

Abstract
Aim
The aim of this study was to characterize the pharmacokinetic and pharmacodynamic profile of donepezil HCl, a chemically distinct and specific acetylcholinesterase (AChE) inhibitor for the treatment of Alzheimer’s disease, following administration of single oral doses.
Methods
This was a double-blind, randomized, single-dose, placebo-controlled, sequential-group, ascending-dose study in healthy male volunteers (n = 48). Six dose levels were investigated, ranging from 0.3 to 6.0 mg. Donepezil concentrations in plasma were determined by HPLC with UV detection. Pharmacodynamic activity was determined by the radioenzymatic measurement of erythrocyte membrane acetylcholinesterase (rbc-AChE) inhibition.
Results
The pharmacokinetic disposition of donepezil was observed to be both linear and dose proportional following single-dose administration. The mean peak plasma concentration (Cmax) of donepezil was observed at 4.1±1.5 h. The mean terminal disposition half-life was 81.5±22.0 h. The post-absorption phase of the plasma concentration–time curves for the 4.0 mg and 6.0 mg doses appeared to be biphasic, but the rate of donepezil clearance was independent of dose. Plasma concentrations for the 0.3, 0.6 and 0.9 mg dose groups were generally below the level of HPLC detection (2.0 ng ml−1), preventing accurate characterization of these doses. A direct correlation was observed between plasma donepezil concentrations and extent of AChE inhibition. For the 4.0 and 6.0 mg donepezil dose groups, maximal AChE inhibition (Emax) ranged from 33% to 35% and there was significant correlation between AChE inhibition and donepezil plasma concentration (P<0.005).
Conclusions
The pharmacokinetics of donepezil were found to be linear and dose proportional following the administration of single doses to healthy volunteers. A direct correlation was also observed between plasma donepezil concentrations and AChE inhibition. The extended half-life of donepezil makes it suitable for once-daily dosing.
Keywords: donepezil, selective acetylcholinesterase inhibitor, Alzheimer’s disease, pharmacokinetics, pharmacodynamics
Introduction
It is generally accepted that the symptoms of Alzheimer’s disease are related to a cholinergic deficit in the cerebral cortex and other areas of the brain [1–4]. The hippocampal formation, in particular, has been shown to play an important role in the processes of memory and learning [5]. The severity of cholinergic loss in the central nervous system (CNS) has been found to correlate with both the extent of neuropathology and the severity of cognitive impairment [3]. These findings have led to the development of various classes of agents designed to augment the activity of the surviving cholinergic system in an attempt to improve cognitive function.
The cholinesterase inhibitors have been studied extensively for the treatment of Alzheimer’s disease. Donepezil HCl (also known as E2020 or Aricept®; the registered trademark of Eisai Co. Ltd, Tokyo, Japan) is a new, piperidine-based molecule that is chemically distinct from other cholinesterase inhibitors. It is the product of a specific research programme designed to produce an agent selective for the acetylcholinesterase (AChE) found in the CNS [6–10].
Donepezil was shown to improve memory and learning performance in normal rats as well as in rats with experimentally induced cholinergic lesions [11]. Preclinical studies indicated that donepezil had markedly greater specificity for brain tissue than physostigmine or tacrine [11]. It also had a longer duration of inhibitory action than either of these agents [11]. Thus, it was expected that this high degree of specificity for AChE and very low affinity for plasma cholinesterases would produce central activity with minimal peripheral cholinergic adverse events. Moreover, unlike acridine-based cholinesterase inhibitors such as tacrine [11, 12], donepezil is not compromised by dose-limiting hepatotoxicity [13–15].
In controlled phase II and III clinical studies of up to 6 months’ duration, donepezil (5 and 10 mg day−1) produced significant improvement in cognitive and global function in patients with mild to moderately severe Alzheimer’s disease [13, 14, 16]. Furthermore, additional clinical trials indicate that these clinical improvements are maintained, relative to untreated historical control subjects, during long-term treatment (over 2 years) [15].
The purpose of the present study was to characterize the pharmacokinetic and pharmacodynamic profiles associated with the administration of single oral doses of donepezil in healthy volunteers.
