

Abstract
Aims
To assess the tolerability and pharmacokinetic profile of single and repeat doses of the oral matrix metalloproteinase inhibitor marimastat in healthy male volunteers.
Methods
A total of 31 subjects participated in two placebo-controlled, rising-dose studies. The first study assessed the pharmacokinetics and tolerability of single doses of marimastat of 25, 50, 100, 200, 400 and 800 mg. In the second study, continuous dosing over 6.5 days with three incremental dose levels of 50, 100 and 200 mg twice daily was assessed. Full pharmacokinetic profiles were obtained on days 0 and 6, and trough concentrations were measured on all days. For each pharmacokinetic profile in the studies, summary measures including Cmax, tmax, elimination half-life and AUC were calculated. Urinary drug weights were also measured. All adverse events were documented, and haematological and biochemical variables, vital signs and ECGs were monitored throughout the study.
Results
Peak plasma concentrations were observed at 1.5–3 h for all subjects at all doses. Peak levels were approximately proportional to dose, as was drug exposure as calculated by AUC. Data from both studies indicate that the terminal elimination half-life is of the order of 8–10 h, and that there is no unexpected drug accumulation. Marimastat was well-tolerated, with adverse effects being mild and occurring with similar frequency to placebo. Small but reversible elevations in liver transaminases were noted with repeat dosing of marimastat, the most significant of these occurring at a dose of 200 mg twice daily.
Conclusion
Single and repeat oral doses of marimastat in healthy male subjects appear to be well-tolerated. The drug is rapidly absorbed with high peak levels achieved. It has a terminal elimination half-life of 8–10 h which would support twice daily dosing in further clinical trials.
Keywords: marimastat, metalloproteinase inhibitors, safety, pharmacokinetics, absorption, half-life
Introduction
Marimastat (BB-2516) is an inhibitor of the family of enzymes known as matrix metalloproteinases (MMPs). These enzymes are considered to be primarily responsible for the degradation of extracellular matrix proteins in processes of tissue formation and remodelling [1]. Under normal conditions the activity of MMPs is controlled at several levels, including their secretion as latent proenzymes and inhibition by endogenous tissue inhibitor of metalloproteinases (TIMPs) [2]. However, excessive MMP activity is now thought to play an important role in the pathogenesis of several diseases including cancer [3], rheumatoid arthritis [4], osteoarthritis [5], inflammatory bowel disease [6], neurodegenerative diseases [7], and cerebral haemorrhage [8]. It is thought that MMP inhibitors may have utility in the treatment of some of these diseases.
The first two MMP inhibitors to be tested in patients were ilomastat (GM6001) and batimastat (BB-94). Neither compound showed good oral bioavailability and indications were sought that allowed alternative routes of administration. Ilomastat was administered as a topical agent in patients with corneal ulceration [9] while batimastat was given as an intraperitoneal or intrapleural suspension in patients with malignant effusions [10]. More recently, marimastat has been identified as one of the first MMP inhibitors to show good absorption following oral administration to animals [11]. It is a broad spectrum reversible inhibitor of MMPs exhibiting IC50s of 5 nm, 6 nm, 3 nm, 16 nm, 230 nm and 5 nm against interstitial collagenase (MMP-1), gelatinase A (MMP-2), gelatinase B (MMP-9), matrilysin (MMP-7), stromelysin-1 (MMP-3) and metalloelastase (MMP-12), respectively.
Safety, pharmacology and toxicology studies suggest that marimastat has low oral and intravenous toxicity in animals. The only chronic target organ toxicity elicited has been inflammation of the tendons and joint ligaments of marmosets. On the basis of these preclinical data, it was thought that marimastat was a suitable compound to introduce into man with a view to further investigation of its role as an anti-cancer agent. The objectives of these first two studies were to assess the tolerability and pharmacokinetic profiles of single and continuous dosing of oral marimastat in healthy male volunteers.
Methods
Subjects
This report reviews the results of two double-blind, placebo-controlled studies of 13 and 18 healthy male subjects, respectively. Both studies were conducted by MDS Harris, Belfast, Northern Ireland, and the final study protocols were approved by the research ethics committee at Queen's University, Belfast, before study commencement. The studies were performed in accordance with the current version of the Declaration of Helsinki and with European guidelines on good clinical practice. Volunteers were considered to be healthy on the basis of medical history, a normal physical examination, and results of clinical laboratory and electrocardiographic (ECG) testing. Subject entry was dependent on the provision of witnessed, written and informed consent.
