

Abstract
Aim
The aim of this study was to evaluate the pharmacokinetics of donepezil HCl (5 mg) in patients with moderately to severely impaired renal function (creatinine clearance: <30 ml min−1 1.73 m−2), following the administration of single oral doses.
Methods
This was an open-label, non-randomized study in patients with compromised renal function (n = 11), and in age- and gender-matched healthy controls (n = 11). Each subject received a single oral dose of 5 mg donepezil. Blood samples for pharmacokinetic measurements were taken at specified intervals for 17 days post-dose. Concentrations of donepezil in plasma were measured by HPLC with UV detection.
Results
There were no statistical differences between the two groups for any of the pharmacokinetic parameters evaluated (ANOVA). Cmax (mean±SD) was 7.7±1.2 ng ml−1 in healthy subjects and 8.3±3.2 ng ml−1 in renally impaired patients. AUC(0–∞) in healthy subjects and in renally impaired patients was 539±115 ng h ml−1 and 640±150 ng h ml−1, respectively. The mean half-life of donepezil was 86.7±23.3 h in healthy subjects and 91.3±40.9 h in the renally impaired patients. The drug was well tolerated by all study participants. There were no clinically significant changes in vital signs, clinical chemistry or ECG parameters.
Conclusions
These findings suggest that the pharmacokinetics of donepezil do not change in patients with moderately to severely impaired renal function.
Keywords: donepezil, Alzheimer’s disease, renal impairment
Introduction
Donepezil HCl (also known as E2020 or Aricept®, the registered trademark of Eisai Co. Ltd, Tokyo, Japan) is a new piperidine-based inhibitor of the enzyme acetylcholinesterase (AChE) that has been approved for the treatment of Alzheimer’s disease [1, 2]. Pre-clinical studies have demonstrated that donepezil possesses a significantly greater degree of specificity for AChE in the central nervous system (CNS) than for butyrylcholinesterase (BuChE) in the periphery [3, 4]. Alzheimer’s disease is characterized by a decrease in levels of the neurotransmitter acetylcholine and neuronal cell death. Donepezil is presumed to produce its clinical effect by enhancing cholinergic function in surviving neurones. Clinical trial results have demonstrated that administration of donepezil (5 or 10 mg, once daily) significantly improves memory and cognitive function in patients with Alzheimer’s disease [5–7]. In addition, these studies have shown a direct relationship between clinical improvement, plasma concentration of the drug and AChE inhibition. Donepezil is well tolerated with the most frequent adverse events being mild, transient gastrointestinal complaints.
The pharmacokinetic profile of donepezil in healthy volunteers is characterized by slow clearance and a long half-life (approximately 70 h). The long plasma half-life is an important feature of the drug’s kinetic profile since it provides stable plasma drug concentrations with once-a-day dosing. Donepezil is metabolized primarily by the P-450 isoenzyme CYP-3A4, and to a lesser extent by 2D6. It is excreted renally both as intact drug as well as several metabolites, most of which have been identified [8]. As renal function declines with increasing age, and as Alzheimer’s disease primarily affects the elderly, this study was designed to examine the effect of impaired renal function on the pharmacokinetics of donepezil following the administration of single oral doses.
Methods
Subjects
This study included 11 patients with a documented history of stable renal impairment, and 11 age- and gender-matched healthy controls. Volunteers between 18 and 70 years of age were eligible. Both male and female subjects could enter the study. Women had been menopausal for at least 2 years or were surgically sterile. Pregnant or lactating women were excluded.
Renal impairment was defined as a creatinine clearance <30 ml min−1 l.73 m−2 body surface area, confirmed at the formal screening visit. Renal transplant patients and subjects requiring dialysis were excluded.
Eligible subjects in either group did not have clinically significant hepatic, gastrointestinal, neurological, endocrine, respiratory, haematological or cardiovascular disease. Subjects who had recently undergone major surgery or who had suffered a myocardial infarction or a cerebrovascular accident within the previous 3 months were excluded from entering the study, as were those with clinically significant biochemical and haematological abnormalities, other than those related to renal disease. Subjects with controlled hypertension or insulin-dependent diabetes could be enrolled.