Methods
Subjects
The study population comprised 48 healthy, ambulatory, non-smoking adult male volunteers, between 19 and 41 years of age. Their body weight ranged between 65.0 and 85.9 kg, and all were within 15% of ideal weight based on the Metropolitan Insurance Company Height and Weight Tables (1983). Upon clinical examination, subjects were found to be free from significant hepatic, gastrointestinal, renal, respiratory, endocrine, haematological, neurological, psychiatric or cardiovascular system abnormalities. None had a known or suspected history of alcohol or drug abuse, and none had taken over-the-counter medications or consumed alcoholic beverages for at least 48 h prior to enrolment. No investigational or prescription medications had been taken within 1 month of entry into the study.
This study was conducted in accordance with the principles stated in the Declaration of Helsinki. The protocol was approved by the Institutional Review Board for Investigations Involving Human Subjects, Harris Laboratories, Lincoln, Nebraska, USA, and all subjects gave written informed consent prior to participation.
Protocol
The study was of a double-blind, randomized, placebo-controlled, single-dose, sequential-group design. Donepezil was administered in single oral doses according to an ascending-dose scheme. The selection of doses (0.3, 0.6, 0.9, 2.0, 4.0 and 6.0 mg), which was based upon results from phase I studies conducted in Japan, included doses below and within the range anticipated to be clinically effective.
Groups of eight subjects were assigned to each dose level. Within each dose group, two subjects were randomized to receive placebo and six to receive donepezil. Progression to the next higher dose was contingent upon demonstration that the previous dose was well tolerated. Baseline measurements were taken on study day 1 and the study medication was taken on study day 2.
The trial was blinded by using a standard dose of three tablets per patient. The dose consisted of donepezil (0.3 mg or 2.0 mg tablets) and/or placebo tablets, the number of each being determined by dose group and randomization code. The tablets were taken with 250 ml of tap water after an 8-h fast. The subjects remained awake and in an upright position (>45° angle from supine) for 4 h after drug administration, after which they were allowed food. Subjects stayed at the investigational site for an additional 6 days until all of the follow-up assessments had been completed. During this time, they abstained from caffeine-containing beverages, and physical exertion was limited to normal walking.
Sample collection and analysis
Venous blood samples for the measurement of donepezil concentrations and AChE activity were collected at 30-min intervals for the first 4 h post-dose, and then at 6, 8, 12, 18, 24 and 36 h. Thereafter, blood samples were taken once daily (48, 72, 96 and 120 h post-dose). A final sample was taken on day 8 (144 h). Samples were placed on ice immediately upon collection.
Characterization of donepezil pharmacokinetics was performed by analysing plasma separated from the cellular components by centrifugation and stored at −20° C until analysis. Plasma donepezil concentrations were measured using a specific and sensitive high-performance liquid chromatography (HPLC) method with ultraviolet (UV) detection [17]. The lowest quantifiable concentration was 2.0 ng ml−1.
Pharmacokinetic analysis
Model-independent pharmacokinetic parameters were determined using standard techniques. The peak plasma concentration (Cmax) and the time at which the peak concentration occurred (tmax) were determined by inspection of individual subject data. For subjects with more than one peak, tmax was defined as the time at which the highest peak occurred. If the multiple peaks were the same height, then tmax was defined for the first peak. A terminal disposition rate constant (λz) was calculated by linear least-squares regression of the log-transformed plasma concentration–time data in the terminal disposition phase. The slope of the terminal disposition phase was calculated from the first time point at which there was no systematic deviation from log-linear decline in plasma concentration to the last quantifiable measurement. The terminal half-life (t½) was calculated as 0.693/λz.
The area under the plasma concentration–time curve (AUC), for the time (t) at which the final measurable concentration was obtained (AUC(0–t)), was calculated by the linear trapezoidal rule. The AUC from the final time point to time infinity was estimated as the ratio of the final measurable concentration to λz. The total area under the concentration–time curve (AUC(0–∞)) was calculated by addition of AUC(0–t) and AUC(t–∞). Time-averaged total body clearance (CL T/F) was calculated as the ratio of the administered dose (D) to AUC(0–∞), where the factor F accounts for bioavailability (D/AUC(0–∞)·F). The apparent volume of distribution (Vλz/F) was estimated as the ratio of CL T/F to λz.
