

Abstract
Aims
The effects of itraconazole on the pharmacokinetics of fluvastatin and lovastatin, two inhibitors of HMG-CoA reductase with different pharmacokinetic properties, were studied.
Methods
Two separate randomized, placebo-controlled, cross-over studies, each involving 10 healthy volunteers, were carried out. The general design was identical in both studies. The subjects took either 100 mg itraconazole or matched placebo orally once daily for 4 days. On day 4, 40 mg fluvastatin or 40 mg lovastatin was administered orally. Plasma concentrations of fluvastatin, lovastatin, lovastatin acid, itraconazole and hydroxyitraconazole were determined up to 24 h.
Results
Itraconazole had no significant effect on the Cmax (190±124 ng ml−1vs 197±189 ng ml−1 (mean±s.d.)) or total AUC (368±153 ng ml−1 h vs 324±155 ng ml−1 h) of fluvastatin compared with placebo. However, the t1/2,z of fluvastatin was slightly prolonged by itraconazole (2.8±0.49 h vs 2.4±0.51 h; P<0.05). The Cmax of lovastatin was increased about 15-fold (P<0.01) and the total AUC more than 15-fold (P<0.01) by itraconazole. Similarly, the Cmax and total AUC of lovastatin acid were increased about 12-fold (95% CI, 5.3 to 17.7-fold; P<0.01) and 15-fold (95% CI, 4.6 to 26.2-fold; P<0.01) by itraconazole, respectively. The t1/2,z of lovastatin averaged 3.7±3.8 h and that of lovastatin acid 4.7±4.0 h during the itraconazole phase; these variables could not be determined in all subjects during the placebo phase.
Conclusions
Itraconazole, even at a small dosage of 100 mg daily, greatly elevated plasma concentrations of lovastatin and its active metabolite, lovastatin acid. Lovastatin should therefore not be used concomitantly with itraconazole and other potent CYP3A4 inhibitors, or the dosage of lovastatin should be greatly reduced while using a CYP3A4 inhibitor. In contrast, fluvastatin concentrations were not significantly increased by itraconazole, indicating that fluvastatin has much less potential than lovastatin for clinically significant interactions with itraconazole and other CYP3A4 inhibitors.
Keywords: fluvastatin, interaction, itraconazole, lovastatin, pharmacokinetics
Introduction
Fluvastatin and lovastatin are inhibitors of 3-hydroxy-3-methylglutaryl-coenzyme A (HMG-CoA) reductase, the rate-limiting step in cholesterol synthesis, which are widely used in the treatment of hypercholesterolaemia. Lovastatin is an inactive lactone pro-drug which is hydrolysed in vivo to lovastatin acid, a competitive inhibitor of HMG-CoA reductase [1]. However, the oxidative metabolism of lovastatin is primarily mediated by CYP3A4 [2]. The pharmacokinetics of fluvastatin differ considerably from those of lovastatin; fluvastatin is not a pro-drug and it appears to be metabolized mainly by CYP2C9 [3, 4].
Concomitant use of lovastatin and, for example, cyclosporine, erythromycin or itraconazole is associated with a considerably increased risk of skeletal muscle toxicity, a rare but potentially serious side-effect of HMG-CoA reductase inhibitors [5–8]. The cause of these interactions was at first unclear, but a recent in vivo study strongly suggests that they result at least partly from inhibition of the CYP3A4-mediated metabolism of lovastatin [9]. The aim of the present study was to characterize the effect of itraconazole on the pharmacokinetics of fluvastatin and, in particular, to evaluate the hypothesis that fluvastatin is less liable to interact with CYP3A4 inhibitors such as itraconazole than lovastatin.
Methods
Subjects
Ten healthy volunteers participated in the fluvastatin study (five women and five men; age range, 20–25 years; weight range, 54–85 kg) and 10 in the lovastatin study (two women and eight men; age range, 19–24 years; weight range, 55–90 kg). All volunteers gave their written informed consent. They were determined to be healthy by a medical history, a physical examination and blood chemistry tests (including blood haemoglobin, serum creatine kinase, creatinine and alanine aminotransferase) before entering the study. None of them had continuous medications, with the exception of one and three females who were using oral contraceptive steroids in the lovastatin and fluvastatin studies, respectively.
