

Abstract
Accumulating evidence indicates that CYP2C9 ranks amongst the most important drug metabolizing enzymes in humans. Substrates for CYP2C9 include fluoxetine, losartan, phenytoin, tolbutamide, torsemide, S-warfarin, and numerous NSAIDs. CYP2C9 activity in vivo is inducible by rifampicin. Evidence suggests that CYP2C9 substrates may also be induced variably by carbamazepine, ethanol and phenobarbitone. Apart from the mutual competitive inhibition which may occur between alternate substrates, numerous other drugs have been shown to inhibit CYP2C9 activity in vivo and/or in vitro. Clinically significant inhibition may occur with coadministration of amiodarone, fluconazole, phenylbutazone, sulphinpyrazone, sulphaphenazole and certain other sulphonamides. Polymorphisms in the coding region of the CYP2C9 gene produce variants at amino acid residues 144 (Arg144Cys) and 359 (Ile359Leu) of the CYP2C9 protein. Individuals homozygous for Leu359 have markedly diminished metabolic capacities for most CYP2C9 substrates, although the frequency of this allele is relatively low. Consistent with the modulation of enzyme activity by genetic and other factors, wide interindividual variability occurs in the elimination and/or dosage requirements of prototypic CYP2C9 substrates. Individualisation of dose is essential for those CYP2C9 substrates with a narrow therapeutic index.
Keywords: CYP2C9, cytochrome P450, drug interactions, drug metabolism, pharmacogenetics
Introduction
Cytochrome P450 (CYP) comprises a superfamily of haemoproteins which function as the terminal oxidase of the mixed function oxidase system. At least 481 CYP genes and 22 pseudogenes are known to exist across all species [1]. These CYP genes are classified into families (designated by an Arabic numeral) and subfamilies (designated by a letter) according to the amino acid identity of the encoded proteins. Of the thirty-five known human CYP genes, currently classified in families 1,2,3,4,5,7,8,11,17,19,21,24,27 and 51 [1], only the eighteen forms comprising families 1–3 appear to substantially contribute to the metabolism of drugs and non-drug xenobiotics [2–4]. The remainder are of importance in the metabolism and/or biosynthesis of endogenous compounds such as bile acids, biogenic amines, eicosanoids, fatty acids, phytoalexins, retinoids and steroids [1].
Xenobiotic metabolising CYP isoforms typically exhibit characteristic, but occasionally overlapping, patterns of substrate specificities and inhibitor profiles [2, 3]. Accordingly, the phenotype of an individual with respect to isoform content at any given time may influence the metabolic clearances of drugs eliminated by CYP catalysed biotransformation. Numerous factors are known to affect CYP isoform activity and, being the products of discrete genes, regulation of expression differs from one isoform to another. Environmental, genetic and physiological influences not uncommonly lead to wide interindividual variability in the pharmacokinetics and hence response of drugs metabolised predominantly by CYP [2–5]. Thus, characterisation of the substrate specificities of individual CYP isoforms and an understanding of their regulation is essential for the prediction of likely alterations in drug metabolic clearance in defined population groups [6, 7].
In recent years there has been increased recognition of the importance of CYP2C9 in drug metabolism. Here we describe features of CYP2C9 of importance to the clinical pharmacology of drugs metabolised by this enzyme.
Structure of CYP2C9
CYP2C9 is one of four known members of the human CYP2C subfamily, although genomic analysis suggests the possible existence of three additional CYP2C enzymes [8]. Other known members of the human CYP2C subfamily include CYP2C8, CYP2C18 and CYP2C19. (While CYP2C10 was originally also considered a discrete isoform, this enzyme is now thought to be a variant of CYP2C9 [1, 9].) Cytochromes P450 2C8, 2C9, 2C18 and 2C19 share >82% amino acid identity [9]. Despite the high level of sequence similarity, the human CYP2C isoforms exhibit relatively little overlap of substrate specificity [2, 9].
CYP2C9 protein has been purified from human liver [10, 11] and a number of variant CYP2C9 cDNAs have been isolated [8, 9, 12–17]. The deduced amino acid sequences of the CYP2C9 variants differ at only a few residues; the significance of the existence of allelic variants on CYP2C9 activity is discussed under ‘Pharmacogenetics’. The CYP2C9 cDNAs encode proteins of 490 amino acids which all have a calculated molecular mass of ≈55.6 KDa. Six substrate recognition sites (SRSs) have been identified for CYP2C9 and other CYP2 gene family proteins based on sequence alignments with CYP101 (a bacterial enzyme for which X-ray cystallographic data are available) and analysis of mutations which alter substrate specificity [18]. These SRSs, which show more non-synonymous substitutions than other regions of the protein, span residues 96–117, 198–205, 233–240, 286–304, 359–369 and 470–477 of the CYP2C9 protein. From studies with chimeric proteins and site-directed mutagenesis it is apparent that the ability of CYP2C9 and closely related isoforms to metabolise a particular substrate or group of compounds may be defined by a few critical residues or even a single amino acid within an SRS. For example, the Ile359Leu substitution in SRS5 of CYP2C9 markedly decreases the hydroxylation of tolbutamide and phenytoin [15] but not that of diclofenac [19]. Similarly, the Phe114Leu substitution (in SRS1) decreases CYP2C9-like warfarin hydroxylation and inhibition by sulphaphenazole without affecting diclofenac hydroxylation [20].
Using amino acid sequence alignment data and the crystal structure of the bacterial enzyme CYP101, a preliminary three dimensional model of CYP2C9 has been constructed [21]. Although potential substrate recognition sites distal to the porphyrin were identified, the resolution of the homology model was considered to be low and of little value in the prediction of substrate binding. Pharmacophore models of the CYP2C9 active site based on the structures of known substrates and inhibitors (up to 1995) have also been developed using molecular modelling approaches [22–24]. Most, but not all, known drug substrates for CYP2C9 are weak acids with pKa values in the range 3.8–8.1 and an electrostatic interaction arising from an electronegative group on the substrate and an electropositive group on the enzyme appears to be of major importance in determining the affinity of these compounds. Whether binding occurs via the charged substrate or due to hydrogen bonding with the protonated form is yet to be established unequivocally. An active site model predicted from three-dimensional quantitative structure-activity relationships for a series of coumarin and non-coumarin inhibitors of CYP2C9 comprised the I helix, a phenyl (hydrophobic) binding site that centres the oxidation site of the substrate over the active oxygen species, the porphyrin, and at least one cationic binding site [24]. Interestingly, dibenzo[a,h]-anthracene and benzo[a]pyrene have been identified recently as CYP2C9 substrates [25, 26]. Unlike most drug substrates for CYP2C9, these neutral, highly hydrophobic compounds do not possess an acidic functional group (or indeed any functional group) and this is consistent with the notion that different SRSs (and their individual amino acids) within any CYP isoform variably contribute to the recognition and binding of discrete classes of substrates.
Drug substrates for CYP2C9
In recent years a variety of approaches have been developed for the identification of the human CYP isoforms involved in the hepatic metabolism of any given drug in vitro [6, 7]. These include: (i) investigation of correlations between the rates of metabolism of the drug and immunoreactive CYP isoform contents or prototypic isoform-specific activities in a ‘panel’ of human liver microsomes; (ii) comparison of the kinetics of metabolism of the drug by human liver microsomes and cDNA-expressed or purified isoforms; (iii) competitive inhibition of the metabolism of isoform specific substrates by the drug, using both human liver microsomes and expressed isoforms, and; (iv) characterisation of the effects of known isoform specific xenobiotic or antibody inhibitors on the metabolism of the drug by human liver microsomes. Although all approaches have been useful in linking substrates to CYP2C9, there is little doubt that application of the inhibitor sulphaphenazole has provided the most useful single tool in defining CYP2C9 drug substrate specificity. It has been demonstrated that sulphaphenazole is both a specific and potent (apparent Ki≈0.1–0.2 μm) inhibitor of human liver microsomal CYP2C9 [27–31]. Furthermore, coadministration of sulphaphenazole and tolbutamide (a prototypic CYP2C9 substrate, see below) results in almost total abolition of the metabolic clearance component tolbutamide elimination in vivo [32–34]. Thus, the extent of sulphaphenazole inhibition of a CYP catalysed reaction provides a measure of the contribution of CYP2C9 to that metabolic pathway.
Structures of selected known CYP2C9 drug substrates and sites of oxidation are shown in Figure 1. Although these compounds appear structurally diverse, many share a number of features. As noted earlier, most, but not all, CYP2C9 substrates are weak acids with the anionic site being ≈7 Å from the carbon oxidised [22, 23]. Evidence supporting the involvement of CYP2C9 in the metabolism of prototypic substrates and other drugs is summarised below. Since several of the drugs (notably phenytoin, S-warfarin and tolbutamide) metabolised by CYP2C9 have narrow therapeutic indices, the activity of this enzyme becomes an important determinant of both the clearance and response (therapeutic and toxic) of such drugs in an individual.
Figure 1.
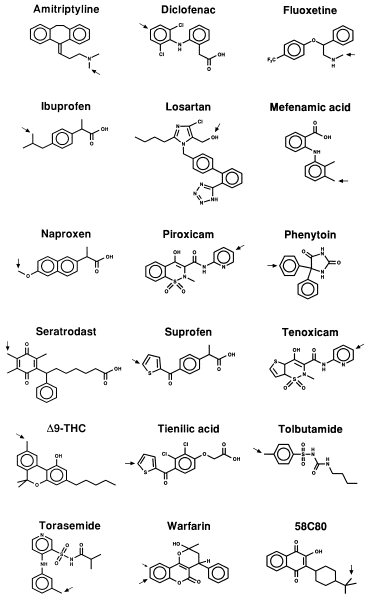
Selected CYP2C9 substrates. The site(s) of oxidation of each substrate is indicated by an arrow(s).
Tolbutamide
In humans tolbutamide is metabolised almost exclusively along a single pathway. Methylhydroxylation to form hydroxytolbutamide is the initial and rate-limiting step; subsequent oxidation of hydroxytolbutamide by alcohol and aldehyde dehydrogenases produces carboxytolbutamide [35, 36]. Overall, this pathway accounts for up to 85% of tolbutamide clearance in humans [32, 35]. Evidence that CYP2C9 is essentially solely responsible for tolbutamide hydroxylation is overwhelming and tolbutamide is accepted widely as a prototypic substrate for the assessment of hepatic CYP2C9 activity, both in vitro and in vivo [30].
Human hepatic tolbutamide hydroxylation in vitro is almost completely abolished by sulphaphenazole [27, 28] and the tolbutamide hydroxylase activity of human liver microsomes is inhibited competitively by other CYP2C9 substrates, with closely matching apparent Ki and Km values [37, 38]. cDNA-expressed CYP2C9 catalyses the hydroxylation of tolbutamide and, importantly, apparent Km values for the recombinant enzyme fall within the range observed with human liver microsomes and the profile of sulphaphenazole inhibition is similar for both enzyme sources (Figure 2) [15, 29]. Although recombinant CYP2C8 and CYP2C18 additionally hydroxylate tolbutamide, affinities are considerably lower than those observed for CYP2C9 [15, 39, 40]. There is also strong evidence supporting the involvement of CYP2C9 in tolbutamide hydroxylation in vivo. Pretreatment of subjects with sulphaphenazole decreases the plasma clearance of tolbutamide by 80% [32–34]. Given the capacity of CYP2C9 to metabolise tolbutamide and the structurally similar compound torasemide (see later), it is possible that this enzyme may also contribute to the elimination of other chemically-related first- and second-generation oral hypoglycaemic agents.