Pharmacodynamic assessments
The pharmacodynamic activity of donepezil was examined by measuring AChE activity in peripheral erythrocyte membranes (rbc-AChE) taken from the samples used for the pharmacokinetic determinations. Following the removal of the plasma for donepezil measurements, the white blood cell/platelet layer and the top 3 mm of erythrocytes (rbc) were extracted, using a Pasteur pipette. The remaining rbcs were gently vortexed before being transferred to a polypropylene Sarstedt storage tube which was stored in an upright position at −20° C until analysis.
To measure AChE activity, a specific and sensitive radioenzyme method was developed in which [3H- acetyl]choline was incubated at 4° C with homogenized rbc containing unknown concentrations of donepezil [18]. After 30 s, the hydrolytic product was extracted into a scintillation cocktail and total radioactivity was counted. Activity from subject samples was quantified using standard curves of percentage rbc-AChE inhibition versus the natural logarithm of the donepezil concentration (ng g−1 erythrocytes). The curves were fit using a third-order polynomial equation. The inter-day precision of this assay was 7.4%. Intra-subject circadian variability in AChE activity was 2.6%. Parameters calculated were maximal AChE inhibition (peak effect), the time at which it occurred, and the area under the effect curve (AUE) from time 0 to time t (AUE(0–t)).
Safety assessments
All adverse events spontaneously reported by a subject or elicited/observed by the investigator were recorded, together with times of onset and cessation, and an assessment of severity and causality.
Vital signs were measured at specified times during the course of the study. Blood pressures were measured according to the American Heart Association recommendations. The point of disappearance of Korotkoff sounds (phase V) was recorded as the diastolic blood pressure.
Routine haematology, clinical chemistry and urinalysis were undertaken following an 8-h fast on days 4 and 8. Haematological assessments included haemoglobin, haematocrit, rbc count, platelet count, white blood cell count and differential. Clinical chemistry comprised assessment of liver function (alanine transaminase, aspartate transaminase, alkaline phosphatase, total bilirubin), renal function (creatinine, blood urea nitrogen), metabolic status (glucose, total protein, albumin, cholesterol, triglycerides), electrolytes (sodium, potassium, chloride, phosphorus, calcium), and cardiac enzymes (lactate dehydrogenase and creatine kinase). Routine urinalysis was performed (pH, glucose, protein, haemoglobin or blood, ketones), together with specific gravity and microscopic examination of the urine sediment.
Statistical analysis
Data obtained from placebo-treated subjects were pooled across doses for statistical comparison. For the pharmacokinetic parameters, comparisons between dose groups were performed using Fisher’s least significant difference procedure.
Three different analytical approaches were taken to evaluate the pharmacodynamic data. These were individual plasma concentration and AChE inhibition versus time profiles; AChE inhibition versus concentration data, pooled both within and across dose levels; and mean AChE inhibition versus concentration profiles within a given dose group, analysed singly or pooled across the dose group. The relationship between plasma donepezil concentration and AChE inhibition was defined using orthogonal least-squares regression analysis.
Fisher’s exact test using two-sided P values was used to compare the incidence of adverse events in the pooled placebo group with that in each donepezil group. Results of haematology, clinical chemistry and routine urinalysis tests were compared with normal ranges, and shift tables were used to identify trends in the data.
Results
Subjects
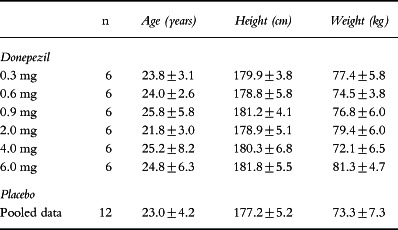
The demographic data for the study population are summarized in Table 1. The dose groups were well balanced with respect to age, height and weight. All of the subjects assigned to treatment with donepezil were Caucasian. The placebo-treated subjects included two black volunteers and two of other ethnic origin. A total of 48 subjects were enrolled in the study, and all 48 subjects completed the study without incident.
Table 1.
Demographic data (mean±SD).
Pharmacokinetics
Plasma concentrations of donepezil in the subjects receiving the 0.3, 0.6 and 0.9 mg doses were below the validated limit of HPLC quantification (2.0 ng ml−1). Pharmacokinetic evaluation was therefore confined to the 2.0, 4.0 and 6.0 mg dose groups.