None of the subjects had a known or suspected history of alcohol or drug abuse, and none were taking drugs or foods considered likely to induce (phenobarbitone, phenytoin, carbamazepine or rifampicin) or inhibit (cimetidine, ketoconazole or grapefruit juice) significantly hepatic drug-metabolizing enzymes.
The trial was conducted in accordance with Good Clinical Practice Guidelines, as issued by the European Commission (1990). The protocol was subjected to local ethics committee approval. All subjects gave written informed consent prior to participation in any study activities.
Protocol
This was an open-label, single-dose, two-centre study designed to assess the phamacokinetics of donepezil HCl in patients with stable, moderate–severe renal impairment. Healthy subjects who fulfilled the entry criteria were admitted to the study site on the evening prior to drug administration (day 0). A drink and a light snack were served after which the subjects fasted for at least 8 h prior to drug administration the following morning. Patients with impaired renal function were allowed to arrive at the study site on the morning of day 1, but had also fasted from food for at least 8 h prior to drug administration.
Donepezil was administered as a single 5 mg tablet with 250 ml of tap water. All subjects continued to fast for an additional 4 h after dosing at which time they received a standard meal. Clear fluids were allowed 2 h after dosing. All participants remained at the study site for at least 24 h after drug administration.
None of the patients or subjects had received any investigational drugs within at least 1 month of entering the study. Healthy subjects were not allowed to take any other medication, including non-prescription preparations, from 7 days before the start of the study until its completion. Patients with renal impairment maintained their existing medication and avoided changes in dosage. Food or drinks containing alcohol or caffeine were prohibited from 12 h prior to the first dose of donepezil until completion of the study.
Sample collection and analysis
Venous blood samples for the determination of plasma levels of donepezil were collected at 0, 1, 2, 2.5, 3, 3.5, 4, 6, 8, 12, 18, 24, 36, 48, 72, 96, 120, 144 and 168 h from all study participants. A 192-h sample was collected in 50% of the study participants, and 240-, 312- and 384-h samples were collected in the remainder. Donepezil in plasma was determined using high-performance liquid chromatography (HPLC) with UV detection [9]. The limit of quantification was 2.0 ng ml−1.
Pharmacokinetic assessments
Characterization of donepezil pharmacokinetics was conducted using plasma drug concentration data from the samples collected during the study. Data from all the volunteers were used in the analysis. Model-independent pharmacokinetic parameters were estimated for donepezil using WinNonLin software (version 1.1, Scientific Consulting Inc, Research Triangle Park, North Carolina, USA). The peak plasma concentration (Cmax) and the time of occurrence of Cmax (tmax) were direct experimental observations. The area under the plasma concentration–time curve from time 0 to time t (AUC(0–t)) was estimated using the linear trapezoidal approximation. The terminal disposition rate constant (λz) was estimated from the terminal phase by log-linear regression. Exact sampling times were used in the estimations. Terminal half-life (t½) was calculated as loge 2 divided by λz. Total area under the plasma concentration–time curve from time 0 to time infinity (AUC(0–∞)) was calculated by adding AUC(0–t) to the extrapolated area beyond the last plasma concentration (calculated as the ratio of the last measurable concentration to λz). Total apparent oral clearance (CL T/F) was calculated as dose divided by AUC(0–∞), and apparent volume of distribution (Vλz/F) was estimated as dose divided by the product of AUC(0–∞) and λz. Serial 24-h urine samples were collected on days 1–17 for the estimation of creatinine clearance.
Safety assessments
All adverse events spontaneously reported by the subjects and/or elicited or observed during physical examination or routine assessments were documented together with times of onset and cessation, and assessments of severity and causality.
Routine haematology, clinical chemistry and urinalysis tests were undertaken at screening, pre- and post-dose. Haematological assessments included haemoglobin, red blood cell count indices, platelet count, white blood cell count and differential. Clinical chemistry assessments included liver function (alanine transaminase, aspartate transaminase, alkaline phosphatase and total bilirubin), renal function (creatinine, blood urea nitrogen), metabolic status (glucose, total protein, albumin) and electrolytes (sodium, potassium, chloride, phosphorus, calcium). Routine urinalysis provided values for urine pH as well as glucose, protein, haemoglobin or blood and ketone levels. Twelve lead ECG monitoring was performed pre- and post-study. Blood pressure and heart rate were recorded according to the guidelines of the British Hypertension Society. Sitting and standing measurements were taken after the subjects had been in the appropriate posture for an adequate period (i.e. 5 min sitting and 2 min standing). Assessments were made pre-study, and at 24-h intervals thereafter.
