

Abstract
Aim
The aim of this study was to evaluate the pharmacokinetic profile of donepezil HCl (5 mg) in patients with impaired hepatic function, following the administration of single oral doses.
Methods
This was an open-label, non-randomized study comparing the pharmacokinetic profile of donepezil in male volunteers with chronic compensated cirrhosis of the liver (n = 10) to that in healthy age- and sex-matched controls (n = 10). Each subject received a single 5 mg oral dose of donepezil. Blood samples for pharmacokinetic analyses were taken at specified intervals up to 120-h post-dose. Concentrations of donepezil in plasma were determined by HPLC with UV detection.
Results
No statistically significant differences in donepezil pharmacokinetics were observed between hepatically impaired patients and normal subjects, with the exception of Cmax. The hepatically impaired patients showed a statistically significant, but not clinically significant, higher mean Cmax value of 6.6 ng ml−1 compared with the control group, which had a mean Cmax value of 4.8 ng ml−1 (P = 0.022). This represented an increase of 37.5%. The observed changes in AUC values were smaller and not statistically different. The AUC(0–120) and AUC(0–∞) were 7% and 21% larger in the hepatically impaired patients than in the normal subjects, respectively, although clearance and volume of distribution were almost identical in the two groups. The t½ increased by 20%, but this change was also not statistically significant. Donepezil was equally well tolerated by all subjects.
Conclusions
This study demonstrates that compromised hepatic function does not produce clinically significant changes in the pharmacokinetics of donepezil following single-dose administration. These results suggest that the administration of donepezil to patients with hepatic disease in clinical practice should not require any dosing modifications.
Keywords: donepezil, Alzheimer’s disease, hepatic impairment
Introduction
Donepezil HCl (also known as E2020 or Aricept®, the registered trademark of Eisai Co. Ltd, Tokyo, Japan), a piperidine, is a highly selective inhibitor of the enzyme acetylcholinesterase (AChE) [1, 2] that is chemically unique from other AChE inhibitors [3,[4]. In vitro and pre-clinical studies have demonstrated that donepezil is approximately 1200 times more selective for AChE in the brain than for butyrylcholinesterase (BuChE) in the periphery [1, 2, 5]. Phase II and III studies conducted in the USA have shown that donepezil (5 mg or 10 mg once daily) produces statistically significant improvements in cognition and global function in patients with Alzheimer’s disease [6–9]. Its clinical efficacy and minimal side-effect profile are thought to be related to its specific inhibition of AChE in the areas of the brain affected by the cholinergic deficit that typifies this disease [1, 2, 5].
Assessment of the potential impact of hepatic dysfunction on the pharmacokinetic and adverse event profiles of donepezil is of primary importance, as donepezil is orally administered and subject to extensive first-pass metabolism. In addition, both pre-clinical and clinical studies have demonstrated that donepezil is metabolized primarily in the liver [6–11]. In vitro studies using human hepatic microsomes have shown that the cytochrome P-450 isoenzyme CYP-3A4 is mostly responsible for the metabolism of donepezil, with CYP-2D6 playing a minor role. With this in mind, this study was designed to assess the effects of compromised hepatic function on the pharmacokinetics of donepezil HCl.
Methods
Subjects
Ten male volunteers with stable, chronic, compensated cirrhosis of the liver and ten age- and sex-matched healthy controls were enrolled in the study. Eligible subjects in both groups were between 18 and 65 years of age and within 30% of ideal weight for their height and body build, based on the Metropolitan Insurance Company Height and Weight Tables (1983). All subjects were required to be free from clinically significant gastrointestinal, renal, respiratory, endocrine, haematological, neurological, psychiatric or cardiovascular system abnormalities. None of the study subjects had a known or suspected recent history of alcohol or drug misuse or a positive urine drug screen at the time of entry into this study.
Healthy subjects were required to have no evidence of hepatic dysfunction in clinical or laboratory evaluations (i.e. all tests and examination results within normal limits). Subjects with liver cirrhosis were selected from patients with a history and clinical evidence of stable, compensated alcoholic cirrhosis of the liver, confirmed by percutaneous biopsy or liver imaging studies. Subjects with clinical evidence of significant ascites, evidence of hepato-renal syndrome, extrahepatic bile duct obstruction, or acute or chronic hepatitis were excluded from entry.