Study design
Two separate randomized, placebo-controlled, cross-over studies with two phases, separated by a wash-out period of 3 weeks, were carried out. The general design was identical in both studies. The subjects were given either 100 mg itraconazole (Sporanox, Janssen, Beerse, Belgium) or matched placebo orally once daily at 08.00 h for 4 days. On day 4, 40 mg fluvastatin (one Canef 40 mg capsule, Astra Ltd, Kirkkonummi, Finland) or 40 mg lovastatin (one Mevacor 40 mg tablet, Merck Sharp & Dohme B.V., Haarlem, Netherlands) was administered orally with 150 ml water at 09.00 h, i.e. 1 h after the last dose of itraconazole. The subjects fasted for 1 h before administration of fluvastatin or lovastatin. A warm standard meal was served 4 h and a light standard meal 8 h after fluvastatin or lovastatin intake. The subjects were not allowed to drink grapefruit juice or alcohol during the study days and the previous 24 h. The study protocol was approved by the Ethics Committee of the Department of Clinical Pharmacology, University of Helsinki, and the Finnish National Agency for Medicines.
Blood sampling and determination of plasma drug concentrations
On day 4, a forearm vein was cannulated and timed blood samples were drawn just before fluvastatin or lovastatin was administered and 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h later. The blood samples (10 ml each) were taken into tubes that contained ethylenediaminetetra-acetic acid (EDTA). Plasma was stored at −40° C until analysis. Plasma concentrations of lovastatin and lovastatin acid were determined by high performance liquid chromatography (h.p.l.c.), as previously described [10]. Simvastatin was used as an internal standard. The limit of quantification was 2.5 ng ml−1 for both compounds. The within-day coefficient of variation (CV) was 4.1% (mean, 9.9 ng ml−1, n=10) for lovastatin and 7.8% (mean, 10.1 ng ml−1, n=10) for lovastatin acid. The between-day CVs were 6.0% (mean, 10.1 ng ml−1, n=4) and 2.7% (mean, 48.1 ng ml−1, n=5) for lovastatin and 8.3% (mean, 9.4 ng ml−1, n=4) and 3.0% (mean, 50.6 ng ml−1, n=5) for lovastatin acid.
Fluvastatin concentrations were determined by h.p.l.c. using automated solid phase extraction and fluorescence detection [11]. The limit of quantification for fluvastatin was 0.4 ng ml−1. The within-day CV for fluvastatin was 4.3% at 0.49 ng ml−1 (n=6) and 2.4% at 43.6 ng ml−1 (n=8). The between-day CV was 2.5% (mean, 21.3 ng ml−1, n=8).
To assess the comparability of plasma itraconazole and hydroxyitraconazole levels between the lovastatin and fluvastatin studies, plasma itraconazole and hydroxyitraconazole concentrations were determined in all samples, i.e. from 1 h after the last itraconazole dose on day 4 up to 25 h. Itraconazole and hydroxyitraconazole concentrations were determined by h.p.l.c. as previously described [12]. The limit of quantification was 10 ng ml−1 for both compounds. The within-day CV was <10% for both compounds at relevant concentrations. The between-day CV was 2.1% (mean, 196 ng ml−1, n=10) for itraconazole and 3.7% (mean, 198 ng ml−1, n=10) for hydroxyitraconazole. At a mean concentration of about 20 ng ml−1, the between-day CV was <10% for both compounds (n=10).
Pharmacokinetics
The pharmacokinetics of fluvastatin, lovastatin and lovastatin acid were characterized, as appropriate, by peak concentration in plasma (Cmax), time to Cmax (tmax), area under the plasma concentration-time curve (AUC) and elimination half-life (t1/2,z). The terminal log-linear phase of the plasma concentration-time curve was identified visually for each subject. The elimination rate constant (λz) was determined by a linear regression analysis of the log-linear part of the plasma concentration-time curve. The t1/2,z was calculated from the equation: t1/2,z=ln 2/λz. The AUC values were calculated by the trapezoidal rule, with extrapolation to infinity by dividing the last measured concentration by λz. The λz for lovastatin and lovastatin acid could not be accurately determined in all subjects during the placebo phase due to low plasma drug concentrations. For itraconazole and hydroxyitraconazole, the AUC (1,25 h) on day 4 was determined by the trapezoidal rule.