Statistical analysis
The pharmacokinetic parameters Cmax, AUC(0–∞), t½,CLT/F and Vλz/F in patients with impaired renal function were compared with those in healthy control subjects by a one-way analysis of variance t-test, using the procedure GLM from SAS for Windows (version 6.11; SAS Institute Inc., Cary, North Carolina, USA). Analysis was undertaken on log-transformed data. The values of tmax in each group were compared by the Wilcoxon rank sums test using the NPAR1WAY procedure. Values of P≤0.05 were considered statistically significant.
Results
Subjects
Twenty-two volunteers were recruited into this study and all of them completed all phases of the study. The volunteers comprised 11 patients with stable renal impairment (defined as creatinine clearance <30 ml min−1 1.73 m−2), and 11 age- and gender-matched healthy controls. Volunteers ranged in age from 22 to 68 years, and in weight from 50 to 88 kg. The demographic data for the two subject groups are shown in Table 1.
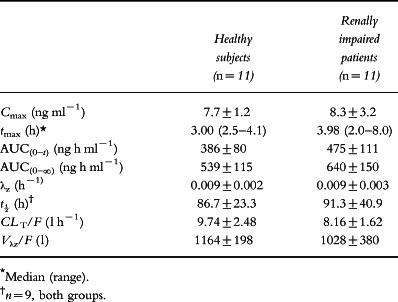
Table 1.
Summary of demographic data.

Mean creatinine clearance (±SD) was 88.0±13.9 ml min−1 1.73 m−2 in the healthy subjects and 18.0±7.2 ml min−1 1.73 m−2 in the patients with renal impairment. Pre-study urinalysis revealed protein levels of 0.3 g l−1 and 1.0 g l−1 in two of the renally impaired patients with trace amounts in two others. Blood was detected in the pre-study or day 1 urine samples of three of those subjects in whom protein was detected in urine as well as in one other. The mean pre-study urine albumin concentration for the renally impaired patients ranged from 38 to 46 g l−1 with a mean of 43 g l−1 compared with 35–50 g l−1 for the normal subjects. Concomitant medications in the patients with renal impairment consisted primarily of beta-blockers, calcium antagonists or diuretics to control co-existent hypertension.
Pharmacokinetics of donepezil
There were no statistical differences in Cmax, AUC(0–∞), t½, CL T/F or Vλz/F between the two subject groups as assessed using one-way analysis of variance (Table 2). In addition, no significant differences were noted when these parameters were regressed against the calculated creatinine clearances for each subject.
Table 2.
Pharmacokinetic parameters (mean±SD) following administration of a single oral dose of donepezil (5 mg) to healthy subjects and to patients with renal impairment.
Mean plasma concentration–time curves for both healthy subjects and subjects with renal impairment are shown in Figure 1. During the first 24 h, mean plasma donepezil concentrations were comparable between the two groups. Thereafter, plasma donepezil concentrations were slightly higher in the patients with renal impairment than in the healthy subjects. However, this difference was not statistically significant. As shown in Figure 1, the terminal phase was reached after approximately 48 h.
Figure 1.
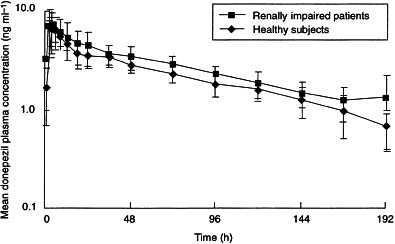
Mean (±SD) plasma concentration–time curves following the administration of single oral doses of donepezil (5 mg) to healthy subjects (n =11) and to patients with moderate–severe renal impairment (n =11).
Half-life data for four of the subjects (two healthy subjects and two renally impaired patients) were not used in the analysis since the values appeared inconsistent with the other pharmacokinetic parameters derived for these subjects. Despite the fact that half-life data were not subjected to formal statistical analysis, there was no evidence to suggest that renal impairment prolonged the half-life of donepezil in plasma (91.3±40.9 h) relative to that seen in healthy subjects (86.7±23.3 h).