The study was conducted in accordance with the Declaration of Helsinki, and the study protocol was approved by the Institutional Review Board of the New Orleans Clinical Research Center. All subjects gave written informed consent prior to participation in any study activities.
Protocol
This was an open-label, non-randomized study of single doses of donepezil HCl. Eligible subjects completed a screening assessment no more than 2 weeks prior to receiving study medication. Subjects were admitted to the study site in the evening approximately 12 h prior to drug administration. A single 5 mg dose of donepezil HCl was administered with 250 ml of water at about 08:00 h, following an 8-h overnight fast. The subjects remained in an upright position (>45° angle from supine) and continued to fast from food and fluids for 4 h after drug administration.
All study participants remained at the study site for 48-h following drug administration. During this time, blood samples were collected at specified intervals for the measurement of donepezil in plasma. The subjects were discharged from the study site on the morning of day 3, after the collection of a blood sample at the 48-h time-point. They returned to the study site each morning for the next 3 days to provide additional blood samples for pharmacokinetic analysis. End-of-study physical and laboratory evaluations were conducted on the morning of day 6, following the 120-h sampling time-point.
Throughout the study, the subjects abstained from physical activity other than normal walking. Healthy subjects had received no prescription, non-prescription or investigational drugs for at least 1 month prior to entering the trial. Over-the-counter products, illicit drugs and alcohol consumption were not permitted from 72 h prior to admission until the end of the treatment period. Subjects with hepatic cirrhosis were allowed to continue receiving medications considered essential for their well-being.
Sample collection and analysis
Venous blood samples for the determination of donepezil concentrations in plasma were collected 1 h prior to, and at 1, 2, 3, 4, 6, 8, 12, 18, 24, 36, 48, 72, 96 and 120 h after drug administration. Blood was collected in 7 ml heparinized, evacuated containers. Immediately after collection, the samples were centrifuged (2000 g) for 15 min at 4° C. The plasma was then removed and transferred into 10 ml polypropylene storage tubes. Samples were stored in an upright position at −20° C until analysis. Plasma concentrations of donepezil HCl were measured using high-performance liquid chromatography (HPLC) with UV detection (315 nm) [12]. The lower detection limit of the assay was 2.0 ng ml−1.
Pharmacokinetic assessments
Characterization of donepezil pharmacokinetics was conducted using plasma drug concentration data from the collected samples. All volunteers who received test medication and who had analytical samples collected were evaluated. The peak concentration (Cmax) and the time of occurrence of Cmax (tmax) were computed from the observed values. In cases where Cmax occurred at multiple time-points, the tmax selected was the earliest. The terminal disposition phase was identified by visual inspection of each subject’s semi-logarithmic donepezil plasma concentration–time curve. The terminal disposition phase was selected for timepoints from 24- to 120-h post-dose. The elimination rate constant (λz) was estimated as −2.303 times the slope of the best-fit linear regression line of the terminal phase. The terminal half-life (t½) was calculated as 0.693/λz. The area under the plasma concentration–time curve from 0 to 120 h (AUC(0–120)) was calculated using the trapezoidal rule and extrapolated to time infinity (AUC(0–∞)) by adding the ratio of the last measurable concentration (at 120 h) to λz. Time-averaged total body clearance (CLT/F) was calculated as the ratio of the administered dose to AUC(0–∞), and apparent volume of distribution (Vλz/F) was calculated as the ratio of CLT/F to λz, where the factor F accounts for bioavailability.
Safety assessments
All adverse events reported spontaneously by the subjects and/or observed or elicited during physical examination were documented together with times of onset and cessation, plus assessments of severity and causality. Vital signs were monitored at regular intervals during the in-house portion of each treatment period. Laboratory evaluations were conducted at the start of the treatment period.
Statistical analysis
One-way analysis of variance (ANOVA) was used to compare the pharmacokinetic parameters of the hepatically impaired and normal subjects. Confidence intervals (95%) were computed for the mean difference between the two subject groups. All statistical tests were two-tailed at the 0.05 significance level.
