharmacokinetic studies found that folinic acid does not influence the metabolism of 5-FU either in vitro or in vivo [20].
A pharmacodynamic interaction has also been found for the combination of a platinum agent (cisplatin or carboplatin) with a taxane (paclitaxel or docetaxel). Carboplatin undergoes primarily renal elimination to a greater extent than cisplatin. Paclitaxel is metabolized by CYP3A4 and CYP2C8 to less active metabolites. Docetaxel is metabolized by CYP3A isoform (s), that have not yet been specifically identified. Studies of the influence of drug sequence on toxicity and activity found that administration of paclitaxel followed by carboplatin led to a decrease in the formation of platinum adducts in patient DNA, which is thought to be the primary lesion in carboplatin antitumour activity [21–23]. Patients treated by this schedule had less haematopoietic toxicity compared with patients treated with carboplatin followed by paclitaxel. No difference in the pharmacokinetics of carboplatin or paclitaxel were found with either administration schedule. A similar pattern of pharmacokinetics was found for the combination of cisplatin and docetaxel, but significantly lower platinum DNA adducts were found in patients treated with docetaxel followed by cisplatin [24]. While the mechanism for this interaction has not been fully elucidated, the decreased markers of activity when taxanes are administered prior to platinum compounds has led to the use of the platinum agent followed by the taxane in the phase II/III studies.
Combined interactions
The relative contributions of pharmacokinetic and pharmacodynamic interactions are not always clearly obvious, as has been demonstrated by several studies attempting to bypass the role of p-glycoprotein in multidrug resistance [25]. The p-glycoprotein has been shown to function as a membrane efflux pump which removes a number of structurally unrelated compounds from the cytosolic compartment. P-glycoprotein has been found to be elevated in drug resistant tumour cells both in vitro and in vivo, leading to the hypothesis that enhanced drug efflux is a mechanism for drug resistance. Many clinical trials have since been performed looking at non cytotoxic p-glycoprotein substrates as a means for modulating the cellular retention of some anticancer agents. This included a trial with a combination of cyclosporin and etoposide [26]. While both agents are p-glycoprotein substrates, both agents are also metabolised by CYP3A4 leading to the potential for either pharmacokinetic or pharmacodynamic interactions. Studies of etoposide administered alone followed by a combination of cyclosporin and etoposide demonstrated a significant increase in plasma AUC, a decrease in systemic clearance and altered volume of distribution with coadministration of cyclosporin. as etoposide plasma concentrations relate closely with haematopoietic toxicity, pharmacodynamic modelling was performed to try to identify the relative contribution of pharmacokinetics or pharmacodynamics to this interaction. Maximum effect modelling demonstrated a sigmoidal relationship between plasma etoposide AUC and decrease in white blood cell count [26]. Closer examination of this relationship in courses with and without cyclosporin found that the interaction was primarily of a pharmacokinetic nature, as the change in white blood cell count in patients with increased etoposide systemic exposure due to coadministration with cyclosporin was well predicted by the same sigmoidal model used for the courses with etoposide alone [25].
Predictive testing for drug interactions
The traditional models for in vitro testing of hepatic drug interactions have been performed for most of the more recently developed anticancer agents. For example, studies in a panel of human livers identified CYP3A4 and CYP2C8 as the enzymes primarily responsible for metabolising paclitaxel [27, 28]. In addition a number of CYP3A4 substrates were shown to directly interact with paclitaxel metabolism in the in vitro systems [27]. However, the systems that are traditionally applied in the preclinical study do not provide information on the relative impact of these drug interactions, nor do they evaluate the role of non P450 enzymes. The potential for acute toxicity and tumourigenesis prevents clinical drug interaction studies in healthy volunteer subjects, impairing the ability to test these questions in a relatively controlled environment. One model that provides a potentially interesting approach for in vitro testing of the impact of drug interactions with antineoplastic agents is the use of lymphocytes transfected with specific human P450 s [29]. While this model does not provide an environment for looking at the role of multiple enzymes in the metabolism of a specific agent, it does allow the evaluation of both metabolism as well as cytotoxicity, in that the viability of lymphocytes after treatment with an anticancer agent and an interacting agent can be tested using the clonogenic assay or similar measures of cytotoxicity. Transfection of human lymphocytes with CYP2A6 led to increased cytotoxicity by cyclophosphamide, an agent which needs activation to have its desired antitumour effect (Figure 5) [30]. The cytotoxicity could be prevented by incubation with the CYP2A6 inhibitor coumarin, confirming the functional significance of this enzyme in this in vitro system (Figure 5). This system provides one tool in which to evaluate the potential for an identified drug interaction to have clinical relevance, at least in terms of haematopoietic toxicity.