Figure 2.
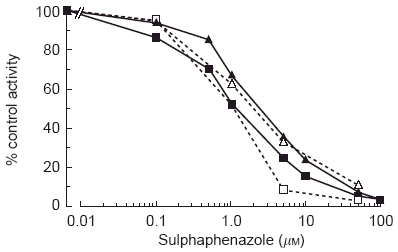
Sulphaphenazole inhibition of phenytoin and tolbutamide hydroxylation in human liver microsomes (HLM) and COS cells transfected with a CYP2C9 (wild type) cDNA. □—□ phenytoin, HLM; ▪—▪ phenytoin, CYP2C9; ▵—▵ tolbutamide, CYP2C9; ▴—▴ tolbutamide, HLM. The concentrations of phenytoin and tolbutamide were 150 μm and 1000 μm, respectively. Data from references 15 and 30.
Phenytoin
The major pathway of phenytoin metabolism is 4′–hydroxylation to form 5-(4 p–hydroxyphenyl)-5-phenylhydantoin (HPPH), which accounts for ≈80% of phenytoin elimination [41]. Like tolbutamide, phenytoin plasma clearance is known to be diminished markedly by sulphaphenazole coadministration [42], indicating likely involvement of CYP2C9 in HPPH formation. Evidence that CYP2C9 is indeed responsible for the conversion of phenytoin to HPPH was provided by investigations in vitro and in vivo. Human liver microsomal HPPH formation was almost abolished by sulphaphenazole (Figure 2) and inhibited competitively by tolbutamide, and Michaelis constants for HPPH formation by human liver microsomes and recombinant CYP2C9 were similar in value [15, 37]. Moreover, rates of HPPH formation correlated highly with rates of tolbutamide methylhydroxylation and S-warfarin 7hydroxylation (see below) in microsomes from panels of human livers [37, 43], and plasma unbound clearances of phenytoin and tolbutamide were coregulated in 18 healthy subjects [44]. As is the case with tolbutamide, these data indicate that CYP2C9 activity is rate-limiting in phenytoin metabolic clearance.
S-Warfarin
Approximately 80 to 85% of the more potent (S)-enantiomer of warfarin is eliminated by CYP catalysed biotransformation to 6- and 7-hydroxy (S)-warfarin, which form in vivo in about a 1:3 ratio [45]. Thus, the activity of the isoform responsible for the formation of these metabolites regulates the steady-state plasma concentration of (S)-warfarin and therefore becomes a critical factor in anticoagulant response. It is now accepted that CYP2C9 is the catalyst for the 6- and 7-hydroxylation of (S)-warfarin at therapeutically relevant substrate concentrations [43, 46, 47]. In particular, sulphaphenazole and tolbutamide are selective inhibitors of these pathways in human liver microsomes and recombinant CYP2C9 (but not other isoforms) displays the same stereoselectivity, regioselectivity and kinetic characteristics associated with human hepatic (S)-warfarin 6- and 7-hydroxylation [46, 47]. Drug-drug interactions involving altered metabolism of (S)-warfarin, phenytoin and tolbutamide show broad similarities (see later), consistent with the involvement of a common isoform in the metabolic clearance of all three drugs.
Non-steroidal anti-inflammatory drugs (NSAIDs)
There is increasing evidence that CYP2C9 is important in the oxidative metabolism of acetic acid, propionic acid, fenemate and oxicam NSAIDs. Sulphaphenazole and/or tolbutamide inhibition along with kinetic studies utilising recombinant CYP2C9 indicate that this isoform is probably solely responsible for the human hepatic 4′–hydroxylation of R- and S-flurbiprofen and is the major contributor to the 2- and 3-hydroxylations of R- and S-ibuprofen [22, 48, 49], the dominant metabolic pathways for each of these drugs. Similar approaches have demonstrated that CYP2C9 is involved in the O-demethylation of S-naproxen and hydroxylation of the thiophene ring of suprofen [22, 50, 51], but these pathways of secondary importance to acyl glucuronide formation [52]. CYP1A2 was also found to contribute to S-naproxen O-demethylation [50, 51], almost to the same extent as CYP2C9. This contrasts to the apparent lack of involvement of CYP1A2 in the human liver microsomal 2- and 3-hydroxylations of ibuprofen and 4′–hydroxylation of flurbiprofen. In vitro studies have similarly linked CYP2C9 to the 4′–hydroxylations of aceclofenac and diclofenac, the 5′–hydroxylations of piroxicam, tenoxicam and lornoxicam (6-chlorotenoxicam), and the 3-hydroxylation of mefenamic acid [53–57], the major metabolic pathways for each of these drugs.
Torasemide
Torasemide is a loop diuretic structurally related to tolbutamide (Figure 1). Like tolbutamide, tolyl hydroxylation and subsequent oxidation to form a carboxylic acid is the major elimination pathway of torsemide in vivo and in vitro [38, 58]. A combination of in vitro procedures, including sulphaphenazole inhibition, was employed to demonstrate that CYP2C9 was almost solely responsible for human liver microsomal torasemide tolyl methylhydroxylation and hence activity of this enzyme would be expected to be rate-limiting in torasemide elimination [38]. Given the higher turnover of torasemide compared with tolbutamide and (S)-warfarin, torasemide represents a useful substrate probe for the assessment of CYP2C9 activity in vitro. Kinetic and inhibitor studies with human liver microsomes and cDNA expressed isoforms also showed that the major metabolic pathway (viz 5-hydroxylation) of another diuretic (and uricosuric agent), tienilic acid, was catalysed predominantly by CYP2C9 [22, 59]. Furthermore, CYP2C9 converts tienilic acid to a reactive species which alkylates CYP2C9 itself and this is the basis of the autoimmune hepatitis observed in some patients administered this drug [59, 60]. Recent studies have mapped the autoantibody binding sites to amino acids 314–322, 345–356 and 439–455 of CYP2C9 [61], which are distinct to the residues involved in substrate recognition and binding (see previous section).
Other drug substrates of CYP2C9 identified using in vitro approaches include amitriptyline, fluoxetine, seratrodast, losartan, Δ9-tetrahydrocannabinol (Δ9-THC) and 2-(4-t-butylcyclohexyl)-3-hydroxy-1,4-naphthoquinone (codenamed 58C80). Application of human liver microsomal kinetic and inhibitor approaches and the use of heterologously expressed CYP isoforms demonstrated that, at therapeutically relevant plasma concentrations, CYP2C9, CYP2D6 and an unidentified enzyme (which was not CYP 1A2, 2C19 or 3A4) variably contribute to the N-demethylation of amitriptyline to form nortriptyline [62]. Microsomal inhibition studies have similarly implicated CP2C9, together with CYP3A4, in the N-demethylation of fluoxetine [63]. Sulphaphenazole inhibited norfluoxetine formation in vitro by more than 50%, suggesting an important role for CYP2C9 in this pathway.
The CYP catalysed metabolism of the angiotensin II receptor antagonist losartan is somewhat unusual; the primary alcohol side-chain attached to the imidazole ring (see Figure 1) is oxidised via an intermediate aldehyde to produce the pharmacologically active carboxylic acid metabolite [64]. CYP2C9 has been implicated in the oxidation of losartan itself and in the oxidation of the aldehyde produced in the two step reaction sequence. However, comparative inhibition experiments with sulphaphenazole, gestodene and ketoconazole demonstrated that CYP3A4 may also partly contribute to both reactions, especially oxidation of the intermediate aldehyde [64]. Microsomal kinetic and inhibition studies and the use of cDNA-expressed enzymes has established a significant role for CYP2C9 in the 5-hydroxylation of seratrodast, a newly developed thromboxane A2 inhibitor [65]. Kinetic experiments further demonstrated that seratrodast had the capacity to inhibit the model CYP2C9 substrate tolbutamide at clinically relevant concentrations. Immunoinhibition, isoform content correlations and catalysis by purified enzyme indicate that CYP2C9 is probably responsible for human liver microsomal Δ9-THC 11-hydroxylation [66], the major metabolic pathway for Δ9-THC in humans. Similar techniques were used to investigate the involvement of CYP2C9 in the hydroxylation of 58C80, an experimental antimalarial compound. Although 58C80 was metabolised by a CYP2C9-like protein and inhibited by an anti-CYP2C9 antibody, the correlation between 58C80 hydroxylation and tolbutamide hydroxylation activities in microsomes from 22 human livers was poor suggesting that CYP2C9 may not be the sole isoform involved in 58C80 hydroxylation [67, 68].
Factors affecting CYP2C9 activity
Enzyme induction
The promoter region of the CYP2C9 gene contains a 15 bp sequence which has high homology to a consensus sequence for barbiturate-induction in CYP102, CYP2B1 and CYP2B2 [9]. There is direct evidence that phenobarbitone upregulates isoforms of the CYP2C subfamily in non-human primates [69], and the results of drug interaction studies in vivo similarly suggest that phenobarbitone, and possibly other barbiturates, may induce CYP2C9 activity in humans, albeit inconsistently. Administration of phenobarbitone to patients on long term warfarin therapy decreased steady-state plasma warfarin concentration and diminished anticoagulant effect [70], suggestive of induction of S-warfarin metabolism. Studies of the effect of therapeutic doses of phenobarbitone on phenytoin kinetics have produced conflicting results [71]. Patients stabilised on phenytoin show a decrease, no change and even occasionally a slight increase in plasma phenytoin concentration. A review of the literature [71] suggested that in most instances phenobarbitone coadministration is likely at best to produce clinically insignificant changes in the plasma concentration of phenytoin. It has, however, been reported [72] that high dose pentobarbitone therapy (in patients with cerebral lesions) reduces phenytoin AUC (unbound) by more than 70%. Similar to phenytoin, phenobarbitone treatment appears to have only a modest effect on losartan clearance. Sub-acute administration of phenobarbitone reduced mean losartan AUC by about 20% [73], but whether this reflects induction of CYP2C9 or CYP3A4, both of which contribute to losartan biotransformation, is unclear.
Like phenobarbitone, investigations of the effects of carbamazepine on phenytoin elimination have produced conflicting results [74]. Although carbamazepine has been reported to increase steady-state plasma phenytoin concentration in some studies, most have demonstrated a trend towards lower phenytoin concentrations during long-term therapy. In most instances this has been attributed to induction of HPPH formation. The clearances of phenytoin, tolbutamide and warfarin, but not aminopyrine, have all been reported to increase in hospitalised alcoholic subjects in the period following ethanol withdrawal [75–77]. Notably, tolbutamide clearance was apparently more than two-fold greater in alcoholic subjects compared with a control group [75]. Interestingly, once tolbutamide clearance had returned to normal in alcoholic subjects following prolonged withdrawal, the higher clearance was difficult to reproduce with pure ethanol. Whether the higher plasma clearance of tolbutamide and other CYP2C9 substrates is due to induction by ethanol itself, congenors in alcoholic beverages, or other dietary and lifestyle factors associated with alcoholism, remains unclear.
