

Abstract
The concept that cancer can be prevented, or its onset postponed, by certain diet-derived substances is currently eliciting considerable interest. Agents which interfere with tumour development at the stage of promotion and progression in particular are of potential clinical value. As chemopreventive agents have to be administered over a long period of time in order to establish whether they possess efficacy in humans, it is of paramount importance to establish their lack of toxicity. The desire to select the best chemopreventive drug candidates for clinical trial, and the necessity to monitor efficacy in the short and intermediate term, render the identification of specific mechanism-based in vivo markers of biological activity a high priority. Antioxidation, inhibition of arachidonic acid metabolism, modulation of cellular signal transduction pathways, inhibition of hormone and growth factor activity and inhibition of oncogene activity are discussed as mechanisms by which the soya constituent genistein, the curry ingredient curcumin and the vitamin A analogue 13-cis retinoic acid exert tumour suppression. A better understanding of these mechanisms will help the establishment of screens for the discovery of new and better chemopreventive agents and the identification of surrogate markers to assess the outcome of clinical chemoprevention trials.
Keywords: cancer chemoprevention, curcumin, genistein, mechanisms, retinoids
Introduction
The notion that the prevention of disease is better than having to cure it is probably as old as the concept of restoration of human health by medical intervention. Many human cancers are preventable because their causes have been identified in the human environment [1]. The finding that regular consumption of certain constituents of fruits and vegetables might protect us from this deadly disease, which was first proposed by Wattenberg [2], has aroused much interest among the medical establishment and the general public. Chemoprevention or chemoprotection can be defined as the use of specific diets, or natural or synthetic chemicals, to reverse, suppress, or prevent carcinogenic progression to invasive cancer. Minimisation of exposure towards carcinogens in the environment (‘primary prevention’) is undoubtedly an effective strategy in cancer prevention. However, most environmental factors which initiate cancer remain to be identified and, once identified, the avoidance of such factors necessitates life-style changes, which may be difficult to implement. Epidemiological data suggesting that cancer is preventable by intervention with chemicals are based on time trends in cancer incidence and mortality, geographic variations and effect of migration, identificaton of specific causative factors and lack of simple patterns of genetic inheritance for the majority of human cancers [3]. In order to understand how chemopreventive agents exert their activity, one has to recall that epithelial carcinogenesis proceeds via multiple discernible steps of molecular and cellular alterations (Figure 1). Invasive cancer is the ultimate product of this sequence of critical events, many of which can theoretically be prevented. These events can be separated into three distinct phases: initiation, promotion and progression. Initiation is rapid, it involves direct carcinogen binding and damage to DNA, and the resulting mutation is irreversible. Promotion follows initiation and involves clonal expansion of initiated cells induced by agents acting as mitogens for the initiated cell. This stage is generally reversible. The progression stage of carcinogenesis is an extension of promotion and results from it in the sense that cell proliferation caused by promotors allows the cellular damage inflicted by initiation to be further propagated. During tumour progression genotypically and phenotypically altered cells gradually emerge. Both promotion and progression phases are prolonged. Depending on which phase of carcinogenesis chemopreventive agents affect, they can be divided into tumour ‘blocking’ agents, which counteract cancer by interfering with initiation, and tumour ‘suppressors’, which intercept promotion or progression [4]. Blocking agents probably play a significant role in reducing the accumulation of initiating mutations, but the fact that initiation can occur very early in life confounds clinical chemoprevention strategies based on anti-initiation only. Hence suppression of the development of the initiated cell to a full-blown tumour is undoubtedly the strategy of choice in human cancer chemoprevention, and tumour-suppressing agents are the focus of this review. Its aim is to outline mechanisms by which such substances suppress tumours and to highlight the importance of the understanding of these mechanisms for the discovery and clinical development of novel and safe chemopreventive substances.
Figure 1.
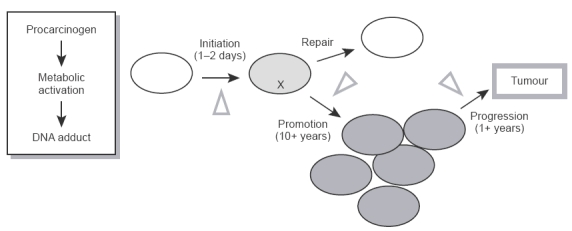
Hypothetical phases of multistep carcinogenesis.