The relationship between creatinine clearance and the AUC(0–∞) for donepezil in plasma is shown in Figure 2. The distribution of Cmax showed slightly more variability in the patients with renal impairment than in subjects with normal renal function (Figure 3). The highest Cmax in the healthy subjects was 10.0 ng ml−1, whereas two subjects with renal impairment had Cmax values of 12.0 ng ml−1 and 14.5 ng ml−1, respectively. These two marginally higher values were not considered to be clinically significant.
Figure 2.
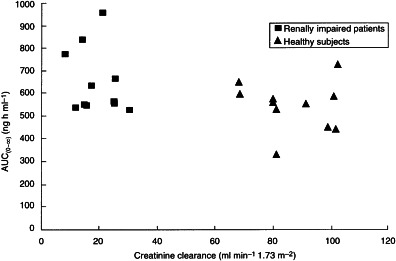
Relationship between creatinine clearance and the AUC(0–∞) of donepezil in plasma in healthy subjects and in patients with renal impairment.
Figure 3.
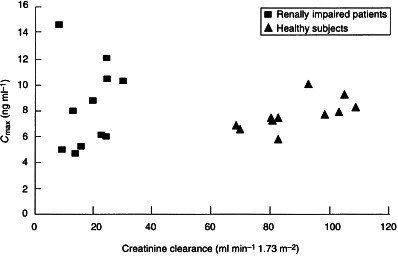
Relationship between creatinine clearance and the Cmax of donepezil in plasma in healthy subjects and in patients with renal impairment.
Safety
The drug was well tolerated by all participants and no serious adverse events were reported during this study. Adverse events that were reported were transient and mild in severity and included headache, nausea, diarrhoea, tiredness and dry eyes. There were no clinically significant changes in any clinical laboratory parameters, vital signs or ECG parameters in either study population.
Discussion
Donepezil HCl is metabolized primarily by the P-450 isoenzymes CYP-3A4 and CYP-2D6 and undergoes significant first-pass metabolism following oral administration [8]. Renal excretion of unchanged drug, glucoronidated metabolites and a hydrolysis product is the primary pathway by which donepezil is eliminated and accounts for about 80% of the administered dose. Faecal elimination of the hydroxylation products and of the N-oxidation product accounts for the remaining 20% of the administered dose. In view of the predominant role of renal excretion in the elimination of donepezil from the body, this study was designed to investigate whether renal impairment had any effect on its pharmacokinetic profile following single-dose administration.
The results of this study indicate that there is no difference in the pharmacokinetics of donepezil in patients with moderate–severe renal impairment and age- and sex-matched healthy controls. In addition, no significant differences were noted when these parameters were regressed against the calculated creatinine clearances for each subject. The pharmacokinetics of donepezil in the healthy controls were consistent with previous data [10]. The plots of AUC(0–∞) and Cmax against normalized creatinine clearance values also emphasize the stability of the pharmacokinetics of this drug in patients with renal impairment. This stability can be attributed to donepezil’s slow clearance (its half-life is approximately 70 h; [11]), which prevents it from becoming saturated. As a result, its pharmacokinetics are unchanged, even in patients with renal impairment. Moreover, donepezil was well tolerated by both groups. No clinically significant changes in clinical or laboratory parameters were observed during the study.
Other phase I studies reported in this supplement demonstrate that co-administration of donepezil with agents commonly prescribed in the elderly (digoxin, warfarin, theophylline and cimetidine) do not produce pharmacokinetic or pharmacodynamic interactions. The results presented here demonstrate that compromised renal function also has no effect on the pharmacokinetics of the drug. Although this was a single-dose study, the results suggest that donepezil pharmacokinetics will be unaffected during repeated dose administration in patients with Alzheimer’s disease.