Unlike carbamazepine and phenobarbitone, treatment with rifampicin has been shown consistently to increase the clearances of drugs eliminated by CYP2C9. The clearances of losartan, phenytoin, tolbutamide and S-warfarin approximately double in healthy volunteers or patients treated with 450–1200 mg of rifampicin daily [78–81]. The magnitude of CYP2C9 induction, and that of a number of other CYP isoforms, due to rifampicin is clearly sufficient to give rise to clinically significant interactions with CYP2C9 substrates.
Enzyme inhibition
Apart from the mutual competitive inhibition which might occur between substrates for the same enzyme, a number of drugs have been reported to inhibit the metabolism of prototypic CYP2C9 substrates in vivo and/or in vitro and these are listed in Table 2. Interactions of potential clinical significance have been documented for a number of the CYP2C9 inhibitors and the more important of these, given current therapeutic trends, are discussed here.
Table 2.
Drugs reported to inhibit the metabolism of CYP2C9 substrates in vivo and/or in vitro.

There are numerous case reports documenting potentiation of the anticoagulant effect of warfarin in patients coadministered amiodarone. In a study conducted in healthy volunteers, pretreatment with amiodarone 400 mg/day for 3 days, was shown to reduce the mean plasma clearances of R- and S-warfarin by 33% and 47%, respectively [83]. A follow-up study [84] demonstrated that all oxidation pathways of the warfarin enantiomers, including S-warfarin 7-hydroxylation, were inhibited by amiodarone. Human liver microsomal studies confirmed the ability of amiodarone to inhibit S-warfarin 6- and 7-hydroxylation [84]. Consistent with inhibition of the CYP2C9 catalysed pathways of S-warfarin metabolism in vivo and in vitro, amiodarone pretreatment (200 mg day−1) increased phenytoin (5 mg kg−1) AUC by an average of 40% and decreased the rate of HPPH formation in seven healthy volunteers [85]. Given the long half-life of amiodarone, inhibition of CYP2C9 activity (and possibly that of other isoforms) may persist for several weeks following withdrawal of the drug.
Cimetidine is a relatively weak inhibitor of human liver microsomal CYP2C9 [27, 28], with an estimated apparent Ki of 327 μm. Not unexpectantly, cimetidine exerts a variable effect on the elimination of CYP2C9 substrates in vivo. Administration of cimetidine, 1 g day−1 did not alter the disposition of tolbutamide but a higher dose (1.6 g day−1) decreased the systemic and metabolic clearances of tolbutamide by ≈40% [33]. Cimetidine generally causes only a modest reduction in the clearance of a single dose of phenytoin, although clinically significant increases in plasma phenytoin concentrations have been reported to occur in patients receiving long term anticonvulsant therapy [71]. As a result of the concentration-dependent metabolic clearance of phenytoin, the effects of cimetidine, and probably other CYP2C9 inhibitors, would be expected to be greater at the higher plasma concentrations achieved with chronic dosing. Pretreatment with cimetidine (1 g day−1) was shown to reduce the plasma clearance of racemic warfarin by over 25% in healthy volunteers and prolong prothrombin time in patients anticoagulated with warfarin [86]. Although cimetidine clearly has the capacity to inhibit CYP2C9 in vivo in a dose-dependent manner, as evidenced by the results of the studies with tolbutamide and phenytoin, the pharmacokinetic and pharmacodynamic changes observed with racemic warfarin appear to arise more from inhibition of the metabolism of R- rather than S-warfarin. Pretreatment with cimetidine (0.8 g day−1) produced a stereoselective reduction in the plasma clearance of the R-enantiomer by inhibition of the formation of R-6 and R-7-hydroxywarfarin formation [87, 88].
Fluconazole has been shown to be a potent inhibitor of a number of CYP2C9 catalysed reactions in vitro, including tolbutamide hydroxylation [28], diclofenac 4′–hydroxylation [89], and S-warfarin 6- and 7-hydroxylation [47]. Inhibition of S-warfarin 7-hydroxylation proceeded according to a mixed mechanism, suggestive of a nonproductive enzyme/substrate/inhibitor complex, with an apparent Ki of 8 μm. This value is below the range of plasma concentrations (15–60 μm) usually associated with fluconazole therapy. Consistent with expectations based on the in vitro data, various studies demonstrated that fluconazole pretreatment (100–800 mg day−1) increased the AUC values of losartan, phenytoin, tolbutamide and S-warfarin by 153% [90], 72–75% [91, 92], 109% [92] and 184% [93], respectively. Based on the calculation of an in vivo apparent Ki for S-warfarin 7-hydroxylation (22 μm; approximately three times higher than that estimated in vitro [90]), a strategy has been developed for relating fluconazole plasma concentration to inhibition of CYP2C9 activity in vivo [94].
Apart from fluconazole, other azole antifungals have been shown to inhibit CYP2C9 activity in vitro and/or in vivo. Miconazole inhibits warfarin 6- and 7-hydroxylation by human liver microsomes according to a mixed model and in vivo reduces S-warfarin clearance by 80% [95]. Clotrimazole is similarly a potent, mixed inhibitor of human liver microsomal CYP2C9 with an apparent Ki of 1 μm [28]. However, since clotrimazole is used topically it is highly unlikely that this compound will cause clinically significant inhibition of CYP2C9 catalysed reactions in vivo. Although ketoconazole inhibits tolbutamide hydroxylation in vitro with an apparent Ki of 8 μm, its affinity for CYP2C9 is more than 10-fold lower than that for CYP3A4 [28, 31, 96]. Ketoconazole has been reported not to affect the clearances of losartan and phenytoin [97, 98].
Fluvastatin, unlike the related HMG-CoA reductase inhibitors lovastatin, pravastatin and simvastatin, is a potent inhibitor of human hepatic CYP2C9 activity in vitro [99]. Both fluvastatin enantiomers inhibit microsomal diclofenac 4′–hydroxylase activities with an apparent Ki value less than 1 μm. However, in vivo fluvastatin pretreatment reduced mean diclofenac plasma clearance and 4′–hydroxydiclofenac urinary metabolic ratio to modest extents only (16% and 25%, respectively) in fourteen volunteers, with several of the subjects showing no response to fluvastatin [100].
Phenylbutazone and oxyphenbutazone are potent inhibitors of tolbutamide metabolism in vivo [34]. Although single doses of these compounds have little or no effect on tolbutamide elimination, pretreatment for 1 week with either compound more than doubled the elimination half-life of tolbutamide. Subsequent in vitro studies [27] showed that phenylbutazone was a competitive inhibitor of human liver microsomal CYP2C9 activity with an apparent Ki close to 10 μm, which is substantially lower than concentrations observed following multiple-dose administration of phenylbutazone. Phenylbutazone pretreatment similarly reduces the clearance of S-warfarin but, at the same time, increases R-warfarin clearance by an almost equivalent extent [101]. Sulphinpyrazone, another pyrazolone, is also known to be an inhibitor of CYP2C9 activity. Pretreatment with sulphinpyrazone for one week reduced tolbutamide plasma clearance by 40% [102]. Tolbutamide clearance was still reduced 24 h after cessation of a 1 week period of sulphinpyrazone treatment, even though sulphinpyrazone was no longer detectable in plasma at the time, suggesting a sulphinpyrazone metabolite(s) contributed to the inhibition of tolbutamide metabolism. In a similar manner to phenylbutazone, sulphinpyrazone pretreatment inhibits S-warfarin plasma clearance (by ≈40%) but increases that of R-warfarin to about the same extent [45, 103]. The capacity of sulphinpyrazone to inhibit human liver microsomal tolbutamide hydroxylase and S-warfarin 7-hydroxylase activity was confirmed subsequently [45, 104]. Interestingly, the respective apparent Ki values for the sulphide and sulphone metabolites of sulphinpyrazone were 13.5- and 3.2-fold lower than the inhibitor constant for the parent drug, confirming a major contribution of these metabolites in CYP2C9 inhibition [104].
Thirty-five years ago, severe hypoglycaemia was observed in patients administered both sulphaphenazole and tolbutamide [105]. In the intervening years, sulphaphenazole and certain other sulphonamides have been characterised as CYP2C9 inhibitors. As noted earlier, sulphaphenazole is a potent inhibitor of the human liver microsomal hydroxylations of tolbutamide, phenytoin and S-warfarin [27–31, 46], with an apparent Ki of 0.1–0.2 μm. In vivo, sulphaphenazole pretreatment reduces the plasma clearance of tolbutamide by ≈80% [32–34]. Indeed, ‘normal’ subjects may be converted to phenotypically ‘poor’ metabolisers of tolbutamide (see following section) by treatment with sulphaphenazole [32]. Sulphaphenazole treatment has also been shown to cause a more than 3-fold prolongation of the half-life of phenytoin [106]. Apparent Ki values for inhibition of human liver microsomal tolbutamide hydroxylase activity by sulphamethoxazole and sulphamethizole are 2–3 orders of magnitude higher than that observed for sulphaphenazole [28]. The lower affinity of these sulphonamides for CYP2C9 is consistent with the modest inhibition of tolbutamide, phenytoin or warfarin elimination observed in vivo [106–108]. Sulphamethoxazole, sulphamethizole and sulphadiazine, but not sulphamethoxypyridazine, sulphadimethoxine or sulphamethoxydiazine, have been reported to prolong phenytoin half-life and reduce the clearances of tolbutamide and racemic warfarin [106, 108]. Trimethoprim pretreatment is also known to decrease the intrinsic clearance of tolbutamide [107] and prolong the half-life of phenytoin [108], indicating that inhibition of CYP2C9 activity in vivo by cotrimoxazole is an additive effect of the two components, sulphamethoxazole and trimethoprim [107].
Available evidence from interaction studies indicates that β-adrenoceptor antagonists [110], dextropropoxyphene [111], diltiazem [112], enoxacin [113], erythromycin [81, 114], metronidazole [115], olanzapine [116] and omeprazole [117, 118] are all unlikely to inhibit CYP2C9 activity in vivo.
Pharmacogenetics
The possible genetic regulation of tolbutamide metabolism was first reported in 1979. A study of tolbutamide pharmacokinetics in fifty subjects showed that elimination rate constant varied almost nine-fold; the corresponding range of elimination half-lives was 2.9–25 h [119]. The elimination rate constant frequency data approximated a trimodal distribution and was used to argue monogenic control of tolbutamide metabolism in humans, with an estimated 30% of the population being classified as poor metabolisers. It is almost certain, however, that none of the subjects in the original study was a true poor metaboliser of tolbutamide [120]. An individual with tolbutamide elimination half-life of 37 h (almost five times the population mean) and a plasma clearance only 20% of the population mean was identified in a later study [121] and subsequently genotyped as homozygous for the recessive (Leu359) CYP2C9 allele [122] (see below). In contrast, another individual with a tolbutamide half-life of 26 h [123] was not homozygous for the recessive allele [122]. The very low plasma clearance of tolbutamide in the phenotypically poor metaboliser [121, 124] is suggestive of almost total abolition of the metabolic clearance component of tolbutamide elimination.