Statistical analysis
Results are expressed as mean values±s.d. or, in case of tmax, as median with range. 95% confidence intervals (CI) were calculated for selected variables. The pharmacokinetic variables between the two pretreatments (itraconazole and placebo) were compared with a paired t-test (two-tailed). The Wilcoxon test was used for analysis of tmax. The data were analysed with the statistical program Systat for Windows, version 5.0 (Systat, Evanston, USA). The level of statistical significance was P<0.05.
Results
Fluvastatin
Pretreatment with itraconazole had no significant effect on the Cmax (190±124 ng ml−1vs 197±189 ng ml−1) or total AUC (368±153 ng ml−1 h vs 324±155 ng ml−1 h) of fluvastatin (Table 1, Figure 1). The total AUC of fluvastatin during the itraconazole phase relative to that during the placebo phase averaged 1.27 (95% CI, 0.89–1.65). The t1/2,z of fluvastatin was slightly prolonged by itraconazole (2.8±0.5 h vs 2.4±0.5 h; P<0.05).
Table 1.
The pharmacokinetic variables of fluvastatin 40 mg (mean±s.d. or median and range) in 10 subjects (study I) and those of lovastatin 40 mg in 10 subjects (study II), following pretreatment with placebo or 100 mg itraconazole once daily for 4 days.
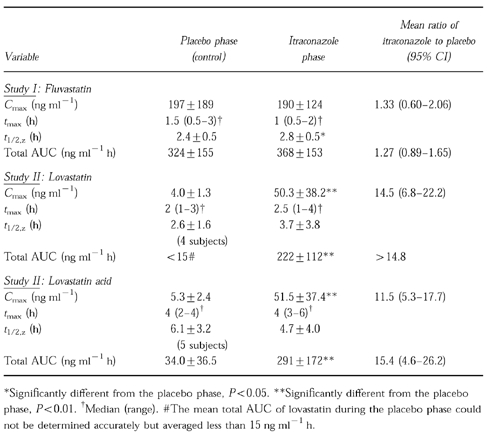
Figure 1.

Plasma concentrations of fluvastatin in 10 healthy volunteers (mean±s.e. mean) after a 40 mg oral dose, following pretreatment with 100 mg itraconazole (closed circles) or placebo (open circles) once daily for 4 days.
Lovastatin and lovastatin acid
Itraconazole considerably increased the plasma concentrations of both lovastatin and lovastatin acid, compared with placebo (Table 1, Figure 2). The Cmax of lovastatin was increased about 15-fold (P<0.01) and the total AUC more than 15-fold (P<0.01) by itraconazole. The t1/2,z of lovastatin averaged 3.7±3.8 h during the itraconazole phase, but during the placebo phase it could be determined in only four subjects (mean, 2.6 h) due to the low plasma lovastatin concentrations.
Figure 2.

Plasma concentrations of lovastatin (a) and lovastatin acid (b) in 10 healthy volunteers (mean±s.e. mean) after a 40 mg oral dose of lovastatin, following pretreatment with 100 mg itraconazole (closed circles) or placebo (open circles) once daily for 4 days.
The Cmax and total AUC of lovastatin acid were increased about 12-fold (95% CI, 5.3-fold to 17.7-fold; P<0.01) and 15-fold (95% CI, 4.6-fold to 26.2-fold; P<0.01) by itraconazole, respectively. The t1/2,z of lovastatin acid was 4.7±4.0 h during the itraconazole phase; in the placebo phase, it could be determined in only five subjects, averaging 6.1 h.