Peak plasma concentrations of donepezil were achieved approximately 4 h after administration of all three doses (Table 2). Mean peak concentrations of donepezil increased proportionately with increasing dose. The AUC(0–∞) values also suggested dose proportionality (Table 2). Dose-normalized Cmax did not differ significantly across the range studied.
Table 2.
Derived pharmacokinetic parameters for orally administered donepezil (mean±SE).

The clearance of donepezil remained fairly constant at all three dose levels. The plasma concentration–time curves for the 4.0 and 6.0 mg dose levels showed characteristics of biphasic elimination, with the initial disposition phase being complete within the first 24 h after drug administration (Figure 1). The disposition of the 2.0 mg dose group could not be adequately characterized because the plasma concentrations of donepezil approached the assay quantification limit (2.0 ng ml−1) by 24 h (Figure 1). Consequently, the terminal half-life (t½) appeared to be shorter in subjects receiving the 2 mg dose (mean 53.8 h) than in those receiving the two higher doses (means 82.8 and 80.1 h, respectively). The same assay limitations are the most likely explanation for the observed differences in Vλz/F (Table 2).
Figure 1.
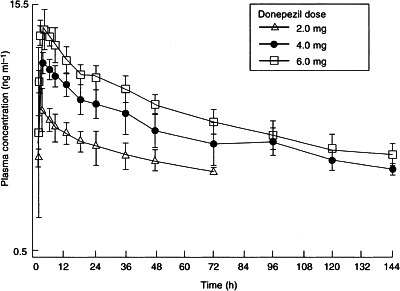
Mean (±SE) plasma concentration–time curves following single oral doses of 2.0, 4.0 and 6.0 mg donepezil to groups of six healthy male volunteers.
Pharmacodynamics
Seventeen of the 18 subjects in the 2.0, 4.0 and 6.0 mg dose groups showed some degree of rbc-AChE inhibition. A single subject in whom no measurable rbc-AChE inhibition was observed had aberrantly low baseline AChE activity.
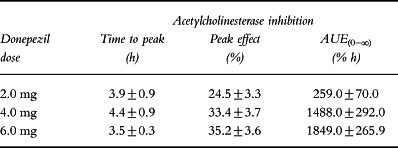
Peak rbc-AChE inhibition was observed within 4 h of administration of the 2.0, 4.0 and 6.0 mg doses of donepezil (Table 3) and coincided with the tmax in plasma. Lower doses of donepezil showed no evidence of pharmacodynamic activity. Mean maximal AChE inhibition (Emax; 33–35%) was comparable in the 4.0 and 6.0 mg dose groups, despite the fact that there was a dose-proportional increase in mean Cmax from 6.9 to 11.6 ng ml−1.
Table 3.
Pharmacodynamics of donepezil (mean±SD).
AChE inhibition lasted less than 24 h in the 2.0 mg dose group. However, the extent of rbc-AChE inhibition declined more gradually in the 4.0 mg and 6.0 mg dose groups, and, on average, remained above 10% for 72 h after donepezil administration (Figure 2). The rapid reversibility of donepezil-mediated inhibition of rbc-AChE is supported by the lack of hysteresis in the relationship between AChE inhibition and plasma concentration of donepezil (Figure 2).
Figure 2.
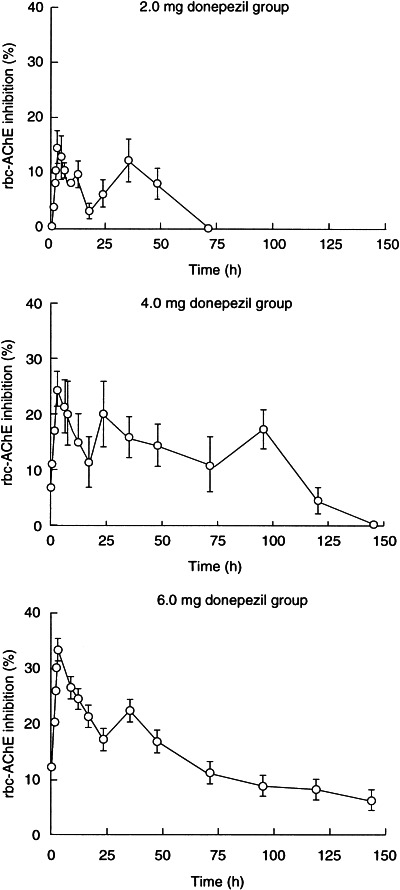
Duration of rbc-AChE inhibition (mean±SE) following administration of single oral doses of 2.0, 4.0 and 6.0 mg of donepezil to healthy male volunteers.