A survey of the tolbutamide hydroxylation capacity of 106 unrelated volunteers from the Australian population failed to detect a single poor metaboliser [124], suggesting that the incidence of the poor metaboliser phenotype is low. Consistent with the tolbutamide population data, the frequency of the poor phenytoin hydroxylation phenotype has been estimated to be 1 in 500 based on the results of a pedigree analysis study [125, 126]. Furthermore, in clinical trials conducted in recent years with losartan, less than 1% of the subjects studied showed markedly diminished conversion of losartan to its active metabolite (a known CYP2C9 catalysed reaction) [127].
As noted earlier, six different CYP2C9 sequences have been identified to date, with another sequence, CYP2C10, originally being classified separately due to differences in the 3′–untranslated region [8, 9, 12–17]. Nine nucleotide substitutions in the coding region of CYP2C9 account for the various sequences reported. Genotyping studies conducted in a number of laboratories suggests, however, that only three sequences appear to represent true CYP2C9 alleles [122, 128–130], the others presumably being cloning artifacts. Polymorphisms in the coding region of the CYP2C9 gene produce variants at amino acid residues 144 and 359 (Figure 3). Three alleles arising from C to T (Arg to Cys) and A to T (Ile to Leu) transversions at codons 416 and 1061, respectively, have been identified; Arg144/Ile359, Cys144/Ile359 and Arg144/Leu359. The respective allele frequencies for the Arg144/Ile359, Cys144/Ile359 and Arg144/Leu359−1 variants (sometimes referred to as CYP2C9*1, CYP2C9*2 and CYP2C9*3, respectively) in Caucasian populations have been reported to range from 0.79–0.86, 0.08–0.125, and 0.03–0.085 [122, 128–130]. Thus, the Arg144/Ile359 allele represents the wild-type CYP2C9. Interestingly, the Cys144 allele is not detected in Chinese or Japanese subjects [122, 130–132] and its frequency is also very low (0.01) in African-Americans [122]. In these populations there is a corresponding increase in the frequency of the Arg144/Ile359 allele to >0.95. No individuals homozygous for the Cys144 or Leu359 alleles were identified in the populations randomly selected for the various genotyping studies.
Figure 3.

Sites of polymorphism in the CYP2C9 protein.
Genotyping of the individual with a tolbutamide half-life of 37 h identified in a previous in vivo study [121] confirmed homozygosity for the Leu359 allele [122, 128]. No other mutations were found in the coding region, intron-exon junctions or upstream region of the CYP2C9 gene of this subject. Sequencing of the coding region of the CYP2C9 gene of two subjects identified as poor metabolisers of losartan similarly confirmed that both were homozygous for the mutation causing the Ile to Leu substitution at residue 359 [127]. Somewhat surprisingly, it therefore appears that the conserved Ile to Leu (both being structurally similar hydrophobic amino acids) substitution gives rise to the poor metaboliser CYP2C9 activity phenotypes. Kinetic studies with the expressed CYP2C9 variants indicate that apparent Km values for tolbutamide [15, 122, 128], S-warfarin [121, 133] and torsemide [J. O. Miners, D. J. Birkett, J. A. Goldstein, unpublished results] are higher while Vmaxvalues are similar or lower for the Arg144/Leu359 variant compared to the wild type enzyme. Overall, intrinsic clearances (Vmax/Km) for the Arg144/Leu359 variant with these three substrates are ≈15–20% of the intrinsic clearances observed with the wild type enzyme, consistent with the magnitude of the decrease in tolbutamide plasma clearance (ca 20% of the population mean) observed in the poor metaboliser with a half-life of 37 h [121]. Phenytoin hydroxylase activity of the Arg144/Leu359 variant has also been shown to be lower for the wild-type enzyme [15]. Application of the Hardy-Weinberg law to the genotyping data indicates that the frequency of individuals homozygous for the Leu359 allele is < 1% [122], in agreement with the estimated frequencies of the tolbutamide and phenytoin poor metaboliser phenotypes [124, 126].
Taken together, available evidence indicates that individuals homozygous for the Leu359 allele are likely to have impaired clearances of losartan, phenytoin, tolbutamide, torsemide, S-warfarin, and possibly other CYP2C9 substrates. This is likely to have consequences for dosage regimens, as has been illustrated recently for warfarin [134]. The warfarin maintenance dose required for anticoagulation of a patient homozygous for the Leu359 allele was 0.5 mg day−1 or less. It has also been reported that the regio- and stereo-selectivity of warfarin hydroxylation may differ between the Ile- and Leu-359 variants [122, 135]. Interestingly, however, the kinetics of diclofenac 4′-hydroxylation are apparently unaffected by the isoleucine to leucine substitution [19]. These data confirm that a single, conserved amino acid substitution in a substrate recognition site may drastically alter the catalytic efficiency and substrate specificity of a CYP isoform.
Whereas homozygosity of the Leu359 allele is likely to have important therapeutic implications, the consequences of expression of the Cys144 and/or Leu359 alleles in heterozygotes is less clear. Unlike residue 359, which is situated in SRS5 of CYP2C9, amino acid 144 does not fall within any of the known substrate recognition sequences and this mutation would not, therefore, be expected to change substrate binding. Intrinsic clearances (Vmax/Km) of the wild type CYP2C9 (Arg144/Ile359) and Cys144/Ile359 variants appear not to differ significantly with tolbutamide [122, 128, 136] and torsemide [J. O. Miners, D. J. Birkett, J. A. Goldstein, unpublished results] as substrates, but the situation is less clear for S-warfarin. Vmax values of the Cys144/Ile359 variant for S-warfarin 7-hydroxylation have been reported to be similar to [122] or lower [136, 137] than that of the wild-type enzyme, although Km values were the same. Different expression systems (baculovirus, COS, lymphoblast, HepG2, yeast) were used in the various studies investigating the activities of the CYP2C9 variants, which could contribute to the different observations. Moreover, it has been reported recently that the magnitude of the difference in the Vmax for S-warfarin (and possibly other substrates) between the Arg- and Cys-144 variants in a number of expression systems is influenced by the CYP to cytochrome P-450 oxidoreductase ratio [137]. It appears that Arg144 may play an important role in the interaction between CYP2C9 and reductase, and a high reductase to CYP2C9 ratio in expression systems is necessary to resemble conditions in human liver microsomes. Also of possible significance to CYP2C9 activity in individuals expressing the Cys144 allele is the observation of relatively greater expression of this allele over the wild-type Arg144 allele in heterozygotes [128].
There was no apparent correlation between genotype and the microsomal hydroxylation of the known CYP2C9 substrates diclofenac, phenytoin, tolbutamide and torsemide in 18 human livers [128]. Activities for a heterozygous Ile359 liver were low, but still within the range of activities observed for the sample population. Tolbutamide hydroxylase activities have been reported not to be influenced by expression of the Cys144 allele in livers from 45 Caucasian subjects [130]. Similarly, tolbutamide hydroxylase activities in livers from both Japanese and Caucasian Ile359/Leu359 heterozygotes tended to low, but were again within the range of activities observed for livers expressing only the wild-type enzyme. In contrast to the in vitro activity data, comparison of the warfarin maintenance dose required for stable anticoagulation in patients who were homozygous wild-type and Arg144Cys heterozygotes revealed that the average dose was 20% lower (P = 0.02) in the heterozygotes [138]. Since CYP2C9 is known to contribute to the metabolism of benzo[a]pyrene [26], a possible association between CYP2C9 genetic polymorphism and lung cancer risk has been addressed recently [139]. No association was found between lung cancer risk and expression of any of the CYP2C9 alleles.
Other factors influencing CYP2C9 expression
CYP2C9 is expressed constitutively in human liver at levels almost as high as CYP2C8, the most abundant CYP2C isoform in that tissue [140]. The CYP2C9 gene has been isolated and shown to be ≈55 kb in size [9, 141]. The promoter region of the gene contains a number of potential transcription factor recognition sites, notably those for AP-1, C/EBP, GRE and HPF-1 [141, 142]. An HPF-1 like motif present in the CYP2C9 promoter appears to be an important cis-acting element contributing to the liver-specific expression of this gene. There is some evidence that CYP2C protein, but not specifically CYP2C9, is expressed at very low levels in human lung [143]. CYP2C9 mRNA is not detectable in full-term placentae [144].
CYP2C9 appears not to be expressed in foetal tissue [16, 145], but CYP2C protein is detectable in neonatal liver [146]. Like a number of other drugs metabolised by CYP, the half-life of phenytoin is shorter and the steady-state plasma concentration lower for a given dose per kilogram in children [147]. Whether this reflects increased activity of CYP2C9 remains unknown. Data concerning the pharmacokinetics of CYP2C9 metabolised drugs in the elderly are conflicting, and frequently complicated by changes in plasma protein binding, but nevertheless tend to suggest that CYP2C9 activity is not decreased by old age [148]. It has been reported recently that the intrinsic clearance of S-warfarin does not differ significantly between young adult and elderly subjects, although differences were noted for the R-enantiomer [149].
Several rat CYP2C isoforms are known to be expressed in a sex-specific manner, but no human CYP isoforms are expressed exclusively in males or females [3]. There appears to be no evidence to suggest that significant gender-related differences exist in the elimination of drugs metabolised by CYP2C9 [150]. It is unclear whether ethnicity is a significant determinant of CYP2C9 activity. The mean Km for phenytoin, estimated from population pharmacokinetic data, has been reported to be lower in Chinese and Japanese than in Europeans, although interindividual variability was substantial [151, 152]. As noted earlier, allele frequencies for the CYP2C9 variants differ in Oriental and Caucasian populations. The Cys144 allele is not detectable in Chinese and Japanese and the frequency of the Leu359 allele may be marginally lower in these populations than in Caucasians [130–132]. It is noteworthy that rates of microsomal tolbutamide hydroxylation in livers from Caucasian and Japanese subjects show wide but comparable ranges of activities. Immunoreactive hepatic CYP2C9 contents also appear to be similar in both races.
Conclusions
With the recent application of in vitro techniques for the identification of the enzymes involved in the metabolism of any given xenobiotic, it has become increasingly clear that CYP2C9 ranks amongst the most important drug metabolising CYP isoforms present in human liver. Many CYP2C9 substrates are weak acids, but CYP2C9 also has the capacity to metabolise neutral, highly lipophilic xenobiotics suggesting substrate specificity may be broader than is currently believed.
CYP2C9 is inducible, notably by rifampicin, and numerous clinically significant inhibitory drug interactions involving CYP2C9 substrates have been described. Genetic polymorphisms, at least one of which is known to be associated with a dramatic change in enzyme activity, result from two mutations in the coding region of the CYP2C9 gene. Given the modulation of CYP2C9 activity by genetic factors, stimulatory and inhibitory interactions, and possibly age effects (viz increased clearance in children), it is hardly surprising that wide interindividual variability has been reported for the elimination and/or dosage requirement of some drugs metabolised by CYP2C9 [120, 124, 126, 134, 153]. For example, tolbutamide hydroxylation capacity varies ≈20-fold in vivo and in vitro [124, 130, 154]. As is current practice with phenytoin, tolbutamide and warfarin, individualisation of dose is clearly necessary for those CYP2C9 substrates with a narrow therapeutic index.
Table 1.
Drugs reported to induce the elimination of CYP2C9 substrates in vivo.