Acknowledgments
We acknowledge the efforts of Drs P. E. Rolan and S. Toon of Medeval Ltd, University of Manchester, Skelton House, Manchester Science Park, Lloyd St North, Manchester M15 6SH, UK, who conducted this clinical trial. The following ethics committees/internal review boards reviewed and approved the study and protocol: Internal Ethics Committee at Medeval Ltd; South Manchester Medical Research Ethics Committee, Pavilion 10, Withington Hospital, West Didsbury, Manchester M20 8LR, UK; Manchester Health Commission, Research Ethics Committee (Central), Gateway House, Piccadilly South, Manchester M60 7LP, UK; Central Health Authority Ethics Committee, Darbishire House, 293a Upper Brook Street, Manchester M13 0FW, UK; South Sheffield Research Ethics Committee, The Royal Hallamshire Hospital, Glossop Road, Sheffield S10 2JF, UK.
References
- 1.Iimura Y, Mishima M, Sugimoto H. Synthesis of 1-benzyl-4-[(5,6-dimethoxy[2−14C]-1-indanon)-2-yl]-methylpiperidine hydrochloride (E2020−14C) J Label Compounds Radiopharm. 1989;XXVII:835–839. [Google Scholar]
- 2.Sugimoto H, Tsuchiya Y, Sugumi H, et al. Novel piperidine derivatives. Synthesis and anti-cholinesterase activity of 1-benzyl-4-[2-(N-benzoylamino)ethyl]piperidine derivatives. J Med Chem. 1990;33:1880–1887. doi: 10.1021/jm00169a008. [DOI] [PubMed] [Google Scholar]
- 3.Rogers SL, Yamanishi Y, Yamatsu K. E2020—the pharmacology of a piperidine cholinesterase inhibitor. In: Becker R, Giacobini E, editors. Cholinergic Basis for Alzheimer Therapy. Boston: Birkhäuser; 1991. pp. 314–320. [Google Scholar]
- 4.Sherman K. Pharmacodynamics of oral E2020 and tacrine in humans: novel approaches. In: Becker R, Giacobini E, editors. Cholinergic Basis for Alzheimer Therapy. Boston: Birkhäuser; 1991. pp. 321–328. [Google Scholar]
- 5.Rogers SL, Friedhoff LT the Donepezil Study Group. The efficacy and safety of donepezil in patients with Alzheimer’s disease: results of a US multicenter, randomized, double-blind, placebo-controlled trial. Dementia. 1996;7:293–303. doi: 10.1159/000106895. [DOI] [PubMed] [Google Scholar]
- 6.Rogers SL, Farlow MR, Doody RS, Mohs R, Friedhoff LT the Donepezil Study Group. A 24-week, double-blind, placebo-controlled trial of donepezil in patients with Alzheimer’s disease. Neurology. 1998;50:136–145. doi: 10.1212/wnl.50.1.136. [DOI] [PubMed] [Google Scholar]
- 7.Rogers SL, Doody RS, Mohs R, Friedhoff LT the Donepezil Study Group. Donepezil improves cognition and global function in Alzheimer’s disease: a 15-week, double-blind, placebo-controlled study. Arch Intern Med. 1998;158:1021–1031. doi: 10.1001/archinte.158.9.1021. [DOI] [PubMed] [Google Scholar]
- 8.Tiseo PJ, Perdomo CA, Friedhoff LT. Metabolism and elimination of 14C-donepezil in healthy volunteers: a single-dose study. Br J Clin Pharmacol. 1998;46(Suppl. 1):19–24. doi: 10.1046/j.1365-2125.1998.0460s1019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee JW, Rogers SL, Friedhoff LT, Stiles MR, Cooper NM. Validation and application of an HPLC method for the determination of 1-benzyl-4-[(5,6-dimethoxy-1-indanon)-2-yl] methyl piperidine HCl (E2020) in human plasma. Pharm Res. 1992;9:350. [Google Scholar]
- 10.Rogers SL, Cooper NM, Sukovaty R, Pederson JE, Lee JN, Friedhoff LT. Pharmacokinetic and pharmacodynamic profile of donepezil HCl following multiple oral doses. Br J Clin Pharmacol. 1998;46(Suppl. 1):7–12. doi: 10.1046/j.1365-2125.1998.0460s1007.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tiseo PJ, Rogers SL, Friedhoff LT. Pharmacokinetic and pharmacodynamic profile of donepezil HCl following evening administration. Br J Clin Pharmacol. 1998;46(Suppl. 1):13–18. doi: 10.1046/j.1365-2125.1998.0460s1013.x. [DOI] [PMC free article] [PubMed] [Google Scholar]