Chemoprevention trials
The clinical evaluation of substances for their chemopreventive characteristics presents a host of challenges which are different from those encountered in clinical trials of new cancer chemotherapeutic agents [5]. First and foremost, the substance under test has to be innocuous from a toxicological standpoint, as it is likely to be administered over a considerable period of time, probably many years and often to ostensibly healthy individuals. Food stuffs are particularly attractive sources of tumour-suppressive substances, as the knowledge of their use has been collected over many generations, which helps gauge the incidence of unwanted effects in humans more easily than is the case for novel synthetic compounds. The conduct of randomized, prospective large-scale clinical trials is the ultimate step in the exploitation of research results for disease prevention. Trials are currently conducted on, for example, tamoxifen in women at high risk of breast cancer and on several retinoids in breast cancer patients at risk of second primary tumours in the contralateral breast, and in men at risk of prostate cancer [6]. Such large trials are expensive and represent major challenges in design, implementation and analysis. They involve thousands of subjects, multi-institutional arrangements, and take years to complete. One of the first diet-derived agents under clinical investigation for chemopreventive efficacy has been the carotenoid β-carotene. Four major intervention trials have been completed and the outcome of two of them, the ‘α-tocopherol, β-carotene prevention study’ [7] and the ‘β-carotene and retinol efficacy trial’ [8], has alarmed proponents of cancer chemopreventive strategies. These studies suggest that in high-risk groups of smokers and/or workers occupationally exposed to asbestos, β-carotene not only failed to protect against lung cancer, it even increased the risk of developing the disease. Subgroup analyses of these two trials [9, 10] revealed that the risk of lung cancer was highest among those individuals who continued to smoke at least 20 cigarettes day−1 and those in the highest quartile of alcohol consumption. It is conceivable that β-carotene suppresses tumours only in those individuals from whom the initiating stimulus has been removed, but not in those who continue to be subjected to it, though the underlying mechanism is currently unknown. This disconcerting outcome underlines the importance of understanding how chemopreventive agents exert their effects, and under which conditions they are beneficial, prior to extensive clinical evaluation. In the light of the problems raised by the β-carotene trials and the enormous costs and effort associated with large trials, the development of chemopreventive agents in the future will incorporate increasingly small preliminary trials designed to identify specific mechanism-based markers of biological activity, to select the best candidates for specific intervention programmes, and to monitor efficacy in the short and intermediate term [11].
Tumour suppressing agents
This review focusses on three representative food-derived agents and the mechanisms which are likely to contribute to their tumour suppressive activity: retinoic acid, genistein and curcumin (for structures see Figure 2). Retinoic acid derivatives are the first agents for which cancer suppressive properties were demonstrated in humans. Genistein is the major isoflavonoid constituent of soya, and curcumin, a polyphenolic ingredient of Curcuma longa, is used as a spice, for example in curry. Both food-derived agents are thought to be responsible for the chemoprotective properties of the foodstuffs in which they occur.
Figure 2.
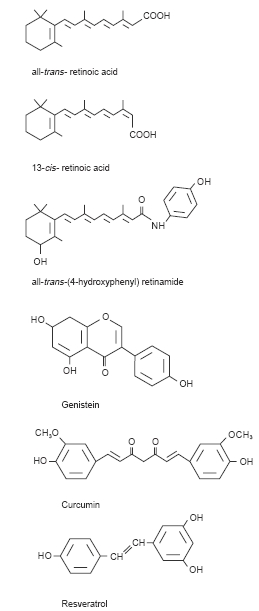
Structures of tumour-suppressive agents.