References
- 1.Nelson DR, Koymans L, Kamataki T, et al. P450 superfamily: Update on new sequences, gene mapping, accession numbers and nomenclature. Pharmacogenetics. 1996;6:1–42. doi: 10.1097/00008571-199602000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Wrighton SA, Stevens JC. The human hepatic cytochromes P450 involved in drug metabolism. Crit Rev Toxicol. 1992;22:1–21. doi: 10.3109/10408449209145319. [DOI] [PubMed] [Google Scholar]
- 3.Gonzalez FJ. Human cytochromes P450: Problems and prospects. Trends Pharmacol Sci. 1992;13:346–352. doi: 10.1016/0165-6147(92)90107-h. [DOI] [PubMed] [Google Scholar]
- 4.Cholerton S, Daly AK, Idle JR. The role of individual human cytochromes P450 in drug metabolism and clinical response. Trends Pharmacol Sci. 1992;13:434–439. doi: 10.1016/0165-6147(92)90140-2. [DOI] [PubMed] [Google Scholar]
- 5.Gonzalez FJ. Molecular genetics of the P450 superfamily. Pharmacol Ther. 1990;45:1–38. doi: 10.1016/0163-7258(90)90006-n. [DOI] [PubMed] [Google Scholar]
- 6.Miners JO, Veronese ME, Birkett DJ. In vitro approaches for the prediction of human drug metabolism. Ann Reports Med Chem. 1994;29:307–316. [Google Scholar]
- 7.Birkett DJ, Mackenzie PI, Veronese ME, Miners JO. In vitro approaches can predict human drug metabolism. Trends Pharmacol Sci. 1993;14:292–294. doi: 10.1016/0165-6147(93)90043-j. [DOI] [PubMed] [Google Scholar]
- 8.Ged C, Umbenhauer DR, Bellew TM, et al. Characterization of cDNAs, mRNAs and proteins related to human liver microsomal cytochrome P450 (S)-mephenytoin 4′–hydroxylase. Biochemistry. 1988;27:6929–6940. doi: 10.1021/bi00418a039. [DOI] [PubMed] [Google Scholar]
- 9.Goldstein JA, de Morais SMF. Biochemistry and molecular biology of the human CYP2C subfamily. Pharmacogenetics. 1994;4:285–299. doi: 10.1097/00008571-199412000-00001. [DOI] [PubMed] [Google Scholar]
- 10.Wang P, Beaune P, Kaminsky LS, et al. Purification and characterization of six cytochrome P450 isozymes from human liver microsomes. Biochemistry. 1983;22:5375–5383. doi: 10.1021/bi00292a019. [DOI] [PubMed] [Google Scholar]
- 11.Shimada T, Misono KS, Guengerich FP. Human liver microsomal cytochrome P450 mephenytoin 4′–hydroxylase, a prototype genetic polymorphism of drug metabolism: Purification and characterization of two similar forms involved in the reaction. J Biol Chem. 1986;261:909–921. [PubMed] [Google Scholar]
- 12.Kimura S, Pastewka J, Gelboin HV, Gonzalez FJ. cDNA and amino acid sequences of two members of the human IIC gene subfamily. Nucleic Acids Res. 1987;15:10053–10054. doi: 10.1093/nar/15.23.10053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Meehan RR, Gosden JR, Rout D, et al. Human cytochrome P450 PB-1: A multigene family involved in mephenytoin and steroid oxidations that maps to chromosome 10. Am J Human Genet. 1988;42:26–37. [PMC free article] [PubMed] [Google Scholar]
- 14.Romkes M, Faletto MB, Blaisdell JA, Raucy JL, Goldstein JA. Cloning and expression of complementary DNAs for multiple members of the human cytochrome P450 IIC subfamily. Biochemistry. 1991;30:3247–3255. doi: 10.1021/bi00227a012. [DOI] [PubMed] [Google Scholar]
- 15.Veronese ME, Doecke CJ, Mackenzie PI, et al. Site-directed mutation studies of human liver cytochrome P450 isoenzymes in the CYP2C subfamily. Biochem J. 1993;289:533–538. doi: 10.1042/bj2890533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Umbenhauer DR, Martin MV, Lloyd RS, Guengerich FP. Cloning and sequence determination of a complementary DNA related to human liver microsomal cytochrome P450 S-mephenytoin 4′–hydroxylase. Biochemistry. 1987;26:1094–1099. doi: 10.1021/bi00378a016. [DOI] [PubMed] [Google Scholar]
- 17.Yasumori TS, Kawano K, Nagata M, Shimada Y, Yamazoe Y, Kato R. Nucleotide sequence of a human liver cytochrome P450 related to the male rat specific form. J Biochem (Tokyo) 1987;102:1075–1082. doi: 10.1093/oxfordjournals.jbchem.a122145. [DOI] [PubMed] [Google Scholar]
- 18.Gotoh O. Substrate recognition sites in cytochrome P-450 family 2 (CYP2) proteins inferred from comparative analyses of amino acid and coding nucleotide sequences. J Biol Chem. 1992;267:83–90. [PubMed] [Google Scholar]
- 19.Wong BK, Goldstein J, Spielberg SP, Rushmore TH. Characterisation of an in vitro model for assessing polymorphic metabolism by CYP2C9: Involvement in the mechanism of deficient losartan metabolism. ISSX Proceedings. 1996;10:236. [Google Scholar]
- 20.Haining RL, Jones JP, Koop DR, Trager WF, Rettie AE. Determinants of substrate and inhibitor specificities of human CYP2C9. ISSX Proceedings. 1996;10:239. [Google Scholar]
- 21.Korzekwa K, Jones JP. Predicting the cytochrome P450 mediated metabolism of xenobiotics. Pharmacogenetics. 1993;3:1–18. doi: 10.1097/00008571-199302000-00001. [DOI] [PubMed] [Google Scholar]
- 22.Mancy A, Broto P, Dijols S, Dansette PM, Mansuy D. The substrate binding site of human liver cytochrome P4502C9: An approach using designed tienilic acid derivatives and molecular modeling. Biochemistry. 1995;34:10365–10375. doi: 10.1021/bi00033a007. [DOI] [PubMed] [Google Scholar]
- 23.Jones BC, Hawksworth G, Horne VA, et al. Putative active site model for cytochrome P4502C9 (tolbutamide hydroxylase) Drug Metab Dispos. 1996;24:260–266. [PubMed] [Google Scholar]
- 24.Jones JP, He M, Trager WF, Rettie AE. Three dimensional quantitative structure-activity relationship for inhibitors of cytochrome P4502C9. Drug Metab Dispos. 1996;24:1–6. [PubMed] [Google Scholar]
- 25.Shou M, Krausz KW, Gonzalez FJ, Gelboin HV. Metabolic activation of the potent carcinogen dibenzo[a,h]anthracene by cDNA-expressed human cytochromes P450. Arch Biochem Biophys. 1996;238:201–207. doi: 10.1006/abbi.1996.0161. [DOI] [PubMed] [Google Scholar]
- 26.Gautier J-C, Lecoeur S, Cosme J, et al. Contribution of human cytochrome P450 to benzo[a]pyrene and benzo[a]pyrene-7, 8-dihydrodiol metabolism, as predicted from heterologous expression in yeast. Pharmacogenetics. 1996;6:489–499. doi: 10.1097/00008571-199612000-00002. [DOI] [PubMed] [Google Scholar]
- 27.Miners JO, Smith KJ, Robson RA, McManus ME, Veronese ME, Birkett DJ. Tolbutamide hydroxylation by human liver microsomes: Kinetic characterisation and relationship to other cytochrome P450 dependent xenobiotic oxidations. Biochem Pharmacol. 1988;37:1137–1144. doi: 10.1016/0006-2952(88)90522-9. [DOI] [PubMed] [Google Scholar]
- 28.Back DJ, Tjia JF, Karbwang J, Colberg J. In vitro inhibition studies of tolbutamide hydroxylase activity of human liver microsomes by azoles, sulphonamides and quinolones. Br J Clin Pharmacol. 1988;25:23–29. doi: 10.1111/j.1365-2125.1988.tb03359.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Veronese ME, Mackenzie PI, Doecke CJ, McManus ME, Miners JO, Birkett DJ. Tolbutamide and phenytoin hydroxylations by cDNA-expressed human liver cytochrome P4502C9. Biochem Biophys Res Commun. 1991;175:1112–1118. doi: 10.1016/0006-291x(91)91680-b. [DOI] [PubMed] [Google Scholar]
- 30.Miners JO, Birkett DJ. The use of tolbutamide as a substrate probe for human hepatic cytochrome P4502C9. Methods Enzymol. 1996;272:139–145. doi: 10.1016/s0076-6879(96)72017-7. [DOI] [PubMed] [Google Scholar]
- 31.Baldwin SJ, Bloomer JC, Smith GJ, Ayrton AD, Clarke SE, Chenery RJ. Ketoconazole and sulphaphenazole as the respective selective inhibitors of P4503A and 2C9. Xenobiotica. 1995;25:261–270. doi: 10.3109/00498259509061850. [DOI] [PubMed] [Google Scholar]
- 32.Veronese ME, Miners JO, Randles D, Birkett DJ. Validation of the tolbutamide metabolic ratio for population screening with use of sulphaphenazole to produce model phenotypic poor metabolisers. Clin Pharmacol Ther. 1990;47:403–411. doi: 10.1038/clpt.1990.46. [DOI] [PubMed] [Google Scholar]
- 33.Back DJ, Tjia J, Ohnhaus EE, Park BK. Selective inhibition of drug oxidation after simultaneous administration of two probe drugs, antipyrine and tolbutamide. Eur J Clin Pharmacol. 1998;34:157–163. doi: 10.1007/BF00614553. [DOI] [PubMed] [Google Scholar]
- 34.Pond SM, Birkett DJ, Wade DN. Mechanisms of inhibition of tolbutamide metabolism: phenylbutazone, oxyphenbutazone, sulphaphenazole. Clin Pharmacol Ther. 1977;22:573–579. doi: 10.1002/cpt1977225part1573. [DOI] [PubMed] [Google Scholar]
- 35.Thomas RC, Ikeda GJ. The metabolic fate of tolbutamide in man and in the rat. J Med Chem. 1966;9:507–510. doi: 10.1021/jm00322a014. [DOI] [PubMed] [Google Scholar]
- 36.Nelson E, O'Reilly I. Kinetics of carboxytolbutamide excretion following tolbutamide and carboxytolbutamide administration. J Pharmacol Exp Ther. 1961;132:103–109. [PubMed] [Google Scholar]
- 37.Doecke CJ, Veronese ME, Pond SM, et al. Relationship between phenytoin and tolbutamide hydroxylations in human liver microsomes. Br J Clin Pharmacol. 1991;31:125–130. doi: 10.1111/j.1365-2125.1991.tb05499.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miners JO, Rees DLP, Valente L, Veronese ME, Birkett DJ. Human hepatic cytochrome P4502C9 catalyzes the rate-limiting pathway of torsemide metabolism. J Pharmacol Exp Ther. 1995;272:1076–1081. [PubMed] [Google Scholar]