As shown in Figure 3, there was a linear relationship between the area under the effect–time profile (AUE(0–t)) and AUC(0–t). The AUE(0–t)/AUC(0–t) ratio was independent of dose, but at the 4.0 mg and 6.0 mg dose levels there was significant positive correlation between the extent of rbc-AChE inhibition and plasma donepezil concentration (P<0.005).
Figure 3.
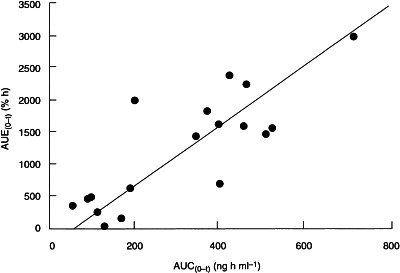
Scatter plot showing the relationship between the area under the plasma concentration–time curve (AUC(0–t)) and area under the rbc-AChE inhibition curve (AUE(0–t)) in individual subjects after single oral doses of 2.0, 4.0 and 6.0 mg of donepezil.
Safety
Donepezil was well tolerated by all subjects with no clinically significant changes in laboratory, ECG or vital sign parameters observed at any dose. Adverse events that were spontaneously reported or observed by the investigator included nausea, diarrhoea, insomnia, vomiting and fatigue. These were all mild in intensity and transient. The difference in the incidence of adverse events between donepezil- and placebo-treated subjects within each dose group was not statistically significant (P >0.05, Fisher’s exact test).
Discussion
The results of this study demonstrate that the pharmacokinetics of donepezil are linear over the 2–6 mg dose range, as shown by the relationship between dose, Cmax and AUC(0–∞). The results also indicate that donepezil is absorbed from the gastrointestinal tract at a moderate rate, with peak concentrations occurring approximately 4 h after drug administration.
Based on the data obtained at the 4.0 mg and 6.0 mg dose levels, it appears that donepezil has a biphasic disposition profile and that distribution equilibrium is achieved within 24 h of administration. Characterization of the disposition profile of the 2.0 mg dose level was incomplete due to assay limitations.
The terminal disposition of donepezil from plasma was relatively slow at the 4.0 and 6.0 mg dose levels, with an apparent half-life of approximately 80 h. The observed long half-life of donepezil is not indicative of irreversible enzyme inhibition, but suggests that the drug is released slowly from its target enzyme or from other binding sites.
Donepezil resulted in a relatively rapid and prolonged inhibition of rbc-AChE activity. The AUE for enzyme inhibition was directly dependent on the AUC of donepezil in plasma. Maximal rbc-AChE inhibition coincided with the Cmax in plasma (i.e. 4 h) and the duration of rbc-AChE inhibition coincided with the long half-life of donepezil. Measurable (>10%) enzyme inhibition persisted for up to 72-h post-dose.
In preclinical studies, donepezil demonstrated greater specificity for rat brain AChE than plasma butyrylcholinesterase (BuChE) (IC50 ratio BuChE/AChE: 1252) than either physostigmine (IC50 ratio: 11.9) or tacrine (IC50 ratio: 0.9) [11]. Furthermore, a study investigating the tissue distribution of cholinesterase inhibition in aged rats has shown that donepezil is specific for AChE in the brain [19].
In this study, the maximal degree of enzyme inhibition achieved after single 4.0 mg or 6.0 mg doses of donepezil was on the order of 35%. Ex vivo studies in patients with Alzheimer’s disease have shown that the maximal pharmacodynamic effect of donepezil (approximately 70% inhibition) occurs in a cumulative fashion over the first 2–3 weeks of long-term administration, consistent with the achievement of steady state in plasma after the same period [20].