Retinoic acid
Retinoic acid analogues, naturally occurring or synthetic vitamin A derivatives, have been tested for chemopreventive activity in clinical trials more intensely than any other class of compound [12]. Examples include all-trans-retinoic acid, 13-cis-retinoic acid and all-trans-N-(4-hydroxyphenyl)retinamide (Figure 2). The rationale for the use of retinoids as chemoprotective agents is based on the strong inverse relationship between dietary vitamin A intake and cancer development which has been established by numerous investigations over the last couple of decades [13]. Vitamin A deficiency in experimental animals has also been associated with a higher incidence of cancer and with increased susceptibility to chemical carcinogens [14]. Natural retinol (vitamin A) has been available for experimental testing and clinical practice for over 50 years. The widespread interest in this compound as a modulator of cell growth was generated by its physiological properties, which are evident from the very early phases of embryogenesis. In higher animals vitamin A is essential for vision, reproduction, and maintenance of differentiated epithelia and mucus secretion [15]. Retinoic acid shares only some of these functions, and animals maintained on retinoic acid as the only source of vitamin A are both blind and sterile [16, 17]. A remarkable early observation in the area of bronchial carcinogenesis demonstrated that bronchial metaplasia could be induced in the hamster tracheal epithelium by vitamin A deprivation or benzo-[a]pyrene instillation, and in both cases it was preventable by retinol administration [18, 19]. These observations led to the hypothesis that physiological levels of retinoids guard the organism against the development of premalignant and malignant lesions. Retinoids show protective effects against lesions associated with cutaneous, oral and bronchial premalignancies in humans [12] and have also been tested in the prevention of second primary tumours. Promising results have been obtained with 13-cis-retinoic acid in Xeroderma pigmentosum, head and neck cancer, and with retinyl palmitate in non-small cell lung cancer [20]. Effects of most retinoids used in clinical trials are reversible, therefore patients have to be treated for prolonged periods of time. However, some of the currently tested retinoids exhibit unacceptable side effects, such as dry skin, cheilitis, conjunctivitis and hypertriglyceridaemia, which limit their use to short periods of time. Another major concern is the teratogenicity of many retinoids [21].
Genistein
Asian diets such as those consumed in China and Japan, are typically lower in total saturated fat and higher in dietary fibre, and these characteristics may contribute to the relatively low rates of breast, prostate and colon cancer in these countries as compared with Western countries. In recent years soy consumption has been implicated in the causation of the lower rates of these cancers in Asian countries. Particularly noteworthy is the fact that soybeans are a rich and relatively unique source of the isoflavonoids genistein and daidzein, considered to be the major constituents responsible for the chemoprotective efficacy of soya [22]. Glycoside conjugates of genistein account for at least two-thirds of the total content of isoflavones in soybean, with conjugates of daidzein and small amounts of other isoflavonoids comprising the remainder [23]. Western diets, which customarily do not include soy products, are almost completely lacking in isoflavonoids: Isoflavonoid intake in people in Britain has been estimated to be <1 mg day−1 [24], compared with ∼16 mg established for Japanese men [25]. A recent review highlighted that in two-thirds of 26 studies on the effect of genistein-containing soy materials on animal models of carcinogenesis, the cancer risk, as measured by incidence, latency or tumour number, was significantly reduced [26]. Furthermore pure genistein delayed mammary tumour development in rats treated with 7,12-dimethylbenz[a]anthracene (DMBA) when administered neonatally, and inhibited aberrant crypt formation in a model of colon cancer [27]. Of all the isoflavones investigated, genistein appears to be the most potent chemoprotective agent, which may reflect the effort which has been, and is currently being, invested in the elucidation of its mechanism of action.
Curcumin
Turmeric, the powdered rhizome from the root of the plant Curcuma longa has long been used as a spice in foods and as a herbal remedy in the treatment of inflammatory diseases. Turmeric has a somewhat bitter taste and gives curry dishes a characteristic yellow colour. Curcumin (diferuloylmethane), the yellow pigment in turmeric, has also been used as spice in foods, as well as in cosmetics and drugs [28]. Curcumin possesses strong anticarcinogenic effects in animals in a variety of tissues [29]. The initial report of its anti-tumour promoting activity was published by Huang et al. [30], in which curcumin applied topically to the skin of mice inhibited phorbol ester-induced tumour promotion in dimethylbenzanthracene-initiated skin. This observation was consistent with the previously reported antioxidant and anti-inflammatory effects of curcumin [28]. Subsequent investigations demonstrated the ability of curcumin to inhibit benzo[a]pyrene-induced forestomach tumours [31], and precancerous lesions, for example DMBA-induced hyperplastic nodules in cultured rat mammary gland tissue [32] and azoxymethane-induced crypts in rat colon [33]. These and other studies clearly demonstrate that curcumin undoubtedly affords protection in various animal cancer model systems.