- 39.Relling MV, Aoyama T, Gonzalez FJ, Meyer UA. Tolbutamide and mephenytoin hydroxylation by human cytochromes P450 in the CYP2C subfamily. J Pharmacol Exp Ther. 1990;252:442–447. [PubMed] [Google Scholar]
- 40.Tassaneeyakul W. 1994. PhD Thesis, Flinders University of South Australia. [Google Scholar]
- 41.Dickinson RG, Hooper WD, Patterson M, Eadie MJ, Maguire B. Extent of urinary excretion of p-hydroxyphenytoin in healthy subjects given phenytoin. Ther Drug Monit. 1985;7:283–289. doi: 10.1097/00007691-198507030-00008. [DOI] [PubMed] [Google Scholar]
- 42.Molholm-Hansen J, Kampmann JP, Siersbaek-Nielsen K, et al. The effect of different sulphonamides on phenytoin metabolism in man. Acta Med Scand. 1979;624(suppl):106–110. doi: 10.1111/j.0954-6820.1979.tb00729.x. [DOI] [PubMed] [Google Scholar]
- 43.Hall SD, Hamman MA, Rettie AE, et al. Relationships between the levels of cytochrome P4502C9 and its prototypic catalytic activities in human liver microsomes. Drug Metab Dispos. 1994;22:975–978. [PubMed] [Google Scholar]
- 44.Tassaneeyakul W, Veronese ME, Birkett DJ, et al. Co-regulation of phenytoin and tolbutamide metabolism in humans. Br J Clin Pharmacol. 1992;34:494–498. [PMC free article] [PubMed] [Google Scholar]
- 45.Toon S, Low LK, Gibaldi M, et al. The warfarin-sulfinpyrazole interaction: stereochemical considerations. Clin Pharmacol Ther. 1986;39:15–24. doi: 10.1038/clpt.1986.3. [DOI] [PubMed] [Google Scholar]
- 46.Rettie AE, Korzekwa KR, Kunze KL, Lawrence RF, Eddy AC, Aoyama T, Gelboin HV, Gonzalez FJ, Trager WF. Hydroxylation of warfarin by human cDNA-expressed cytochrome P450: A role for P4502C9 in the etiology of (S)-warfarin drug interactions. Chem Res Toxicol. 1992;5:54–59. doi: 10.1021/tx00025a009. [DOI] [PubMed] [Google Scholar]
- 47.Kunze KL, Wienkers LC, Thummel KF, Trager WF. Warfarin-fluconazole I. Inhibition of human cytochrome P450 dependent metabolism of warfarin by fluconazole: in vitro studies. Drug Metab Dispos. 1996;24:414–421. [PubMed] [Google Scholar]
- 48.Hamman MA, Thompson GA, Hall SD. Regioselective and stereoselective metabolism of ibuprofen by human cytochrome P4502C. Biochem Pharmacol. 1997;54:33–41. doi: 10.1016/s0006-2952(97)00143-3. [DOI] [PubMed] [Google Scholar]
- 49.Tracy TS, Rosenbluth BW, Wrighton SA, Gonzalez FJ, Korzekwa KR. Role of cytochrome P4502C9 and an allelic variant in the 4′–hydroxylation of (R)- and (S)-flurbiprofen. Biochem Pharmacol. 1995;49:1269–1275. doi: 10.1016/0006-2952(95)00048-5. [DOI] [PubMed] [Google Scholar]
- 50.Miners JO, Coulter S, Tukey RH, Veronese ME, Birkett DJ. Cytochromes P450 1A2 and 2C9 are responsible for the human hepatic O-demethylation of (R)- and (S)-naproxen. Biochem Pharmacol. 1996;51:1003–1008. doi: 10.1016/0006-2952(96)85085-4. [DOI] [PubMed] [Google Scholar]
- 51.Rodrigues AD, Kukulka MJ, Roberts EM, Ouellet D, Rodgers TR. [O-methyl14C]naproxen O-demethylase activity in human liver microsomes. Evidence for the involvement of cytochrome P4501A2 and P4502C9/10. Drug Metab Dispos. 1996;24:126–136. [PubMed] [Google Scholar]
- 52.Miners JO, Mackenzie PI. Drug glucuronidation in humans. Pharmacol Ther. 1991;51:347–369. doi: 10.1016/0163-7258(91)90065-t. [DOI] [PubMed] [Google Scholar]
- 53.Leemann T, Transom C, Dayer P. Cytochrome P450TB (CYP2C): A major monooxygenase catalyzing diclofenac 4′–hydroxylation in human liver. Life Sci. 1993;52:29–34. doi: 10.1016/0024-3205(93)90285-b. [DOI] [PubMed] [Google Scholar]
- 54.Bonnabry P, Leemann T, Dayer P. Role of human liver microsomal CYP2C9 in the biotransformation of lornoxicam. Eur J Clin Pharmacol. 1996;49:305–308. doi: 10.1007/BF00226332. [DOI] [PubMed] [Google Scholar]
- 55.Zhao J, Leemann T, Dayer P. In vitro oxidation of oxicam NSAIDs by a human liver cytochrome P450. Life Sci. 1992;51:575–581. doi: 10.1016/0024-3205(92)90226-f. [DOI] [PubMed] [Google Scholar]
- 56.Bonnabry P, Leemann T, Dayer P. Biotransformation by hepatic P450TB (CYP2C) controls mefenamic acid elimination. Clin Pharmacol Ther. 1994;55:139. [Google Scholar]
- 57.Bort R, Ponsoda E, Carrasco E, Gomez-Lechon MJ, Castell JV. Metabolism of aceclofenac in humans. Drug Metab Dispos. 1996;24:834–841. [PubMed] [Google Scholar]
- 58.Neugebauer G, Bessenfelder E, von Mollendorff E. Pharmacokinetics and metabolism of torsemide in man. Arzneim.-Forsch. 1988;38:164–166. [PubMed] [Google Scholar]
- 59.Lopez Garcia MP, Dansette PM, Valadon P, et al. Human liver cytochromes P450 expressed in yeast as tools for reactive metabolite formation studies. Oxidative activation of tienilic acid by cytochromes P450 2C9 and 2C10. Eur J Biochem. 1993;213:223–232. doi: 10.1111/j.1432-1033.1993.tb17752.x. [DOI] [PubMed] [Google Scholar]
- 60.Dansette PM, Amar C, Valadon P, Pons C, Beaune PH, Mansuy D. Hydroxylation and formation of electrophilic metabolites of tienilic acid and its isomer by human liver microsomes. Catalysis by a cytochrome P450 IIC different from that responsible for mephenytoin hydroxylation. Biochem Pharmacol. 1991;41:553–560. doi: 10.1016/0006-2952(91)90627-h. [DOI] [PubMed] [Google Scholar]
- 61.Lecouer S, Andre C, Beune PH. Tienilic-induced autoimmune hepatitis: Anti-liver and -kidney microsomal type 2 autoantibodies recognise a three-site conformational epitope on cytochrome P4502C9. Mol Pharmacol. 1996;50:326–333. [PubMed] [Google Scholar]
- 62.Ghahramani P, Ellis SW, Lennard MS, Ramsay LE, Tucker GT. Cytochromes P450 mediating the N-demethylation of amitriptyline. Br J Clin Pharmacol. 1997;43:137–144. doi: 10.1046/j.1365-2125.1997.05382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.von Moltke LL, Greenblatt DJ, Duan SX, Schmider J, Harmatz JS, Shader RI. Human cytochromes mediating N-demethylation of fluoxetine in vitro. Clin Pharmacol Ther. 1997;61:177. doi: 10.1007/s002130050362. [DOI] [PubMed] [Google Scholar]
- 64.Stearns RA, Chakravarty PK, Chen R, Chui S-HL. Biotransformation of losartan to its active carboxylic metabolite in human liver microsomes. Role of cytochrome P450 2C and 3A subfamily members. Drug Metab Dispos. 1995;23:207–215. [PubMed] [Google Scholar]
- 65.Kumar GN, Dubberke E, Rodrigues AD, Roberts E, Dennisen JF. Identification of cytochromes P450 involved in the human liver microsomal metabolism of the thromboxane A2 inhibitor seratrodast (ABT-001) Drug Metab Dispos. 1997;25:110–115. [PubMed] [Google Scholar]
- 66.Bornheim LM, Lasker JM, Raucy JL. Human hepatic microsomal metabolism of Δ1-tetrahydrocannabinol. Drug Metab Dispos. 1992;20:241–246. [PubMed] [Google Scholar]
- 67.Weaver RJ, Dickins M, Burke MD. Cytochrome P4502C9 is responsible for hydroxylation of the naphthoquinone antimalarial drug 58C80 in human liver. Biochem Pharmacol. 1993;46:1183–1197. doi: 10.1016/0006-2952(93)90467-b. [DOI] [PubMed] [Google Scholar]
- 68.Weaver RT, Dickins M, Burke MD. Hydroxylation of the antimalarial drug 58C80 by CYP2C9 in human liver microsomes: Comparison with mephenytoin and tolbutamide hydroxylations. Biochem Pharmacol. 1995;49:997–1004. doi: 10.1016/0006-2952(94)00457-w. [DOI] [PubMed] [Google Scholar]
- 69.Jones CR, Guengerich FP, Rice JM, Lubet RA. Induction of various cytochromes CYP2B, CYP2C and CYP3A by phenobarbitone in non-human primates. Pharmacogenetics. 1992;2:160–172. doi: 10.1097/00008571-199208000-00003. [DOI] [PubMed] [Google Scholar]
- 70.Serlin MJ, Breckinridge AM. Drug interactions with warfarin. Drugs. 1983;25:610–620. doi: 10.2165/00003495-198325060-00004. [DOI] [PubMed] [Google Scholar]
- 71.Nation RL, Evans AM, Milne RW. Pharmacokinetic drug interactions with phenytoin. Clin Pharmacokinet. 1990;18:37–60. doi: 10.2165/00003088-199018010-00003. [DOI] [PubMed] [Google Scholar]
- 72.Oda Y, Yoshida N, Nishi S, et al. Effect of high dose barbiturate therapy on phenytoin pharmacokinetics. Clin Pharmacol Ther. 1992;51:187. [Google Scholar]
- 73.Goldberg MR, Lo MW, Deutsch PJ, Wilson SE, McWilliams EJ, McCrea JB. Phenobarbital minimally alters plasma concentrations of losartan and its active metabolite E-3174. Clin Pharmacol Ther. 1996;59:268–274. doi: 10.1016/S0009-9236(96)80004-X. [DOI] [PubMed] [Google Scholar]
- 74.Baciewicz AM. Carbamazepine drug interactions. Therapeutic Drug Monit. 1986;8:305–317. doi: 10.1097/00007691-198609000-00012. [DOI] [PubMed] [Google Scholar]
- 75.Iber FL. Drug metabolism in heavy consumers of ethyl alcohol. Clin Pharmacol Ther. 1977;22:735–742. doi: 10.1002/cpt1977225part2735. [DOI] [PubMed] [Google Scholar]
- 76.Kater RMH, Roggin G, Tobon F, Zieve P, Iber FL. Increased rate of clearance of drugs from the circulation of alcoholics. Am J Med Sci. 1969;258:35–39. doi: 10.1097/00000441-196907000-00005. [DOI] [PubMed] [Google Scholar]
- 77.Sandor P, Sellers EM, Dumbrell M, Khouw V. Effect of short- and long-term alcohol use on phenytoin kinetics in chronic alcoholics. Clin Pharmacol Ther. 1981;30:390–397. doi: 10.1038/clpt.1981.178. [DOI] [PubMed] [Google Scholar]