Study design
The first study investigated six single oral rising doses of marimastat over 6 weeks. There were two groups of subjects; group A dosed on weeks 1,3 and 5 receiving 25, 100 and 400 mg of marimastat, and group B dosed on weeks 2, 4 and 6 receiving 50, 200 and 800 mg marimastat. The second study investigated three oral incremental dose levels of marimastat given for 6.5 days. There were three groups of six subjects; group I received marimastat 50 mg twice daily (week 1), group II received marimastat 100 mg twice daily (week 3), and group III received marimastat 200 mg twice daily (week 5). In both studies, progression to the next dose level could only take place if adequate safety had been demonstrated at the preceding dose level. At each dose level, four of six subjects received marimastat and two received placebo in a double-blind, randomized fashion.
Assay methodology
Pharmacokinetic analyses were made on sequential plasma and urine samples. Venous blood was collected into lithium heparin tubes, put on ice and centrifuged within 30 min of collection. The supernatant plasma was collected and frozen. Frozen plasma and 10 ml urine samples were sent to British Biotech, where all analytical work was conducted in accordance with the European guidelines on good laboratory practice (GLP).
Concentrations of marimastat were determined in human plasma samples using a liquid chromatography-mass spectrometric (LC-MS) assay method. Calibration standards and quality control samples in the nominal concentration range 5 to 1000 μg l−1 were prepared by spiking control human plasma with marimastat and stored at −20° C until required.
The extraction procedure involved addition of an internal standard, BB-1090, (a close structural analogue of marimastat) and extraction of the analytes into ethyl acetate at pH 6.6 followed by back extraction into 0.1 m ammonia solution. The ethyl acetate layer was discarded and the ammonia fraction neutralized by the addition of 0.15 m hydrochloric acid prior to injection onto an LC-MS system.
The chromatographic system comprised a C18 reversed phase h.p.l.c. column and a mobile phase of methanol/water (55/45/v/v). The mass spectrometer used was a Finnigan SSQ 710C instrument fitted with an atmospheric pressure chemical ionisation (APCI) interface. Marimastat and the internal standard were detected by selected ion monitoring (SIM).
The analytical procedure was validated using published guidelines [12]. The validation package comprised analytical runs designed to examine assay variation, accuracy, precision and stability of marimastat and demonstrated that the assay performed acceptably during validation.
During validation, inter-batch accuracy was between 89% and 105%. Inter-batch precision (c.v. %) was between 9.4% and 11.9%. The assay was specific with no endogenous interferences present in any of the control plasma screened and the limit of detection of the assay was 3 μg l−1.
Assessments
In the single dose study, plasma drug concentrations were profiled for each patient on each study day. Samples were taken at 15 (except for 400 mg), 30, and 45 min, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 15, 24, 36 (400 and 800 mg only), 48, and 60 (800 mg only) h, and 3 (800 mg only) and 4 days after dosing. In the repeat dose study, full pharmacokinetic profiles were obtained on days 0 and 6, and trough (pre-dose) concentrations were measured from days 0 to 6. Thus samples were taken at 0.5, 1, 1.5, 2, 3, 4, 5, 6, 7, 8, 10, 12, 12.5, 13.5, 14, 15, 24, 48, 72, 96, 120, 144, 144.5, 145, 145.5, 146, 147, 148, 149, 150, 151, 152, 154, 156, 159, 168, 180, 204, 216, and 240 h. For each pharmacokinetic profile, appropriate summary measures, including Cmax, tmax, elimination half-life, AUC (calculated by the linear trapezoidal rule) were calculated. Urinary drug weights were measured and summary measures calculated.
All adverse events were documented in the case report forms. Events were elicited voluntarily, and by open questioning. The severity of the adverse event (mild, moderate, severe), dates and time of onset/offset, relationship to treatment (unrelated, possible, probable, definite) and whether or not the drug was withdrawn were recorded. Haematology and plasma and urine biochemistry were monitored as determined in the study protocol, as were vital signs (pulse, blood pressure, temperature and respiratory rate) and 12-lead ECGs. All safety data were clinically reviewed and tabulated.