Mechanisms of tumour suppression
Potential mechanisms of tumour suppression for promising chemopreventive agents are listed in Table 1. It is pertinent to stress that the importance of each of these mechanisms for cancer prevention is not yet fully understood. Research into cancer chemotherapeutic agents in the past 30 years has demonstrated that the mechanisms by which they elicit their antineoplastic effects are multiple and complex, and our understanding is often a function of the length of time for, and degree of intensity at, which they have been studied. It is not surprising that in order to combat effectively a disease as complicated and multi-factorial as cancer, a good chemotherapeutic agent needs to possess a variety of mechanistically distinct but complementary properties germane to events which regulate cell growth, differentiation and survival. For cancer chemopreventive agents the situation is likely to be similar: compounds such as the retinoids, genistein and curcumin, affect a number of biochemical and physiological cell parameters potentially associated with carcinogenesis. One should bear in mind that these agents possess not only tumour-suppressing but also tumour-blocking activity. This review concentrates on five tumour suppressive mechanisms—antioxidation, inhibition of arachidonic acid metabolism, modulation of cellular signal transduction pathways, inhibition of hormone and growth factor activity and inhibition of oncogene activity—because they are thought to be of particular importance for tumour suppression caused by retinoids, genistein and curcumin. Information about the pharmacokinetic behaviour of this type of agent is scarce. It is therefore not yet possible to assess whether the concentrations at which these agents are known to elicit biochemical responses considered germane to their mechanisms, are achieved in vivo after consumption of foods which contain them.
Table 1.
Possible mechanisms of tumour suppression and examples of tumour suppressive agents.
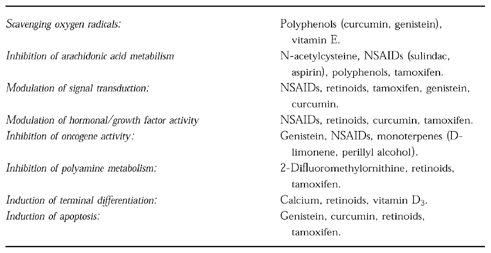
Antioxidant activity
There is abundant evidence that activated oxygen species, i.e. singlet oxygen, peroxy radicals, superoxide anion, and hydroxyl radicals, are involved in carcinogenesis. Potentially they act both in the initiation and promotion and progression stages of carcinogenesis. Their involvement in promotion and progression is based on the following evidence [reviewed in 34]: 1. Oxygen radical generating systems, e.g. superoxide anion generation via xanthine oxidase, show activities in vitro similar to those of known tumour promotors. These activities include increasing transformation frequencies of fibroblasts and keratinocytes and increasing transcription of genes associated with early steps in cell proliferation including c-fos, c-myc, c-jun, and ornithine decarboxylase (ODC). 2. Inflammatory cells produce a range of reactive oxygen species; and there is evidence associating inflammation with cancers in various tissues including stomach, oesophagus, colon/rectum and bladder. 3. Tumour promotors stimulate the endogenous production of oxygen radicals in inflammatory cells and keratinocytes. 4. Tumour promotors inhibit endogenous activities which protect against oxidative damage, such as those of glutathione peroxidase, catalase and superoxide dismutase. 5. Free radical-generating agents such as benzoyl peroxide and butylated hydroxytoluene hydroperoxide are tumour promotors in mouse skin. Consistent with the notion that reactive oxygen species are important for growth stimulation, a recent report suggests that constitutive production of superoxide in Ras-transformed cells activates mitogenic pathways probably distinct from those activated by extracellular growth factors [35]. Consequently, scavenging reactive oxygen species is a chemoprotective mechanism. For example, thiols such as N-acetylcysteine are known to react with hydroxyl radicals, and phenolic antioxidants, to which genistein and curcumin belong, scavenge peroxy radicals [34, 36, 37].