Donepezil was well tolerated by the healthy men in this study and there were no dose-dependent or clinically significant changes in any of the safety variables. The incidence of adverse events was comparable with that in placebo-treated subjects and there was no evidence of hepatotoxicity as measured by standard liver function tests, suggesting that the specificity for AChE may indeed reduce the incidence of peripheral cholinergic side-effects. Indeed, in over 400 000 patient-days of drug exposure in the clinical development programme, other than transient gastrointestinal complaints, there have been no specific adverse events considered likely to be related to donepezil treatment.
These results confirm the findings of earlier studies performed in Japan [21, 22] and suggest that the dose-proportional pharmacokinetic and pharmacodynamic profile of donepezil seen in this study will provide the basis for a predictable and specific clinical response in patients with Alzheimer’s disease. Its long half-life, which allows once-daily dosing, and its apparent lack of hepatotoxicity at pharmacodynamically effective doses suggest that donepezil may be used safely and conveniently in the treatment of Alzheimer’s disease.
Acknowledgments
We acknowledge the efforts of Dr James Kisicki, Harris Laboratories Inc, 624 Peach Street, Box 80827, Lincoln, NE 68501, USA, who conducted this clinical trial, and Harris Laboratories Institutional Review Board, who reviewed and approved the study and protocol.
References
- 1.Whitehouse PJ, Price DL, Clarke AW, Coyle JT, DeLong MR. Alzheimer’s disease: evidence for selective loss of cholinergic neurons in the nucelus basalis. Ann Neurol. 1981;10:122–126. doi: 10.1002/ana.410100203. [DOI] [PubMed] [Google Scholar]
- 2.Giacobini E. Pharmacotherapy of Alzheimer’s disease: new drugs and novel strategies. Prog Brain Res. 1993;98:447–454. doi: 10.1016/s0079-6123(08)62431-0. [DOI] [PubMed] [Google Scholar]
- 3.Davis RE, Doyle PD, Carroll RT, Emmerling MR, Jaen J. Cholinergic therapies for Alzheimer’s disease. Arzneimittelforschung. 1995;45:425–431. [PubMed] [Google Scholar]
- 4.Davis P, Maloney AJ. Selective loss of central cholinergic neurons in Alzheimer’s disease. Lancet. 1976;ii:1403. doi: 10.1016/s0140-6736(76)91936-x. [DOI] [PubMed] [Google Scholar]
- 5.Dunnette SB, Low WC, Iversen SD, Stenevi U, Björklund A. Septal transplants restore maze learning in rats with fornix-fimbria lesions. Brain Res. 1982;251:335–348. doi: 10.1016/0006-8993(82)90751-x. [DOI] [PubMed] [Google Scholar]
- 6.Cardozo MG, Iimura Y, Sugimoto H, Yamanishi Y, Hopfinger AJ. QSAR analysis of the substituted indanone and benzylpiperidine rings of a series of indanone-benzylpiperidine inhibitors of acetylcholinesterase. J Med Chem. 1992;35:584–589. doi: 10.1021/jm00081a022. [DOI] [PubMed] [Google Scholar]
- 7.Cardozo MG, Kawai T, Iimura Y, Sugimoto H, Yamanishi Y, Hopfinger AJ. Conformational analysis and molecular shape comparisons of a series of indanone-benzylpiperidine inhibitors of acetylcholinesterase. J Med Chem. 1992;35:590–601. doi: 10.1021/jm00081a023. [DOI] [PubMed] [Google Scholar]
- 8.Sugimoto H, Iimura Y, Yamanishi Y, Yamatsu K. Synthesis and anti-acetylcholinesterase activity of 1-benzyl-4-[(5,6-dimethoxy-1-indanon-2-yl)methyl]piperidine hydrochloride (E2020) and related compounds. Biorg Med Chem Lett. 1992;2:871–876. doi: 10.1021/jm00024a009. [DOI] [PubMed] [Google Scholar]
- 9.Pang YP, Kozikowski AP. Prediction of the binding site of 1-benzyl-4-[(5,6-dimethoxy-1-indanon-2-yl)methyl]piperidine in acetylcholinesterase by docking studies with the SYSDOC program. J Comput Aided Mol Design. 1994;8:683–693. doi: 10.1007/BF00124015. [DOI] [PubMed] [Google Scholar]