Results
Subjects
A total of 31 subjects were recruited into the two studies, 13 into the single rising dose study, and 18 into the repeat dose study. Subject 3 in the first study received one treatment (25 mg) only after which he failed to attend. He was replaced by subject 13 who received the second and third scheduled treatments for subject 3. All subjects in the second study completed the dosing schedule. The 31 subjects were white males and ranged in age from 18 to 47 years.
In both studies, each dose of marimastat was received by four subjects, and at each dose level two subjects received placebo. One of four patients randomized to receive 200 mg in the single dose study only received 150 mg. The results from this subject are included with those from subjects who actually received 200 mg, with the exception of the calculation of pharmacokinetic parameters. All assessments were completed for all subjects.
Pharmacokinetics
Mean plasma concentrations of marimastat for the single rising dose study and the repeat dose study are shown in Figures 1 and 2, respectively. Pharmacokinetic parameters for the two studies are summarized in Tables 1 and 2.
Figure 1.
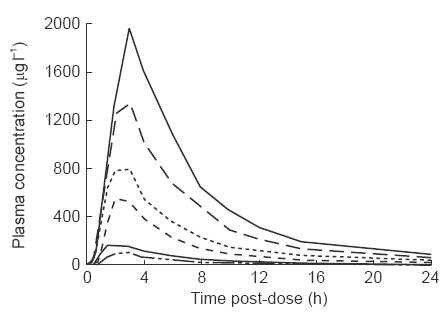
Mean marimastat plasma concentration-time profiles following single oral administration of 25, 50, 100, 200, 400 or 800 mg of marimastat to male volunteers. For clarity, error bars have not been included. Standard errors for Cmaxare representative; 16.4, 47.8, 44.7, 118.6, 149.3, and 164.7 for the 25 mg, 50 mg, 100 mg, 200 mg, 400 mg and 800 mg doses respectively.
Figure 2.
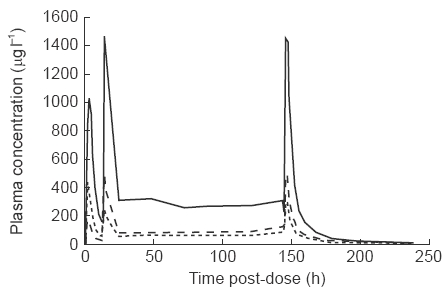
Mean marimastat plasma concentration-time profiles following repeat oral administration of 50, 100 and 200 mg of marimastat to male volunteers. For clarity, error bars have not been included. Standard errors for Cmax and Cminare representative; 97.5, 67.9 and 339.2 for Cmaxat last dose, 16.1, 11.7 and 42.4 for Cminat last dose, for 50 mg, 100 mg and 200 mg doses respectively.
Table 1.
Pharmacokinetic parameters for single doses of marimastat.
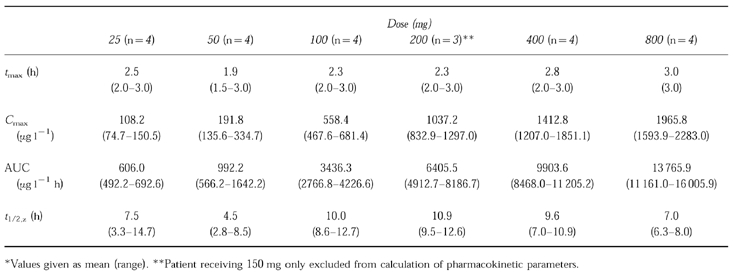
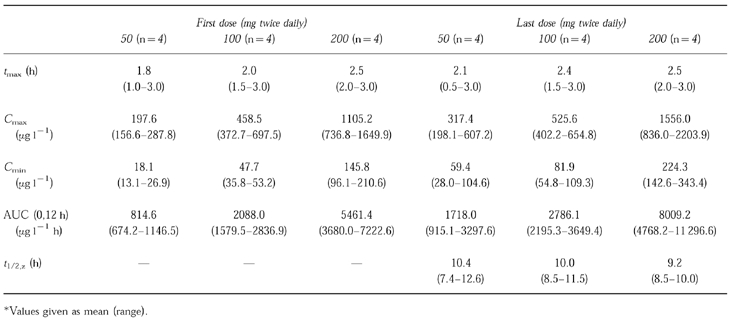
Table 2.