Results
Subjects
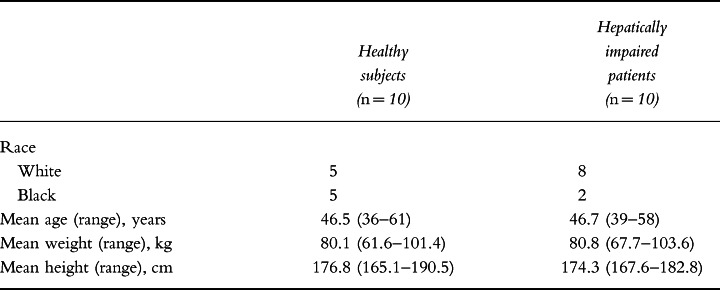
Ten male patients with stable hepatic cirrhosis and ten healthy, age- and sex-matched controls were enrolled into this study. The two treatment groups were similar with respect to mean age, weight and height (Table 1). However, liver function tests in the hepatically impaired patients revealed elevated levels of liver enzymes at baseline compared with the healthy controls. Their baseline concentrations of alanine aminotransferase, aspartate aminotransferase and bilirubin were 74.3 ± 21.9 IU l−1, 92.1 ± 18.0 IU l−1 and 2.02 ± 0.45 mg dl−1, respectively, compared with 19.5 ± 1.4 IU l−1, 23.5 ± 1.9 IU l−1 and 0.71 ± 0.07 mg dl−1, respectively, in the normal subjects. All 20 volunteers completed the study without incident.
Table 1.
Summary of demographic data.
Pharmacokinetics of donepezil
The mean plasma concentration–time curves following administration of a single 5-mg oral dose of donepezil to healthy subjects and to patients with cirrhosis are shown in Figure 1. Data from one hepatically impaired subject was excluded from the pharmacokinetic analysis as a consequence of his HPLC plasma analysis in which a large extraneous peak eluted with a similar retention time as donepezil. The peak was subsequently found to be caused by spironolactone, which was being concomitantly administered for the treatment of peripheral oedema.
Figure 1.
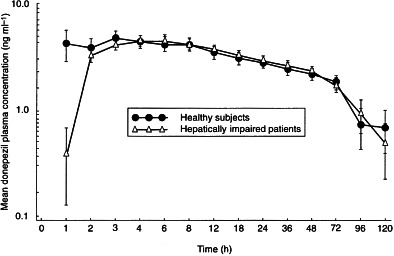
Mean ( ± SE) plasma concentration–time curves following administration of a single 5-mg oral dose of donepezil to healthy subjects (n = 10) and to patients with compromised hepatic function (n = 9).
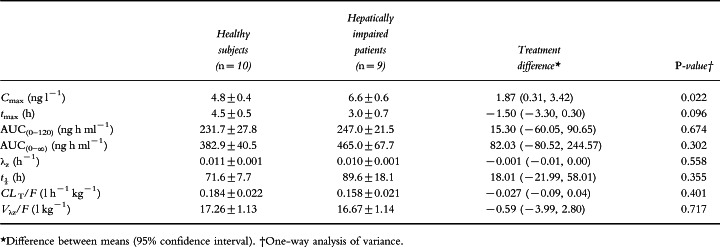
No statistically significant differences in donepezil pharmacokinetics were observed between the hepatically impaired patients and the normal subjects, with the exception of Cmax (Table 2). The hepatically impaired group showed a statistically higher mean Cmax value of 6.6 ng ml−1 compared with the control group, which had a mean Cmax value of 4.8 ng ml−1 (P = 0.022), an increase of 37.5%. The AUC values of the hepatically impaired group were also higher than those of the control group, but the differences were not statistically significant. The AUC(0–120) and AUC(0–∞) of the hepatically impaired group were found to be 7% and 21% greater, respectively, than for the normal subjects. The value of t½ was about 20% greater in the hepatically impaired group than in the control group, but again this change was not statistically significant. CLT/F, Vλz/F and λz were essentially identical in the two groups. In the majority of normal and hepatically impaired subjects, donepezil was no longer detectable in plasma 120 h after drug administration.