- 78.Heimark LD, Gibaldi M, Trager WF, O'Reilly RA, Goulart DA. The mechanism of the warfarin-rifampin drug interaction in humans. Clin Pharmacol Ther. 1987;42:388–394. doi: 10.1038/clpt.1987.168. [DOI] [PubMed] [Google Scholar]
- 79.Zilly W, Breimer DD, Richter E. Induction of drug metabolism in man after rifampicin treatment measured by increased hexobarbital and tolbutamide clearance. Eur J Clin Pharmacol. 1975;9:219–227. doi: 10.1007/BF00614021. [DOI] [PubMed] [Google Scholar]
- 80.Kay L, Kampmann JP, Svendsen TL, et al. Influence of rifampicin and isoniazid on the kinetics of phenytoin. Br J Clin Pharmacol. 1985;20:323–326. doi: 10.1111/j.1365-2125.1985.tb05071.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Williamson KM, Patterson JH, Pieper JA, McQueen RH, Adams KF. Effects of erythromycin or rifampin on steady state losartan kinetics in healthy volunteers. Clin Pharmacol Ther. 1997;61:202. doi: 10.1016/S0009-9236(98)90163-1. [DOI] [PubMed] [Google Scholar]
- 82.Nolan PE, Marcus FI, Hoyer GL, Bliss M, Gear K. Pharmacokinetic interaction between intravenous phenytoin and amiodarone in healthy volunteers. Clin Pharmacol Ther. 1989;46:43–50. doi: 10.1038/clpt.1989.104. [DOI] [PubMed] [Google Scholar]
- 83.O'Reilly RA, Trager WF, Rettie AE, Goulart DA. Interaction of amiodarone with racaemic warfarin and its separated enantiomorphs in humans. Clin Pharmacol Ther. 1987;42:290–294. doi: 10.1038/clpt.1987.149. [DOI] [PubMed] [Google Scholar]
- 84.Heimark LD, Wienkers L, Kunze K, et al. The mechanism of the interaction between amiodarone and warfarin in humans. Clin Pharmacol Ther. 1992;51:398–407. doi: 10.1038/clpt.1992.39. [DOI] [PubMed] [Google Scholar]
- 85.Skovsted L, Kristensen M, Hansen JM, Siersbaek-Nielsen K. The effect of different oral anticoagulants on diphenylhydration and tolbutamide metabolism. Acta Med Scand. 1976;199:513–515. doi: 10.1111/j.0954-6820.1976.tb06772.x. [DOI] [PubMed] [Google Scholar]
- 86.Serlin MJ, Mossman S, Sibeon RG, et al. Cimetidine: Interaction with oral anticoagulants in man. Lancet. ii:317–319. doi: 10.1016/s0140-6736(79)90340-4. [DOI] [PubMed] [Google Scholar]
- 87.Toon S, Hopkins KJ, Garstang FM, Rowland M. Comparative effects of ranitidine and cimetidine on the pharmacokinetics and pharmacodynamics of warfarin in man. Eur J Clin Pharmacol. 1987;32:165–172. doi: 10.1007/BF00542190. [DOI] [PubMed] [Google Scholar]
- 88.Niopas I, Toon S, Rowland M. Further insight into the stereoselective interaction between warfarin and cimetidine in man. Br J Clin Pharmacol. 1991;32:508–511. doi: 10.1111/j.1365-2125.1991.tb03940.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hargreaves JA, Jezequel S, Houston JB. Effect of azole antifungals on human microsomal metabolism of diclofenac and midazolam. Br J Clin Pharmacol. 1994;38 [Google Scholar]
- 90.Kazierad DJ, Martin DE, Tenero D, et al. Fluconazole significantly alters the pharmacokinetics of losartan but not eprosartan. Clin Pharmacol Ther. 1997;61:203. doi: 10.1016/S0009-9236(97)90120-X. [DOI] [PubMed] [Google Scholar]
- 91.Blum RA, Wilton JH, Hilligoss DM, et al. Effect of fluconazole on the disposition of phenytoin. Clin Pharmacol Ther. 1991;49:420–425. doi: 10.1038/clpt.1991.49. [DOI] [PubMed] [Google Scholar]
- 92.Lazar JD, Wilner KD. Drug interactions with fluconazole. Rev Infec Dis. 1990;12(Suppl 3):S327–S333. doi: 10.1093/clinids/12.supplement_3.s327. [DOI] [PubMed] [Google Scholar]
- 93.Black DJ, Kunze KL, Wienkers LC, et al. Warfarin-fluconazole II. A metabolically based drug interaction. Drug Metab Dispos. 1996;24:422–428. [PubMed] [Google Scholar]
- 94.Kunze KL, Trager WF. Warfarin-fluconazole III. A rational approach to management of a metabolically based drug interaction. Drug Metab Dispos. 1996;24:429–435. [PubMed] [Google Scholar]
- 95.O'Reilly RA, Goulart DA, Kunze KL, et al. Mechanisms of the stereoselective interaction between miconazole and racemic warfarin in human subjects. Clin Pharmacol Ther. 1992;51:656–667. doi: 10.1038/clpt.1992.78. [DOI] [PubMed] [Google Scholar]
- 96.Back DJ, Stevenson P, Tjia JF. Comparative effects of two antimycotic agents, ketoconazole and terbinafine, on the metabolism of tolbutamide, ethinyloestradiol, cyclosporin and ethoxycoumarin by human liver microsomes. Br J Clin Pharmacol. 1989;28:166–170. doi: 10.1111/j.1365-2125.1989.tb05410.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McCrea JB, Lo MW, Furtek CI, et al. Ketoconazole does not affect the systemic conversion of losartan to E-3174. Clin Pharmacol Ther. 1996;59:169. [Google Scholar]
- 98.Touchette MA, Chandrasekar PH, Milad MA, Edwards DJ. Contrasting effects of fluconazole and ketoconazole on phenytoin and testosterone disposition in man. Br J Clin Pharmacol. 1992;34:75–78. doi: 10.1111/j.1365-2125.1992.tb04111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Transon C, Leeman R, Dayer P. In vitro comparative inhibition profiles of major drug metabolising cytochrome P450 isozymes (CYP2C9, CYP2D6, CYP3A4) by HMG-CoA reductase inhibitors. Eur J Clin Pharmacol. 1996;50:209–215. doi: 10.1007/s002280050094. [DOI] [PubMed] [Google Scholar]
- 100.Transon C, Leeman R, Vogt N, Dayer P. In vivo inhibition profile of cytochrome P450TB (CYP2C9) by (±)-fluvastatin. Clin Pharmacol Ther. 1995;58:412–417. doi: 10.1016/0009-9236(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 101.Lewis RJ, Trager WF, Chan KE, et al. Warfarin—Stereochemical aspects of its metabolism and the interaction with phenylbutazone. J Clin Invest. 1974;53:1607–1617. doi: 10.1172/JCI107711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Miners JO, Foenander T, Wanwimolruk S, Gallus AS, Birkett DJ. The effect of sulphinpyrazone on oxidative drug metabolism in man: Inhibition of tolbutamide elimination. Eur J Clin Pharmacol. 1982;22:321–326. doi: 10.1007/BF00548400. [DOI] [PubMed] [Google Scholar]
- 103.Miners JO, Foenander T, Wanwimolruk S, Gallus AS, Birkett DJ. Interaction of sulphinpyrazone with warfarin. Eur J Clin Pharmacol. 1982;22:327–331. doi: 10.1007/BF00548401. [DOI] [PubMed] [Google Scholar]
- 104.He M, Kunze KL, Trager WF. Inhibition of S-warfarin metabolism by sulfinpyrazone and its metabolites. Drug Metab Dispos. 1995;23:659–663. [PubMed] [Google Scholar]
- 105.Christensen LK, Hansen JM, Kristensen M. Sulphaphenazole-induced hypoglycaemic attacks in tolbutamide-treated diabetics. Lancet. 1963;2:1298–1301. doi: 10.1016/s0140-6736(63)90847-x. [DOI] [PubMed] [Google Scholar]
- 106.Hansen JM, Kampmann JP, Siersbaek-Nielsen K, et al. The effect of different sulfonamides on phenytoin metabolism in man. Acta Med Scand. 1979;24(Suppl 6):106–110. doi: 10.1111/j.0954-6820.1979.tb00729.x. [DOI] [PubMed] [Google Scholar]
- 107.Wing LMH, Miners JO. Cotrimoxazole as an inhibitor of oxidative drug metabolism: Effects of trimethoprim and sulphamethoxazole separately and combined on tolbutamide disposition. Br J Clin Pharmacol. 1985;20:482–485. doi: 10.1111/j.1365-2125.1985.tb05102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lumholtz B, Siersbaek-Nielsen K, Skovsted L, Kampmann J, Molholm-Hansen J. Sulfamethizole-induced inhibition of diphenylhydratoin, tolbutamide and warfarin metabolism. Clin Pharmacol Ther. 1975;17:731–734. doi: 10.1002/cpt1975176731. [DOI] [PubMed] [Google Scholar]
- 109.Suttle AB, Vargo DL, Wilkinson LA, Birmingham BK, Lasseter K. Effect of zafirlukast on the pharmacokinetics of R- and S-warfarin in healthy men. Clin Pharmacol Ther. 1997;61:186. [Google Scholar]
- 110.Miners JO, Wing LMH, Lillywhite KJ, Smith KJ. Failure of therapeutic doses of ß-adrenoreceptor antagonists to alter the disposition of tolbutamide and lignocaine. Br J Clin Pharmacol. 1984;18:853–860. doi: 10.1111/j.1365-2125.1984.tb02555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Robson RA, Miners JO, Whitehead AG, Birkett DJ. Specificity of the inhibitory effect of dextropropoxyphene on oxidative drug metabolism: Lack of effect on theophylline and tolbutamide disposition. Br J Clin Pharmacol. 1987;23:772–776. doi: 10.1111/j.1365-2125.1987.tb03115.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Abernethy DR, Kaminsky LS, Dickinson TH. Selective inhibition of warfarin metabolism by diltiazem in humans. 257:411–416. [PubMed] [Google Scholar]
- 113.Toon S, Hopkins KJ, Garstang FM, Aarons L, Sedman A, Rowland M. Enoxacin-warfarin interaction: Pharmacokinetic and stereochemical aspects. Clin Pharmacol Ther. 1987;42:33–41. doi: 10.1038/clpt.1987.104. [DOI] [PubMed] [Google Scholar]
- 114.Milne RW, Coulthard K, Nation RL, Penna AC, Roberts G, Sansom LN. Lack of effect of erythromycin on the pharmacokinetics of single oral doses of phenytoin. Br J Clin Pharmacol. 1988;26:330–333. doi: 10.1111/j.1365-2125.1988.tb05285.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Jensen JC, Gugler R. Interaction between metronidazole and drugs eliminated by oxidative metabolism. Clin Pharmacol Ther. 1985;37:407–410. doi: 10.1038/clpt.1985.63. [DOI] [PubMed] [Google Scholar]