Pharmacokinetic parameters for repeat doses of marimastat.
In the single dose study, marimastat was first detected at 15 to 60 min after dosing, and the highest mean plasma concentrations were observed at between 1.5 and 3 h. At doses of 200 mg and above, marimastat was detectable up to day 2, however, by day 5 marimastat was not detectable in any subject. Within each dose, there was considerable variation between subjects with respect to AUC and Cmax. Up to 200 mg, mean AUC and Cmax were approximately proportional to the dose, although at the higher doses departure from linearity was apparent. Elimination half-life was also variable but was not dose-dependent. The mean half-life overall was 8.26 h.
In the repeat dose study, maximum mean plasma concentrations were seen between 1.5 and 3 h after dosing on both the first and last doses. At 12 h after the last dose (Cmin ), mean concentrations were 59.4, 81.9 and 224.3 μg l−1 for those subjects receiving 50, 100 and 200 mg twice daily, respectively. This compares with Cmin of 18.1, 47.7 and 145.8 μg l−1 after the first dose, suggesting some accumulation upon repeated dosing, as would be expected in attaining steady state. There was no increase in accumulation at the high doses. Marimastat was detectable in small quantities (<10 μg l−1 ) in four patients up to day 10.
The mean terminal elimination half-life (after the last dose) was 10.38, 9.96 and 9.17 h for those subjects treated with 50, 100 and 200 mg twice daily, respectively. There was relatively little inter-subject variability in this parameter, with values ranging from a minimum of 7.4 h, to a maximum of 12.6 h. AUC after the last dose was 3072.0, 4346.2 and 11 525.8 μg l−1 h, respectively, in subjects receiving 50, 100 and 200 mg twice daily. A linear relationship was observed between AUC and dose (Figure 3).
Figure 3.
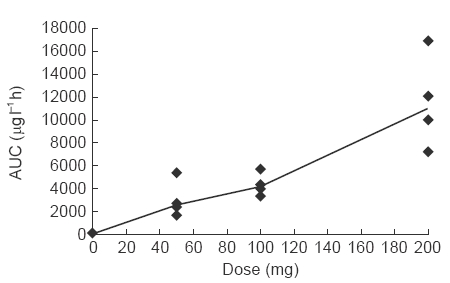
Relationship between dose and overall drug exposure (AUC). Line joins median values.
In the single dose study, between 1.7 and 3.2% of the administered dose of marimastat appeared unchanged in the urine in the 24 h after dosing. Maximum urinary concentrations were seen in the period 0 to 6 h after dosing, with mean values increasing in an approximately linear fashion from 578.7 to 15 083.6 μg l−1 between the 25 and 800 mg doses, respectively. Between 2.4 and 4.2% of the drug appeared in the urine in the 24 h after day 6 dosing in the repeat dose study. Day 6 urinary drug concentrations increased with dose, from 3208.0 to 12 478.9 μg l−1 between the 50 and 200 mg twice daily doses, respectively, although did not increase between the first and the last dose.
Tolerability
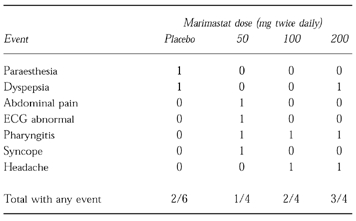
Adverse events occurring in the single and repeat dose studies are summarized in Tables 3 and 4, respectively. All events were mild in severity and none was described as being more than possibly related to treatment. Overall, the incidence of adverse events in subjects receiving marimastat was similar to that in subjects receiving placebo. There were no serious adverse events recorded, and no adverse events lead to study drug withdrawal.
Table 3.
Summary of adverse events occurring in single dose study.
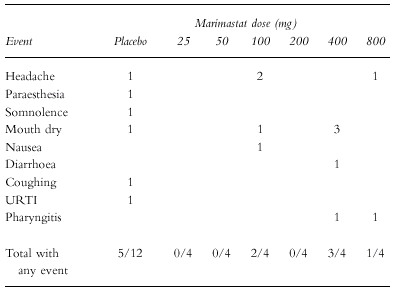
Table 4.