Itraconazole and hydroxyitraconazole
The mean plasma concentrations of itraconazole and its active metabolite hydroxyitraconazole on the day of fluvastatin or lovastatin administration are shown in Figure 3. The AUC (1,25 h) of itraconazole was 1921±849 ng ml−1 h in the fluvastatin study and 2018±966 ng ml−1 h in the lovastatin study and that of hydroxyitraconazole was 5497±2083 and 5557±2335 ng ml−1 h, respectively. There was no significant difference in the AUC (1,25 h) of itraconazole or hydroxyitraconazole between the two studies.
Figure 3.

Plasma concentrations of itraconazole (open circles, fluvastatin study; closed circles, lovastatin study) and hydroxyitraconazole (open triangles, fluvastatin study; closed triangles, lovastatin study) after intake of 100 mg itraconazole orally once daily for 4 days. Each curve represents mean±s.e. mean in 10 healthy volunteers. The time zero (0) refers to the administration of fluvastatin or lovastatin (i.e. 1 h after the last dose of itraconazole).
Discussion
The results of this study demonstrate that plasma concentrations of the pro-drug lovastatin and its active metabolite, lovastatin acid, are greatly increased by a daily dose of 100 mg itraconazole. By contrast, itraconazole had no significant effect on plasma fluvastatin concentrations. However, the half-life of fluvastatin was slightly prolonged by itraconazole, but this should not have any clinical relevance. The increase in the total AUC of lovastatin acid by itraconazole was, on an average, 15-fold, but showed marked interindividual variation. Itraconazole appeared to have a similar effect on the total AUC of lovastatin; however, this interaction could not be accurately quantified, since the lovastatin concentrations during the placebo phase were often below the limit of quantification.
These results are in good agreement with the knowledge of the biotransformation of lovastatin and fluvastatin. The pro-drug lovastatin is partly hydrolysed to the active metabolite, lovastatin acid; this metabolic pathway is not dependent on the CYP enzymes. In contrast, formation of the other major metabolites of lovastatin is catalysed by CYP enzymes, primarily by CYP3A4 [2]. Thus, although the hydrolysis of lovastatin to lovastatin acid is not dependent on CYP3A4, inhibition of CYP3A4 by itraconazole results in greatly elevated plasma lovastatin concentrations, leading to a corresponding increase in lovastatin acid levels. Unlike lovastatin, fluvastatin seems to be metabolized primarily by CYP2C9; CYP3A4 does not play a significant role in fluvastatin biotransformation [3, 4].
Lovastatin has a low oral bioavailability due to incomplete absorption and extensive first-pass metabolism [13]. Considering that the Cmax and AUC of lovastatin and lovastatin acid were greatly increased by itraconazole, with only a minor effect on their t1/2,z, as well as the fact that significant quantities of CYP3A4 are present in the gut wall and liver, it is likely that the interaction between lovastatin and itraconazole results mainly from inhibition of the CYP3A4-mediated first-pass metabolism of lovastatin.
In a previous study, a 4-day pretreatment with itraconazole, 200 mg daily, increased the AUC of lovastatin and lovastatin acid about 20-fold [9]. One aim of the present study was to evaluate the effect of a lower itraconazole dosage, 100 mg daily, on lovastatin pharmacokinetics. Although the lower itraconazole dosage appeared to have a slightly smaller effect on the plasma levels of lovastatin and lovastatin acid, it also is associated with a clinically significant risk of interaction with lovastatin.
Considering the marked effect of itraconazole 100 mg daily on plasma lovastatin and lovastatin acid concentrations as well as its lack of effect on plasma fluvastatin, it is unlikely that a clinically significant interaction would occur between fluvastatin and higher dosages of itraconazole. In a recent study, AUC values for fluvastatin were about 2-fold higher in hypercholesterolaemic renal transplant patients receiving fluvastatin 20 mg day−1 concomitantly with cyclosporine than in hypercholesterolaemic patients who had not undergone renal transplantation and were not receiving cyclosporine [14]. However, this difference in the AUC of fluvastatin observed between the two groups may be related more to underlying diseases, such as decreased renal function, in the transplant patients than to cyclosporine therapy. In any event, it should be noted that apart from inhibiting CYP3A4, cyclosporine and itraconazole may increase plasma concentrations of HMG-CoA reductase inhibitors and other drugs by inhibiting P-glycoprotein mediated drug elimination [15, 16].