Figure 5.
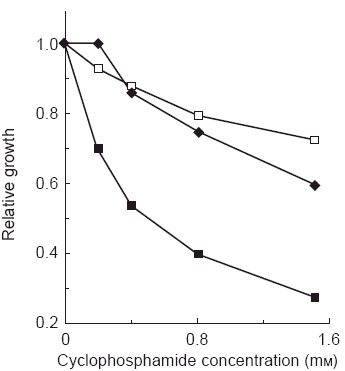
Cytotoxicity of cyclophosphamide in cultured human B-lymphoblastoid cells expressing CYP2A6, with (♦) or without (▪) the inhibitor coumarin (50 μm). Control cells (□) were transformed with vector alone. Reproduced from reference 30, with permission.
The clinical use of drug interactions
Further investigations are also taking place into using identified drug interactions to decrease variability or pharmacy expenditure for anticancer agents. This approach is following the description by several groups that coadministration of ketoconazole or other CYP3A4 inhibitors (e.g. grapefruit juice) in solid organ transplant patients receiving immunosuppresive therapy with cyclosporin led to a decreased drug dosage required to achieve therapeutic drug levels and an overall cost savings for the particular regimen [31, 32]. The combination of allopurinol and azathioprine has already been proposed in an attempt to increase the systemic availability of azathioprine in a small number of kidney transplant recipients [33]. This approach may also be applicable to the use of 6-mercaptopurine in children with leukaemia. As the high cost of many of the more recently developed anticancer agents are a burden on healthcare systems world wide, the approach could be taken in which inhibitors of CYP3 A4, for example, could be coadministered with drugs such as paclitaxel. The current limitation to direct application of this approach to anticancer agents is the relative lack of information of the contribution of most metabolites to systemic toxicity or more importantly antitumour activity. Until this information is available, the use of drug interactions to optimize anticancer therapy should not be performed outside of the experimental setting.
In conclusion, drug interactions among commonly used anticancer agents have been identified. Their clinical significance can have more impact than many other classes of medications due to the narrow therapeutic index of antineoplastic agents and the potential for lethal side-effects. It is only through prospective, preclinical and early clinical evaluation that the presence of clinically significant drug interactions can be identified and the information used to provide better therapy for this significant health problem.
Table 2.
Areas for potential drug interactions.

Acknowledgments
Many thanks to Maria Johnson in the preparation of this manuscript. Support of the author's laboratory from the Aberdeen Royal Hospitals Research & Development Office and the University of Aberdeen Faculty of Medicine is greatly appreciated.