Summary of adverse events in repeat dose study.
Laboratory measures in the single dose study revealed a number of incidental departures from normal ranges, although none were of a magnitude that would be considered clinically significant. Recordings of bilirubinaemia and albuminuria for one subject at 25 mg, and an elevated ALT for another subject at 50 mg, were just outside normal ranges and not repeated in those same individuals at higher doses. A single recording of lymphocytosis was observed in one subject 24 h after receiving a 100 mg dose, and a further subject displayed eosinophilia before and after marimastat dosing. Small decreases in mean platelet count were observed at doses above 50 mg.
In the repeat dose study, 2/6 placebo-treated and 4/12 marimastat-treated subjects recorded unsustained hyperglycaemia and/or glycosuria at some point in the study. The abnormalities in the placebo group comprised one subject with blood glucose levels of 9.6 and 7.3 mmol l−1 on two separate occasions, and a single episode of glycosuria in another subject. Abnormalities in the marimastat-treated subjects comprised single recorded blood glucose levels of 7.7, 7.9 and 7.5 mmol l−1 in three patients and glycosuria on three occasions in one other subject.
A serial rise in ALT was observed in one subject receiving 200 mg twice daily; ALT at screening was 42 iu l−1, rising to 122 iu l−1 5 days after the final dose and returning to normal over the subsequent 2 weeks. The three other subjects receiving 200 mg twice daily showed small rises in ALT over the dosing period, and two subjects receiving 50 mg twice daily recorded ALT values exceeding the upper limit of the normal range (55 and 56 iu l−1 ). Small but consistent increases in AST were also noted, although there were no rises in bilirubin or γGT. Examination of other laboratory parameters did not reveal any changes occurring in a consistent dose-related manner. In contrast with the first study, there was no clear effect on platelet count.
There were no obvious treatment or dose-related changes in vital signs or physical examination in the single or repeat dose studies. Similarly, there was no indication of an effect of marimastat on ECGs, although T wave inversion was recorded as an adverse event in a subject receiving 50 mg twice daily in the repeat dose study.
Discussion
After oral administration in healthy volunteers, marimastat rapidly appears in the plasma, with the majority of subjects achieving detectable levels within one hour of dosing. Peak plasma concentrations were observed at between 1.5 and 3 h, and these peaks were approximately proportional to dose. Terminal elimination half-life in the single dose study varied considerably between subjects, although the overall mean half-life of 8.26 h was consistent with the overall mean half-life observed in the repeat dose study of 9.84 h, in which there was relatively little inter-subject variability. There was no evidence that the terminal elimination half-life varied with dose.
Calculated overall drug exposure (AUC) was approximately proportional to dose in both studies. In the single dose study AUC and Cmax did not increase proportionally at higher doses, possibly indicating a reduction in bioavailability of marimastat at doses of 400 mg and higher. However, there are no currently available data on intravenous administration of marimastat to clarify this. In the repeat dose study, a degree of drug accumulation was noted as steady state was achieved, although comparisons of Cmax, Cmin and AUC(0,12 h) after the first and last doses showed that the accumulation was not extensive. The observed accumulation ratio was slightly less in subjects receiving 100 and 200 mg twice daily, than in those receiving 50 mg twice daily.
Marimastat was well-tolerated by all subjects. Only mild events were recorded, and the overall incidence of adverse events occurring in subjects receiving marimastat was similar to that seen in placebo-treated subjects. In the single dose study, physical examination, recording of vital signs, and ECGs and laboratory tests did not reveal any changes suggestive of important drug toxicity. A small fall in mean platelet counts is probably not of significance in light of the stable platelet counts observed in the repeat dose study.
Small but consistent elevations in liver transaminases, particularly ALT, were observed with repeat dosing of marimastat. All changes were reversible and no other changes in liver function were noted, including measures of plasma bilirubin and γGT. Finally, in one subject receiving 50 mg twice daily, electrocardiographic T wave inversion was observed. This was not considered to be clinically significant by a reviewing cardiologist and no ECG abnormalities in other subjects were noted. Other measures of safety, including physical examination and vital signs did not reveal any areas of concern.