Table 2.
Pharmacokinetic parameters (mean ± SE) following administration of a single oral dose of donepezil (5 mg) to healthy subjects and to patients with hepatic impairment.
Safety
Donepezil was well tolerated by all subjects and no changes in vital sign parameters were observed during the course of the study. Several hepatically impaired patients had ECG characteristics at baseline consistent with their medical condition. These were primarily non-specific ST–T wave changes. None of the changes was clinically significant or related to treatment.
Discussion
The involvement of cytochrome P-450 isoenzymes in the metabolism of donepezil combined with first-pass metabolism following oral dosing has highlighted the importance of investigating whether or not hepatic impairment produces any changes in the pharmacokinetic profile of the drug.
The pharmacokinetic results summarized here suggest that compromised hepatic function appears to increase plasma levels of donepezil relative to those achieved in healthy age- and sex-matched control subjects. However, of the pharmacokinetic parameters examined, only Cmax increased significantly. The observed difference (37.5%) was greater than the 20% range that is generally considered to represent clinical significance. This may in part reflect the variability associated with the small subject groups used in this study. This finding is also tempered because the differences observed following single-dose administration are expected to become less prominent once steady state is reached. In addition, the pharmacokinetic stability of donepezil in hepatically impaired patients is further supported by the fact that the AUC and t½ values showed much smaller and non-statistically significant changes from those in healthy subjects.
The stability of donepezil’s pharmacokinetic profile is explained by its relatively slow clearance from plasma, primarily by hepatic metabolic pathways [10, 11], which results in a long half-life of approximately 70 h. As a result, concentrations of this drug in the liver are maintained at relatively low levels, with the consequence that its pharmacokinetics are unaltered, even in patients with compromised liver function.
The absence of hepatotoxicity following long-term administration of donepezil to patients with Alzheimer’s disease [13, 14] offers a clear benefit over tacrine. In clinical practice, the potential benefits of tacrine have been offset by its propensity to increase transaminase levels, resulting in clinically significant dose-limiting hepatotoxicity in up to 25% of patients [15–17]. Tacrine is contraindicated in patients with hepatic disease, and safety monitoring is required even in patients with normal liver function.
Donepezil, which is metabolized by the P-450 isoenzymes CYP-3A4 and CYP-2D6, has a low potential for drug–drug interactions, as shown by the studies published in this supplement. The minimal changes in the pharmacokinetic profile of donepezil in hepatically impaired patients observed in the present study provide further support for the safety profile of this drug. The results of the present study suggest that compromised hepatic function does not produce clinically significant changes in the pharmacokinetics of donepezil and hence dose modifications should not be required when treating this cohort of patients in clinical practice.
Acknowledgments
We acknowledge the efforts of Dr William B. Smith of Louisiana Cardiovascular Research Center, 2820 Canal Street, New Orleans, LA 70119, USA, who conducted this clinical trial, and the Institutional Review Board of Louisiana Cardiovascular Research Center who reviewed and approved the study and protocol.