Modulation of arachidonic acid metabolism
Arachidonic acid (AA) is metabolised to prostaglandins, thromboxanes, leukotrienes and hydroxyeicosatetraenoic acids (HETEs) via oxidative enzymes [38, 39]. Activated oxygen species and alkylperoxy species are formed throughout this process. AA metabolism is increased during inflammation. Two aspects of AA metabolism are associated strongly with carcinogenicity, and both are inhibited by antioxidants and anti-inflammatory agents. The first is the prostaglandin (PG) synthetic pathway, which involves the enzyme prostaglandin H synthase (PHS). This enzyme has two activities—cyclooxygenase (COX), which catalyses the formation of prostaglandin G2 (PGG2 ) from AA, and hydroperoxidase, which catalyses the reduction of PGG2 to PGH2. PHS activation is germane to both initiation and promotion. On the one hand PHS hydroperoxidase can activate procarcinogens, which act as reducing cosubstrates, to their ultimate carcinogens [40]. On the other hand PGH2 and other prostaglandins, products of the reaction catalysed by this enzyme, are involved with signal transduction pathways related to promotion (see below) [39, 41, 42]. The second aspect of AA metabolism associated with carcinogenesis is the burst of PHS and lipoxygenase activity that is seen during inflammation and is stimulated by the tumour promotor, 12-O-tetradecanoylphorbol-13-acetate (TPA). The available evidence suggests that the immediate products of lipoxygenase activity, the HETEs and their hydroperoxy precursors are at least as important to tumour promotion as are PGs. Compounds that inhibit lipoxygenase, such as vitamin E and curcumin, inhibit tumour promotion in mouse skin [30]. The release of AA from membrane phospholipids catalysed by phospholipases is another control point in the AA metabolic pathway. AA can also be released from phospholipids via diacylglycerol (DAG) lipase. Control of AA release by these enzymes is likely to be mediated via signal transduction pathways. Compounds which block signal transduction at the membrane level, such as genistein, may inhibit AA metabolism and exert chemprotection by this mechanism. Thus inhibition of AA metabolism may play a role in controlling carcinogenesis both directly and indirectly.
PHS exists as two isoforms, COX1 and COX2. COX1 is involved in mediating the physiological functions of prostaglandins, whereas COX2 is primarily associated with pathological states such as inflammation. COX2 has been shown to be selectively overexpressed in human colon, gastric and breast cancer, and may also have a role in skin cancer [43, 44]. That COX2 plays an important role in colon carcinogenesis has been demonstrated in COX2 knock-out mice, which were resistant to the development of colon tumours caused by a knock-out mutation in the adenomatous polyposis coli (APC) tumour suppressor gene [45]. Overexpression of COX2 in normal colon epithelial cells alters the cellular phenotype by rendering them resistant to apoptosis and changing their cell adhesion properties [46]. Direct inhibition of COX2 may be an important mechanism by which non-steroidal anti-inflammatory agents (NSAID) such as aspirin exert chemoprevention. The long term use of aspirin has been associated with a ∼50% reduction in risk for colon cancer [47]. Inhibition of COX2 expression may also be an important chemopreventive mechanism. Adenoma and carcinoma formation in the MIN mouse, an animal model for familial adenomatous polyposis coli (FAP) in humans, is blocked by sulindac [48]. This block correlates with downregulation of COX2 gene expression and restoration of ‘normal’ levels of apoptosis in the colon epithelium. Curcumin has been shown to inhibit the induction of COX2 mRNA by TNF-α and fecapentaene-12 in human colon epithelial cells in vitro [49] and retinoids suppress phorbol ester-mediated COX2 induction in human oral epithelial cells [50].
Modulation of hormonal and growth factor activity
Chemicals may inhibit cell growth associated with carcinogenesis by directly regulating the induction and activity of specific hormones and growth factors which initiate steps in signal transduction. This regulation can occur at the level of membrane receptors for growth factors, peptide hormones, neurotransmitters, or via cytoplasmic and nuclear receptors for the steroid superfamily consisting of oestrogen, progesterone, retinoid, glucocorticoid and vitamin D [51]. For example, antioestrogens such as tamoxifen bind to nuclear oestrogen receptors, preventing the binding and activity of oestrogens [52, 53]. Phytoestrogenic isoflavonoids such as genistein have weak oestrogenic or antioestrogenic activity, depending on levels. Studies in human breast cancer-derived MCF-7 cells indicate that the antioestrogenic effect of genistein may result from slowed translocation of genistein-bound receptor from the cytoplasm to the nucleus compared with that of oestradiol-bound receptor [54]. Transforming growth factor β (TGF-β) has antiproliferative activity in both normal and neoplastic cells in vitro [55] and in mammary glands and liver in vivo [56, 57, 48]. Neoplastic cells produce TGF-β, but usually in a latent form that cannot bind to its receptor; these cells are responsive to antiproliferative effects of activated TGF-β [55]. There is evidence from studies in rat intestinal crypt epithelial cells that TGF-β may promote differentiation [58]. These observations suggest that chemicals which activate TGF-β could also control proliferation in carcinogenesis. Retinoic acid, which inhibits tumour promotion in mouse skin, induced TGF-β2 in mouse keratinocytes and in mouse skin in vivo after topical administration [57]. The increase in TGF-β2 occurs post-transcriptionally. In vitamin A deficient rats treated with retinoic acid, the level of expression of TGF-β correlated with levels of retinoids in skin, intestine, and respiratory epithelia [59].