It is concluded from single rising dose and 1 week continuous dosing of marimastat, that the drug is well-tolerated in healthy male subjects. The only biochemical changes observed were small, reversible increases in liver transaminases. The drug is rapidly absorbed with peak levels achieved within 3 h of administration. There is no unexpected accumulation of marimastat at the doses studied, and the terminal elimination half-life would suggest twice daily dosing is appropriate for use in clinical trials. On the basis of these studies, it is estimated that at a dose of 25 mg twice daily trough plasma concentrations of 40 μg l−1 would be achieved, producing free drug levels of approximately six times the IC50 for collagenase, gelatinase A, gelatinase B and matrilysin. It is important to note, however, that the recorded data in this healthy volunteer population may differ substantially from that in patients with cancer. This latter group represents the principal target population for marimastat, in whom increased age, reduced organ function, and changes in plasma protein binding and volume of distribution may have significant impact on the drug's pharmacokinetic profile.
References
- 1.Matrisian LM. Metalloproteinases and their inhibitors in matrix remodelling. Trends In Genetics. 1990;6:121–125. doi: 10.1016/0168-9525(90)90126-q. [DOI] [PubMed] [Google Scholar]
- 2.Kleiner DJ, Stetler-Stevenson WG. Structural biochemistry and activation of matrix metalloproteinases. Curr Op Cell Biol. 1993;5:891–897. doi: 10.1016/0955-0674(93)90040-w. [DOI] [PubMed] [Google Scholar]
- 3.Liotta LA, Stetler-Stevenson WG. Metalloproteinases and cancer invasion. Sem Cancer Biol. 1990;1:99–106. [PubMed] [Google Scholar]
- 4.Gordon JL, Drummond AH, Galloway WA. Metalloproteinase inhibitors as therapeutics. Clin Exp Rheum. 1993;11:91–94. [PubMed] [Google Scholar]
- 5.O'Byrne EM, Parker DT, Roberts ED, et al. Oral administration of a matrix metalloproteinase inhibitor, CGS 27023A, protects the cartilage proteoglycan matrix in a partial meniscectomy model of osteoarthritis in rabbits. Inflamm Res. 1995;44:117–118. doi: 10.1007/BF01778290. [DOI] [PubMed] [Google Scholar]
- 6.Saarialho-Kere UK, Vaalamo M, Puolakkainen P, Airola K, Parks WC, Karjalainen-Lindsberg ML. Enhanced expression of matrilysin, collagenase and stromelysin-1 in gastrointestinal ulcers. Am J Pathol. 1996;148:519–526. [PMC free article] [PubMed] [Google Scholar]
- 7.Gijbels K, Masure S, Carton H, Opdenakker G. Gelatinase in cerebrospinal fluid of patients with multiple sclerosis and other inflammatory neurological disorders. J Neuroimmunol. 1992;41:29–34. doi: 10.1016/0165-5728(92)90192-n. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg GA. Matrix metalloproteinases in brain injury. J Neurotrauma. 1995;12:833–842. doi: 10.1089/neu.1995.12.833. [DOI] [PubMed] [Google Scholar]
- 9.Galardy RE, Cassabone ME, Giese C, et al. Low molecular weight inhibitors in corneal ulceration. Ann NY Acad Sci. 1994;732:315–323. doi: 10.1111/j.1749-6632.1994.tb24746.x. [DOI] [PubMed] [Google Scholar]
- 10.Macaulay VM, O'Byrne KJ, Saunders MP, et al. Phase I study of matrix metalloproteinase (MMP) inhibitor batimastat (BB-94) in patients with malignant pleural effusions. Br J Cancer. 1995;71:11. (Abstract). [Google Scholar]
- 11.Drummond AH, Beckett P, Bone EA, et al. BB-2516: An orally bioavailable matrix metalloproteinase inhibitor with efficacy in animal cancer models. Proc Am Assoc Cancer Res. 1995;36:100. (Abstract). [Google Scholar]
- 12.Shah VP, Midha KK, Dighe S, et al. Analytical methods validation: bioavailability, bioequivalence and pharmacokinetic studies. Conference report. Eur J Drug Metab Pharmacokin. 1991;16:249–255. doi: 10.1007/BF03189968. [DOI] [PubMed] [Google Scholar]