Concomitant administration of the CYP3A4 inhibitors cyclosporine and erythromycin has been shown to increase the incidence and severity of the skeletal muscle toxicity of lovastatin [5, 6, 8]. In addition, a severe case of rhabdomyolysis occurring in a patient receiving both itraconazole (200 mg day−1) and lovastatin has been reported [7]. These interactions probably result from the greatly elevated blood and tissue concentrations of lovastatin acid; normally, monotherapy with lovastatin is well tolerated due to the low levels of lovastatin acid.
The biotransformation of the pro-drug simvastatin is similar to that of lovastatin [13] and it might therefore be expected that simvastatin would also interact with itraconazole and other potent CYP3A4 inhibitors to a clinically significant degree. Indeed, several recent case reports indicate that concomitant therapy with simvastatin and itraconazole, cyclosporine or other CYP3A4 inhibitors predisposes to myositis and rhabdomyolysis [17–20]. These case-reports conform well with our recent data which indicate that simvastatin acid concentrations are greatly increased by itraconazole [21].
In conclusion, the results of this study indicate that itraconazole, even at a dosage of 100 mg daily, greatly elevates plasma concentrations of lovastatin and its active metabolite, lovastatin acid. Lovastatin (and simvastatin) should not be used concomitantly with potent CYP3A4 inhibitors, or the dosage of these statins should be greatly reduced while using CYP3A4 inhibitors. By contrast, fluvastatin concentrations were not significantly increased by itraconazole, indicating that fluvastatin has much less potential than lovastatin for clinically significant interactions with itraconazole and other CYP3A4 inhibitors.
Acknowledgments
The skilful technical assistance of Mr Jouko Laitila, Mrs Kerttu Mårtensson, Mrs Lisbet Partanen and Mrs Eija Mäkinen-Pulli is greatly appreciated. Plasma fluvastatin concentrations were kindly determined by Astra Hässle Ltd. This study was supported by a grant from the Helsinki University Central Hospital Research Fund.
References
- 1.Duggan DE, Chen IW, Bayne WF, et al. The physiological disposition of lovastatin. Drug Metab Dispos. 1989;17:166–173. [PubMed] [Google Scholar]
- 2.Wang RW, Kari PH, Lu AYH, Thomas PE, Guengerich FP, Vyas KP. Biotransformation of lovastatin. IV. Identification of cytochrome P450, 3A proteins as the major enzymes responsible for the oxidative metabolism of lovastatin in rat and human liver microsomes. Arch Biochem Biophys. 1991;290:355–361. doi: 10.1016/0003-9861(91)90551-s. [DOI] [PubMed] [Google Scholar]
- 3.Transon C, Leemann T, Dayer P. Selective in vitro P450 inhibition profile by fluvastatin indicates its potential in vivo drug interactions. Clin Pharmacol Ther. 1993;53:188. [Google Scholar]
- 4.Transon C, Leemann T, Vogt N, Dayer P. In vivo inhibition profile of cytochrome P450TB (CYP2C9) by (±) -fluvastatin. Clin Pharmacol Ther. 1995;58:412–417. doi: 10.1016/0009-9236(95)90054-3. [DOI] [PubMed] [Google Scholar]
- 5.Norman DJ, Illingworth DR, Munson J, Hosenpud J. Myolysis and acute renal failure in a heart-transplant patient receiving lovastatin. N Engl J Med. 1988;318:46–47. doi: 10.1056/NEJM198801073180110. [DOI] [PubMed] [Google Scholar]
- 6.Tobert JA. Efficacy and long-term adverse effect pattern of lovastatin. Am J Cardiol. 1988;62:28–34. doi: 10.1016/0002-9149(88)90004-5. [DOI] [PubMed] [Google Scholar]