Other receptors which may be be important for the chemopreventive activity of this class of compound are the retinoid receptors. They are members of the steroid receptor superfamily and mediate effects of retinoids on gene expression, growth and differentiation in normal and tumour cells. There are two types of retinoid receptors, retinoic acid receptors (RARs) and retinoid X receptors (RXRs) and they possess different ligand binding properties. RARs and RXRs form heterodimers which bind to specific DNA sequences, the retinoic acid response elements, and enhance the transcription of retinoid-responsive genes. Changes in the expression of these receptors can abrogate the retinoid signalling pathway and result in enhanced carcinogenesis. Application of the tumour promotor TPA to the skin decreased expression of RARs with a concomitant decrease of retinoic acid binding to RARs [60]. Decreased expression of retinoic acid receptors may be associated with development and/or progression of carcinomas. The expression of RAR-β, which is suppressed selectively at early stages of carcinogenesis in the oral cavity and bronchial epithelium, is upregulated by retinoids [61]. Thus RAR-β might serve as an intermediate biomarker, because it is upregulated by the chemoprotective agent, and this upregulation is associated with clinical response. Another activity of nuclear retinoid receptors is to antagonise the activity of other transcription factors. For example, they can, in the presence of all-trans-retinoic acid, antagonise the action of the transcription factor activator protein 1 (AP-1), which is activated by TPA and regulates various genes involved in proliferation, differentiation and invasion [62]. One cytokine via which chemoprotective agents may exert their action is tumour necrosis factor α (TNF-α), which plays a central role in carcinogenesis, probably as an endogenous tumour promotor. Recently the chemoprotective tea constituent epigallocatechin gallate was shown to inhibit TNF-α mRNA expression and TNF-α release in BALB/3T3 cells after induction by a tumour promotor [63].
Modulation of signal transduction
One of the most significant advances in cancer research in recent years has been the increased understanding of the biochemical control mechanisms involved in the regulation of cell growth and development. Cells respond to signals from extracellular stimuli via a complicated network of highly regulated events, collectively referred to as signal transduction pathways (Figure 3). Stimulation of these pathways results in changes in transcriptional activity of genes. Whilst normal cells respond appropriately to extracellular stimuli, many precancerous and cancerous cells have lost this ability and display aberrant signalling [64]. A variety of steps in deregulated signalling pathways could theoretically be targeted as potential sites for chemopreventive intervention. Key components of these pathways are the protein tyrosine kinases (PTKs) which catalyse the transfer of the γ-phosphate of ATP to the hydroxyl group of tyrosine on numerous proteins. Loss of PTK-regulatory mechanisms has been implicated in neoplastic growth; indeed many oncogenes code for PTKs [65]. Two general classes of PTKs are currently recognised: receptor tyrosine kinases, which receive signals directly through their extracellular domains, and cytosolic tyrosine kinases, which are downstream signal transducing elements. Specific PTKs activated during the development of many human neoplasias have been identified. Examples of aberrant expression of receptor PTKs are epidermal growth factor receptor (EGFR)-associated PTK in head and neck cancers [66], p185c-erb B2 in breast and ovarian cancers, and platelet-derived growth factor receptor-associated PTK in glioblastomas and breast cancers [67]. Based on the redundancy of growth factor networks, selective inhibition of signalling pathways activated in precancerous and cancerous cells is considered to be possible. Proliferation of normal cells is dependent on more than one growth factor, and one growth factor activates multiple intracellular signalling pathways. Genetic knockout experiments have established that, if a particular growth factor-signalling pathway is inactivated, an alternative pathway takes over [65, 67]. One of the most extensively studied PTKs associated with cancer is the EGFR, the product of the c-erbB-1 proto-oncogene [68]. Among the presently recognised EGFR ligands, apart from EGF, are TGF-α and amphiregulin [69]. The importance of EGF and TGF-α in controlling cell growth has been well established [69, 70]. Activation of EGFR can occur via autocrine, paracrine or juxtacrine mechanisms [71]. On ligand binding, EGFR dimerises with neighbouring receptors and is autophosphorylated at three major tyrosine residues [72]. Subsequently the receptor interacts with a number of proteins that are elements of signal transduction pathways, including phospholipase C-γ, phosphatidylinositol-3′-kinase, growth factor-mediated binding protein 2 (grb2), Src family kinases, mitogen-activated protein kinase (MAPK) and components of the Jak/STAT pathway [73]. Genistein is a good inhibitor of PTKs with some specificity for EGFR PTK [74]. Disruption of the EGFR-mediated signalling pathway has been suggested as an important mechanism by which genistein protects against cancer. Genistein is also a reasonably potent inhibitor of cell proliferation [75]. Interestingly, it has been recently shown that growth-inhibitory concentrations of genistein in cells in vitro do not cause measurable inhibition of tyrosine phosphorylation, which casts doubt on the notion that EGFR PTK inhibition is an important mechanistic element in genistein-induced effects on proliferation [76, 77].