References
- 1.McLeod HL. Therapeutic drug monitoring opportunities in cancer therapy. Pharmacol Ther. 1997;74:39–54. doi: 10.1016/s0163-7258(96)00201-x. [DOI] [PubMed] [Google Scholar]
- 2.Calvert AH. Dose optimization of carboplatin in adults. Anticancer Res. 1994;14:2273–2277. [PubMed] [Google Scholar]
- 3.Relling MV, Fairclough D, Ayers D, et al. Patient characteristics associated with high-risk methotrexate concentrations and toxicity. J Clin Oncol. 1994;12:1667–1672. doi: 10.1200/JCO.1994.12.8.1667. [DOI] [PubMed] [Google Scholar]
- 4.van Meerten E, Verweij J, Schellens JHM. Antineoplastic agents: Drug interactions of clinical significance. Drug Safety. 1995;12:168–182. doi: 10.2165/00002018-199512030-00003. [DOI] [PubMed] [Google Scholar]
- 5.Lennard L. The clinical pharmacology of 6-mercaptopurine. Eur J Clin Pharmacol. 1992;43:329–339. doi: 10.1007/BF02220605. [DOI] [PubMed] [Google Scholar]
- 6.Poplack DG, Balis FM, Zimm S. The pharmacology of orally administered chemotherapy. A reappraisal. Cancer. 1986;58:473–480. doi: 10.1002/1097-0142(19860715)58:2+<473::aid-cncr2820581311>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 7.Kennedy DT, Hayney MS, Lake KD. Azathioprine and allopurinol: The price of an avoidable drug-interaction. Ann Pharmacother. 1996;30:951–954. doi: 10.1177/106002809603000906. [DOI] [PubMed] [Google Scholar]
- 8.Balis FM, Holcenberg JS, Zimm S, et al. The effect of methotrexate on the bioavailability of oral 6-mercaptopurine. Clin Pharmacol Ther. 1987;41:384–387. doi: 10.1038/clpt.1987.45. [DOI] [PubMed] [Google Scholar]
- 9.Innocenti F, Danesi R, Dipaolo A, et al. Clinical and experimental pharmacokinetic interaction between 6-mercaptopurine and methotrexate. Cancer Chemother Pharmacol. 1996;37:409–414. doi: 10.1007/s002800050405. [DOI] [PubMed] [Google Scholar]
- 10.Riva R, Albani F, Contin M, Baruzzi A. Pharmacokinetic interactions between antiepileptic drugs—Clinical considerations. Clin Pharmacokin. 1996;31:470–493. doi: 10.2165/00003088-199631060-00005. [DOI] [PubMed] [Google Scholar]
- 11.Baker DK, Relling MV, Pui CH, et al. Increased teniposide clearance with concomitant anticonvulsant therapy. J Clin Oncol. 1992;10:311–315. doi: 10.1200/JCO.1992.10.2.311. [DOI] [PubMed] [Google Scholar]
- 12.Rodman JH, Murry DJ, Madden T, Santana VM. Altered etoposide pharmacokinetics and time to engraftment in pediatric patients undergoing autologous bone marrow transplantation. J Clin Oncol. 1994;12:2390–2397. doi: 10.1200/JCO.1994.12.11.2390. [DOI] [PubMed] [Google Scholar]
- 13.Grem JL, Chu E, Boarman D, et al. Biochemical modulation of fluorouracil with leucovorin and interferon: preclinical and clinical investigations. Semin Oncol. 1992;19(S3):36–44. [PubMed] [Google Scholar]
- 14.Seymour MT, Patel N, Johnston A, Joel SP, Slevin ML. Lack of effect of interferon alpha-2A upon fluorouracil pharmacokinetics. Br J Cancer. 1994;70:724–728. doi: 10.1038/bjc.1994.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Swinbanks D. Deaths bring clinical trials under scrutiny in Japan. Nature. 1994;369:697. doi: 10.1038/369697b0. [DOI] [PubMed] [Google Scholar]
- 16.Watabe T. Strategic proposals for predicting drug–drug interactions during new drug development: Based on sixteen deaths caused by interactions of the new antiviral sorivudine with fluorouracil prodrugs. J Toxicol Sci. 1996;21:299–300. doi: 10.2131/jts.21.5_299. [DOI] [PubMed] [Google Scholar]
- 17.Brown MC. Multisite mucous membrane bleeding due to a possible interaction between warfarin and 5-fluorouracil. Pharmacotherapy. 1997;17:631–633. [PubMed] [Google Scholar]