- 10.Yamanishi Y, Ogura H, Kosasa T, Araki S, Sawa Y, Yamatsu K. Inhibitory action of donepezil, a novel acetylcholinesterase inhibitor, on cholinesterase. Comparison with other inhibitors. In: Nagatsu T, Fisher A, Yoshida M, editors. Basic, Clinical, and Therapeutic Aspects of Alzheimer’s and Parkinson’s Diseases. Vol. 2. New York: Plenum Press; 1990. pp. 409–413. [Google Scholar]
- 11.Rogers SL, Yamanishi Y, Yamatsu K. E2020—the pharmacology of a piperidine cholinesterase inhibitor. In: Becker R, Giacobini E, editors. Cholinergic Basis for Alzheimer Therapy. Boston: Birkhäuser; 1991. pp. 314–320. [Google Scholar]
- 12.Watkins PB, Zimmerman HJ, Knapp MJ, et al. Hepatotoxic effects of tacrine administration in patients with Alzheimer’s disease. JAMA. 1994;271:992–998. [PubMed] [Google Scholar]
- 13.Rogers SL, Friedhoff LT the Donepezil Study Group. The efficacy and safety of donepezil in patients with Alzheimer’s disease: results of a US multicenter, randomized, double-blind, placebo-controlled trial. Dementia. 1996;7:293–303. doi: 10.1159/000106895. [DOI] [PubMed] [Google Scholar]
- 14.Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT the Donepezil Study Group. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology. 1998;50:136–145. doi: 10.1212/wnl.50.1.136. [DOI] [PubMed] [Google Scholar]
- 15.Rogers SL, Friedhoff LT. Long-term efficacy and safety of donepezil in the treatment of Alzheimer’s disease: an interim analysis of the results of a US multicentre open-label extension study. Eur Neuropsychopharmacol. 1998;8:67–75. doi: 10.1016/s0924-977x(97)00079-5. [DOI] [PubMed] [Google Scholar]
- 16.Rogers SL, Doody RS, Mohs R, Friedhoff LT the Donepezil Study Group. Donepezil improves cognition and global function in Alzheimer’s disease: a 15-week, double-blind, placebo-controlled study. Arch Intern Med. 1998;158:1021–1031. doi: 10.1001/archinte.158.9.1021. [DOI] [PubMed] [Google Scholar]
- 17.Lee JW, Rogers SL, Friedhoff LT, Stiles MR, Cooper NM. Validation and application of an HPLC method for the determination of 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methyl piperidine HCl (E2020) in human plasma. Pharm Res. 1992;9:350. [Google Scholar]
- 18.Hulse JD, Rogers SL, Friedhoff LT, Sukovaty R, Pedersen JE, Lee JW. A radioenzyme assay of acetylcholinesterase activity in red blood cells and its correlation with 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methyl piperidine HC1 (E2020) Pharm Res. 1992;9:338. [Google Scholar]
- 19.Yamanishi Y, Ogura H, Kosasa T, et al. Inhibitory action of E2020, a novel acetylcholinesterase inhibitor, on cholinesterase in aged rats. Eur J Pharmacol. 1990;183:1935. [Google Scholar]
- 20.Rogers SL, Cooper NM, Sukovaty R, Pederson JE, Lee JN, Friedhoff LT. Pharmacokinetic and pharmacodynamic profile of donepezil HCl following multiple oral doses. Br J Clin Pharmacol. 1998;46(Suppl. 1):7–12. doi: 10.1046/j.1365-2125.1998.0460s1007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ohnishi A, Mihara M, Kamakura H, et al. Comparison of the pharmacokinetics of E2020, a new compound for Alzheimer’s disease, in healthy young and elderly subjects. J Clin Pharmacol. 1993;33:1086–1091. doi: 10.1002/j.1552-4604.1993.tb01945.x. [DOI] [PubMed] [Google Scholar]
- 22.Mihara M, Ohnishi A, Tomono Y, et al. Pharmacokinetics of E2020 a new compound for Alzheimer’s disease, in healthy male volunteers. Int J Clin Pharmacol Ther Toxicol. 1993;31:223–229. [PubMed] [Google Scholar]