- 7.Lees RS, Lees AM. Rhabdomyolysis from the coadministration of lovastatin and the antifungal agent itraconazole. N Engl J Med. 1995;333:664–665. doi: 10.1056/NEJM199509073331015. [DOI] [PubMed] [Google Scholar]
- 8.Marinella MA. More on lovastatin. West J Med. 1995;162:176–177. [PMC free article] [PubMed] [Google Scholar]
- 9.Neuvonen PJ, Jalava K-M. Itraconazole drastically increases plasma concentrations of lovastatin and lovastatin acid. Clin Pharmacol Ther. 1996;60:54–61. doi: 10.1016/S0009-9236(96)90167-8. [DOI] [PubMed] [Google Scholar]
- 10.Stubbs RJ, Schwartz M, Bayne WF. Determination of mevinolin and mevinolinic acid in plasma and bile by reversed-phase high-performance liquid chromatography. J Chromatogr. 1986;383:438–443. doi: 10.1016/s0378-4347(00)83492-1. [DOI] [PubMed] [Google Scholar]
- 11.Toreson H, Eriksson B-M. Liquid chromatographic determination of fluvastatin and its enantiomers in blood plasma by automated solid-phase extraction. Chromatographia. 1997;45:29–34. [Google Scholar]
- 12.Allenmark S, Edebo A, Lindgren K. Determination of itraconazole in serum with high-performance liquid chromatography and fluorescence detection. J Chromatogr. 1990;532:203–206. doi: 10.1016/s0378-4347(00)83770-6. [DOI] [PubMed] [Google Scholar]
- 13.Lennernäs H, Fager G. Pharmacodynamics and pharmacokinetics of the HMG-CoA reductase inhibitors. Clin Pharmacokinet. 1997;32:403–425. doi: 10.2165/00003088-199732050-00005. [DOI] [PubMed] [Google Scholar]
- 14.Goldberg R, Roth D. Evaluation of fluvastatin in the treatment of hypercholesterolemia in renal transplant recipients taking cyclosporine. Transplantation. 1996;62:1559–1564. doi: 10.1097/00007890-199612150-00005. [DOI] [PubMed] [Google Scholar]
- 15.Kivistö KT, Kroemer HK, Eichelbaum M. The role of human cytochrome P450 enzymes in the metabolism of anticancer agents: implications for drug interactions. Br J Clin Pharmacol. 1995;40:523–530. doi: 10.1111/j.1365-2125.1995.tb05796.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bertz RJ, Granneman GR. Use of in vitro and in vivo data to estimate the likelihood of metabolic pharmacokinetic interactions. Clin Pharmacokinet. 1997;32:210–258. doi: 10.2165/00003088-199732030-00004. [DOI] [PubMed] [Google Scholar]
- 17.Meier C, Stey C, Brack T, Maggiorini M, Risti B, Krahenbuhl S. Rhabdomyolysis in patients treated with simvastatin and cyclosporin: role of the hepatic cytochrome P450 enzyme system activity. Schweiz Med Wochenschr. 1995;125:1342–1346. [PubMed] [Google Scholar]
- 18.Horn M. Coadministration of itraconazole with hypolipidemic agents may induce rhabdomyolysis in healthy individuals. Arch Dermatol. 1996;132:1254. doi: 10.1001/archderm.1996.03890340120028. [DOI] [PubMed] [Google Scholar]
- 19.Segaert MF, De Soete C, Vandewiele I, Verbanck J. Drug-interaction-induced rhabdomyolysis. Nephrol Dial Transplant. 1996;11:1846–1847. [PubMed] [Google Scholar]
- 20.Jacobson RH, Wang P, Glueck CJ. Myositis and rhabdomyolysis associated with concurrent use of simvastatin and nefazodone. JAMA. 1997;277:296–297. doi: 10.1001/jama.277.4.296. [DOI] [PubMed] [Google Scholar]
- 21.Neuvonen PJ, Kantola T, Kivistö KT. Simvastatin but not pravastatin is very susceptible to interaction with the CYP3A4 inhibitor itraconazole. Clin Pharmacol Ther. 1998;63:332–341. doi: 10.1016/S0009-9236(98)90165-5. [DOI] [PubMed] [Google Scholar]