References
- 1.Yamanishi Y, Ogura H, Kosasa T, Araki S, Sawa Y, Yamatsu K. Inhibitory action of E2020, a novel acetylcholinesterase inhibitor, on cholinesterase: comparison with other inhibitors. In: Nagatsu T, Yoshida M, Fisher A, editors. Basic, Clinical, and Therapeutic Aspects of Alzheimer’s and Parkinson’s Diseases. Vol. 2. New York: Plenum Press; 1990. pp. 409–413. [Google Scholar]
- 2.Rogers SL, Yamanishi Y, Yamatsu K. E2020—the pharmacology of a piperidine cholinesterase inhibitor. In: Becker R, Giacobini E, editors. Cholinergic Basis for Alzheimer Therapy. Boston: Birkhäuser; 1991. pp. 314–320. [Google Scholar]
- 3.Sugimoto H, Iimura Y, Yamanishi Y, Yamatsu K. Synthesis and antiacetylcholinesterase activity of 1-benzyl-4-[(5,6-dimethoxy-1-indanon-2-yl)methyl]piperidine hydrochloride (E2020) and related compounds. Bioorg Med Chem Lett. 1992;2:871–876. [Google Scholar]
- 4.Iimura Y, Mishima M, Sugimoto H. Synthesis of 1-benzyl-4-[(5,6-dimethoxy[2−14C]-1-indanon)-2-yl]-methylpiperidine hydrochloride (E2020−14C) J Label Compounds Radiopharm. 1989;XXVII:835–839. [Google Scholar]
- 5.Sherman K. Pharmacodynamics of oral E2020 and tacrine in humans: novel approaches. In: Becker R, Giacobini E, editors. Cholinergic Basis for Alzheimer Therapy. Boston: Birkhäuser; 1991. pp. 321–328. [Google Scholar]
- 6.Rogers SL, Friedhoff LT. The efficacy and safety of donepezil in patients with Alzheimer’s disease: results of a US multicenter, randomized, double-blind, placebo-controlled trial. Dementia. 1996;7:293–303. doi: 10.1159/000106895. [DOI] [PubMed] [Google Scholar]
- 7.Rogers SL, Doody RS, Mohs R, Friedhoff LT. E2020 produces both clinical global and cognitive test improvement in patients with mild to moderately severe Alzheimer’s disease (AD): results of a 30-week Phase III trial. Neurology. 1996;46:A217. S14.001. [Google Scholar]
- 8.Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT the Donepezil Study Group. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology. 1998;50:136–145. doi: 10.1212/wnl.50.1.136. [DOI] [PubMed] [Google Scholar]
- 9.Rogers SL, Doody RS, Mohs R, Friedhoff LT the Donepezil Study Group. Donepezil improves cognition and global function in Alzheimer’s disease: a 15-week, double-blind, placebo-controlled study. Arch Intern Med. 1998;158:1021–1031. doi: 10.1001/archinte.158.9.1021. [DOI] [PubMed] [Google Scholar]
- 10.Mihara M, Ohnishi A, Tomono Y, et al. Pharmacokinetics of E2020, a new compound for Alzheimer’s disease, in healthy male volunteers. Int J Clin Pharmacol Ther Toxicol. 1993;31:223–229. [PubMed] [Google Scholar]
- 11.Ohnishi A, Mihara M, Kamakura H, et al. Comparison of the pharmacokinetics of E2020, a new compound for Alzheimer’s disease, in healthy young and elderly subjects. J Clin Pharmacol. 1993;33:1086–1091. doi: 10.1002/j.1552-4604.1993.tb01945.x. [DOI] [PubMed] [Google Scholar]
- 12.Lee JW, Rogers SL, Friedhoff LT, Stiles MR, Cooper NM. Validation and application of an HPLC method for the determination of 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methyl piperidine hydrochloride (E2020) in human plasma. Pharm Res. 1992;9:350. [Google Scholar]
- 13.Rogers SL, Perdomo C, Friedhoff LT. Clinical benefits are maintained during long-term treatment of Alzheimer’s disease with the acetylcholinesterase inhibitor, E2020. Eur Neuropsychopharmacol. 1995;5:386. P-8–21. [Google Scholar]
- 14.Rogers SL, Friedhoff LT. Long-term efficacy and safety of donepezil in the treatment of Alzheimer’s disease: an interim analysis of the results of a US multicentre open label extension study. Eur Neuropsychopharmacol. 1998;8:67–75. doi: 10.1016/s0924-977x(97)00079-5. [DOI] [PubMed] [Google Scholar]
- 15.Wagstaff AJ, McTavish D. Tacrine: a review of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy in Alzheimer’s disease. Drugs Aging. 1994;4:510–540. doi: 10.2165/00002512-199404060-00006. [DOI] [PubMed] [Google Scholar]
- 16.Crimson ML. Tacrine: first drug approved for Alzheimer’s disease. Ann Pharmacother. 1994;28:744–751. doi: 10.1177/106002809402800612. [DOI] [PubMed] [Google Scholar]
- 17.Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA. 1994;271:992–998. [PubMed] [Google Scholar]