- 116.Maya JF, Callaghan JT, Bergstrom RF, Cerimele BJ, Kassahun K, Nyhart EH. Olanzapine and warfarin drug interaction. Clin Pharmacol Ther. 1997;61:182. [Google Scholar]
- 117.Andersson T, Lagerstrom P-O, Unge P. A study of the interaction between omeprazole and phenytoin in epileptic patients. Ther Drug Monit. 1990;12:329–333. doi: 10.1097/00007691-199007000-00005. [DOI] [PubMed] [Google Scholar]
- 118.Sutfin T, Balmer K, Bostrom H, Eriksson S, Hoglund P, Paulsen O. Stereoselective interaction of omeprazole with warfarin in healthy men. Ther Drug Monit. 1989;11:176–184. doi: 10.1097/00007691-198903000-00010. [DOI] [PubMed] [Google Scholar]
- 119.Scott J, Poffenbarger PL. Pharmacogenetics of tolbutamide metabolism in humans. Diabetes. 1979;28:41–51. [PubMed] [Google Scholar]
- 120.Back DJ, Orme ML’E. Genetic factors influencing the metabolism of tolbutamide. Pharmacol Ther. 1989;44:147–155. doi: 10.1016/0163-7258(89)90064-8. [DOI] [PubMed] [Google Scholar]
- 121.Miners JO, Wing LMH, Birkett DJ. Normal metabolism of debrisoquine and theophylline in a slow tolbutamide metaboliser. Aust NZ J Med. 1985;15:348–349. doi: 10.1111/j.1445-5994.1985.tb04052.x. [DOI] [PubMed] [Google Scholar]
- 122.Sullivan-Klose TH, Ghanayem BI, Bell DA, et al. The role of the CYP2C9-Leu (359) allelic variant in the tolbutamide polymorphism. Pharmacogenetics. 1996;6:341–349. doi: 10.1097/00008571-199608000-00007. [DOI] [PubMed] [Google Scholar]
- 123.Page MA, Boutagy JS, Shenfield GM. A screening test for slow metabolisers of tolbutamide. Br J Clin Pharmacol. 1991;31:649–654. doi: 10.1111/j.1365-2125.1991.tb05587.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Veronese ME, Miners JO, Rees DLP, Birkett DJ. Tolbutamide hydroxylation in humans: Lack of biomodality in 106 healthy subjects. Pharmacogenetics. 1993;3:86–93. doi: 10.1097/00008571-199304000-00004. [DOI] [PubMed] [Google Scholar]
- 125.Vermeij P, Ferrari MD, Buruma JS, Veenema H, deWolff FA. Inheritance of poor phenytoin parahydroxylation capacity in a Dutch family. Clin Pharmacol Ther. 1988;44:588–593. doi: 10.1038/clpt.1988.198. [DOI] [PubMed] [Google Scholar]
- 126.Inaba T. Phenytoin: Pharmacogenetic polymorphism of 4′-hydroxylation. Pharmacol Ther. 1990;46:341–347. doi: 10.1016/0163-7258(90)90022-t. [DOI] [PubMed] [Google Scholar]
- 127.Spielberg S, McCrea J, Cribb A, et al. A mutation in CYP2C9 is responsible for decreased metabolism of losartan. Clin Pharmacol Ther. 1996;59:215. [Google Scholar]
- 128.Bhasker CR, Miners JO, Coulter S, Birkett DJ. Allelic and functional variability of cytochrome P4502C9. Pharmacogenetics. 1997;7:51–58. doi: 10.1097/00008571-199702000-00007. [DOI] [PubMed] [Google Scholar]
- 129.Stubbins MJ, Harries LW, Smith G, Tarbit MH, Wolf CR. Genetic analysis of the human cytochrome P4502C9 locus. Pharmacogenetics. 1996;6:429–439. doi: 10.1097/00008571-199610000-00007. [DOI] [PubMed] [Google Scholar]
- 130.Inoue K, Yamazaki H, Imiya K, Akasaka S, Guengerich FP, Shimada T. Relationship between CYP 2C9 and 2C19 genotypes and tolbutamide methylhydroxylation and S-mephenyotin 4′–hydroxylation activities in livers of Japanese and Caucasian populations. Pharmacogenetics. 1997;7:103–113. doi: 10.1097/00008571-199704000-00003. [DOI] [PubMed] [Google Scholar]
- 131.Wang S-L, Huang J-D, Lai M-D, Tsai J-J. Detection of CYP2C9 polymorphism based on the polymerase chain reaction in Chinese. Pharmacogenetics. 1995;5:37–42. doi: 10.1097/00008571-199502000-00004. [DOI] [PubMed] [Google Scholar]
- 132.Nasu K, Kubota T, Ishizaki T. Genetic analysis of CYP2C9 polymorphism in a Japanese population. Pharmacogenetics. 1997;7:405–409. doi: 10.1097/00008571-199710000-00011. [DOI] [PubMed] [Google Scholar]
- 133.Haining RL, Hunter AP, Veronese ME, Trager WF, Rettie AE. Allelic variants of human cytochrome P4502C9: Baculovirus-mediated expression, purification, structural characterization, substrate stereoselectivity, and prochiral selectivity of the wild-type and I359L mutant forms. Arch Biochem Biophys. 1996;333:447–458. doi: 10.1006/abbi.1996.0414. [DOI] [PubMed] [Google Scholar]
- 134.Steward DJ, Haining RL, Henne KR, et al. Genetic association between sensitivity to warfarin and expression of CYP2C9*3. Pharmacogenetics. 1997;7:361–367. doi: 10.1097/00008571-199710000-00004. [DOI] [PubMed] [Google Scholar]
- 135.Kaminsky LS, deMorais S, Faletto MB, Dunbar DA, Goldstein JA. Correlation of human cytochrome P4502C substrate specificities with primary structure: Warfarin as a probe. Mol Pharmacol. 1993;43:234–239. [PubMed] [Google Scholar]
- 136.Rettie AE, Wienkers LC, Gonzalez FJ, Trager WF, Korzekwa KR. Impaired (S)-warfarin metabolism catalysed by the R144C allelic variant of CYP2C9. Pharmacogenetics. 1994;4:39–42. doi: 10.1097/00008571-199402000-00005. [DOI] [PubMed] [Google Scholar]
- 137.Crespi CR, Miller VP. The R144C change in the CYP2C9*2 allele alters interaction of the cytochrome P450 with NADPH: cytochrome P450 oxidoreductase. Pharmacogenetics. 1997;7:203–210. doi: 10.1097/00008571-199706000-00005. [DOI] [PubMed] [Google Scholar]
- 138.Furuya H, Fernandez-Salguero P, Gregory W, et al. Genetic polymorphism of CYP2C9 and its effect on warfarin maintenance dose requirement in patients undergoing anticoagulation therapy. Pharmacogenetics. 1995;5:389–392. doi: 10.1097/00008571-199512000-00008. [DOI] [PubMed] [Google Scholar]
- 139.London SJ, Sullivan-Klose T, Daly AK, Idle JR. Lung cancer risk in relation to the CYP2C9 genetic polymorphism among Caucasians in Los Angeles County. Pharmacogenetics. 1997;7:401–404. doi: 10.1097/00008571-199710000-00010. [DOI] [PubMed] [Google Scholar]
- 140.Furuya H, Meyer UA, Gelboin HV, Gonzalez FJ. Polymerase chain reaction directed identification, cloning and quantitation of CYP2C18 mRNA. Mol Pharmacol. 1991;40:375–382. [PubMed] [Google Scholar]
- 141.deMorais SMF, Schweiki H, Blaisdell J, Goldstein JA. Gene structure and upstream regulatory regions of human CYP2C9 and CYP2C18. Biochem Biophys Res Commun. 1993;194:194–201. doi: 10.1006/bbrc.1993.1803. [DOI] [PubMed] [Google Scholar]
- 142.Ibeanu GC, Goldstein JA. Transcriptional regulation of human CYP2C genes: Functional comparison of CYP2C9 and CYP2C18 promoter regions. Biochemistry. 1995;34:8028–8036. doi: 10.1021/bi00025a008. [DOI] [PubMed] [Google Scholar]
- 143.Shimada T, Yamazaki H, Mimura M, et al. Characterisation of microsomal cytochrome P450 enzymes involved in the oxidation of xenobiotic chemicals in human fetal livers and adult lungs. Drug Metab Dispos. 1996;24:515–522. [PubMed] [Google Scholar]
- 144.Hakkola J, Pasanen M, Hukkanen J, et al. Expression of xenobiotic-metabolising cytochrome P450 forms in human full-term placenta. Biochem Pharmacol. 1996;51:403–411. doi: 10.1016/0006-2952(95)02184-1. [DOI] [PubMed] [Google Scholar]
- 145.Hakkola J, Pasanen M, Purkunen R, et al. Expression of xenobiotic metabolising cytochrome P450 forms in human adult and fetal liver. Biochem Pharmacol. 1994;48:59–64. doi: 10.1016/0006-2952(94)90223-2. [DOI] [PubMed] [Google Scholar]
- 146.Ratanasavanh D, Beaune P, Morel F, Flinois J-P, Guengerich FP, Guillouzo A. Intralobular distribution and quantitation of cytochrome P450 enzymes in human liver as a function of age. Hepatology. 1991;13:1142–1151. [PubMed] [Google Scholar]
- 147.Morrow JI, Richens A. Disposition of anticonvulsants in children. Clin Pharmacokinet. 1989;17(Suppl 1):89–104. doi: 10.2165/00003088-198900171-00007. [DOI] [PubMed] [Google Scholar]
- 148.Loi C-M, Vestal RE. Drug metabolism in the elderly. Pharmacol Ther. 1988;36:131–149. doi: 10.1016/0163-7258(88)90115-5. [DOI] [PubMed] [Google Scholar]
- 149.Horn JR, Wittkowsky AK, Linenberger M, Greenberg D, Meade DM, Lam A. Effect of age on disposition of warfarin enantiomers. Clin Pharmacol Ther. 1997;61:175. [Google Scholar]
- 150.Harris RZ, Benet LZ, Schwartz JB. Gender effects in pharmacokinetics and pharmacodynamics. Drugs. 1995;50:222–239. doi: 10.2165/00003495-199550020-00003. [DOI] [PubMed] [Google Scholar]
- 151.Chan E, Ti TY, Lee HS. Population pharmacokinetics of phenytoin in Singapore Chinese. Eur J Clin Pharmacol. 1990;39:177–181. doi: 10.1007/BF00280055. [DOI] [PubMed] [Google Scholar]
- 152.Grasela TH, Sheiner LB, Rambeck B, et al. Steady-state pharmacokinetics of phenytoin from routinely collected patient data. Clin Pharmacokinet. 1983;8:355–364. doi: 10.2165/00003088-198308040-00006. [DOI] [PubMed] [Google Scholar]
- 153.James AH, Britt RP, Raskino CL, Thompson SG. Factors affecting the maintenance doses of warfarin. J Clin Pathol. 1992;45:704–706. doi: 10.1136/jcp.45.8.704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Chen L, Yasumori T, Yamazoe Y, Kato R. Hepatic microsomal tolbutamide hydroxylation in Japanese: In vitro evidence for rapid and slow metabolisers. Pharmacogenetics. 1993;3:77–85. doi: 10.1097/00008571-199304000-00003. [DOI] [PubMed] [Google Scholar]