- 18.Aherne GW, Piall E, Marks V, et al. Prolongation and enhancement of serum methotrexate concentrations by probenecid. Br Med J. 1978;1:1097–1099. doi: 10.1136/bmj.1.6120.1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Basin KS, Escalante A, Beardmore TD. Severe pancytopenia in a patient taking low-dose methotrexate and probenecid. J Rheumat. 1991;18:609–610. [PubMed] [Google Scholar]
- 20.Machover D. A comprehensive review of 5-fluorouracil and leucovorin in patients with metastatic colorectal carcinoma. Cancer. 1997;80:1179–1187. doi: 10.1002/(sici)1097-0142(19971001)80:7<1179::aid-cncr1>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 21.Baker SD. Drug interactions with the taxanes. Pharmacotherapy. 1997;17:S126–S32. [PubMed] [Google Scholar]
- 22.Calvert AH. A review of the pharmacokinetics and pharmacodynamics of combination carboplatin/paclitaxel. Semin Oncol. 1997;24(S2):S85–S90. [PubMed] [Google Scholar]
- 23.Kearns CM, Egorin MJ. Considerations regarding the less than expected thrombocytopenia encountered with combination paclitaxel/carboplatin chemotherapy. Semin Oncol. 1997;24(S2):S91–S96. [PubMed] [Google Scholar]
- 24.Schellens JHM, Ma J, Bruno R, et al. Pharmacokinetics of cisplatin and taxotere (docetaxel) and WBC DNA-adduct formation of cisplatin in the sequence taxotere/cisplatin and cisplatin/taxotere in a phase I/II study in solid tumor patients. Proc Am Soc Clin Oncol. 1994;13:132. [Google Scholar]
- 25.McLeod HL. Clinical reversal of the multidrug resistance phenotype: True tumor modulation or pharmacokinetic interaction. Eur J Cancer. 1994;30A:2039–2041. doi: 10.1016/0959-8049(94)00423-3. [DOI] [PubMed] [Google Scholar]
- 26.Lum BL, Kaubisch S, Yahanda AM, et al. Alteration of etoposide pharmacokinetics and pharmacodynamics by cyclosporine in a phase I trial to modulate multidrug resistance. J Clin Oncol. 1992;10:1635–1642. doi: 10.1200/JCO.1992.10.10.1635. [DOI] [PubMed] [Google Scholar]
- 27.Harris JW, Rahman A, Kim BR, Guengerich FP, Collins JM. Metabolism of taxol by human hepatic microsomes and liver slices: Participation of cytochrome P450, 3A4 and an unknown P450 enzyme. Cancer Res. 1994;54:4026–4035. [PubMed] [Google Scholar]
- 28.Rahman A, Korzekwa KR, Grogan J, Gonzalez FJ, Harris JW. Selective biotransformation of taxol to 6-alpha-hydroxytaxol by human cytochrome P450, 2C8. Cancer Res. 1994;54:5543–5546. [PubMed] [Google Scholar]
- 29.Crespi CL, Penman BW, Gonzalez FJ, Gelboin HV, Galvin M, Langenbach R. Genetic toxicology using human cell lines expressing human P-450. Biochem Soc Trans. 1993;21:1023–1028. doi: 10.1042/bst0211023. [DOI] [PubMed] [Google Scholar]
- 30.Chang TKH, Weber GF, Crespi CL, Waxman DJ. Differential activation of cyclophosphamide and ifosphamide by cytochrome P450, 2B and cytochrome; P450, 3A: in human liver microsomes. Cancer Res. 1993;53:5629–5637. [PubMed] [Google Scholar]
- 31.Jones TE. The use of other drugs to allow a lower dosage of cyclosporin to be used—Therapeutic and pharmacoeconomic considerations. Clin Pharmacokinet. 1997;32:357–367. doi: 10.2165/00003088-199732050-00002. [DOI] [PubMed] [Google Scholar]
- 32.Sobh M, Elagroudy A, Moustafa F, Harras F, Elbedewy M, Ghoneim M. Coadministration of ketoconazole to cyclosporine treated kidney transplant recipients: A prospective randomized study. Am J Nephrol. 1995;15:493–499. doi: 10.1159/000168892. [DOI] [PubMed] [Google Scholar]
- 33.Chocair P, Duley J, Simmonds HA, et al. Low-dose allopurinol plus azathioprine cyclosporine prednisolone, a novel immunosuppressive regimen. Lancet. 1993;342:83–84. doi: 10.1016/0140-6736(93)91287-v. [DOI] [PubMed] [Google Scholar]