Figure 3.
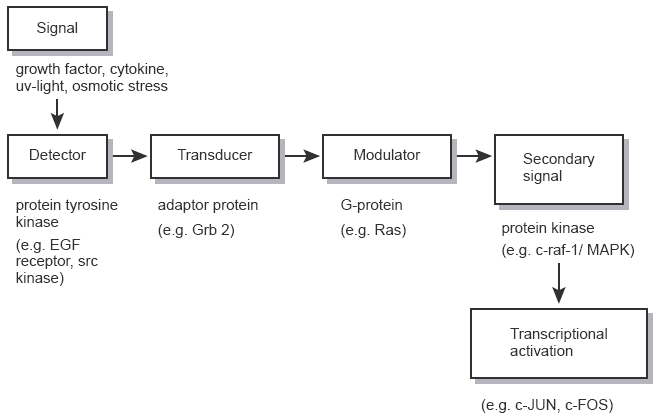
Signalling pathways which are known to be subject to pharmacological manipulation [modified from 99]. In this hypothetical scheme the initial signal is considered to be a growth factor or other ligand interacting with a cell surface receptor. The receptor acts as the originating detector for the signal, which is subsequently transduced to produce second messengers to bring about biological responses. Specific examples of components of signalling pathways which can be influenced by cancer chemopreventive agents and which are mentioned in the text are shown below the boxes.
Many steps in signal transduction downstream of receptor- linked PTKs might be affected by chemopreventive agents and thus contribute to chemopreventive efficacy. One such step is, for example, the activation of protein kinase C (PKC) by diacylglycerol (DAG). There is evidence that carcinogenesis may be suppressed by inhibiting this enzyme. Phorbol ester tumour promotors such as TPA can replace DAG in activating PKC [78]. Curcumin inhibits PKC [79], TPA-induced tumour promotion in mouse skin (see above) and also EGFR tyrosine kinase activity in murine fibroblasts [80]. Inhibition of TPA-induced ODC activity by genistein seems to involve several signal transduction mechanisms which affect both transcription and translation of ODC [81]. At high concentrations (4×104) genistein inhibited TPA-induced MAPK tyrosine phosphorylation, whereas at lower concentrations (2.5×106) it interfered with TPA-induced phosphorylation of p70 S6 kinase, a protein kinase involved in the control of translational efficiency.
Inhibition of oncogene activity
During the course of cell proliferation in carcinogenesis, numerous oncogenes are expressed abnormally—possibly functioning as intermediates in signal transduction pathways, e.g. protein kinases [82]. In the case of the ras oncogene, there are several steps during its activation which can be inhibited, and some data relate such inhibition to chemopreventive activity. The EGFR PTK is involved in ras activation, and kinase inhibitors would be expected to prevent ras activation. To be activated ras must undergo farnesylation. ras oncogenes are involved in mammary gland carcinogenesis induced by methylnitrosourea and, to a lesser extent, by dimethylbenzanthracene [83]. The citrus fruit constituent -limonene and its cogener perillyl alcohol inhibit the progression of mammary tumours induced in rats by these carcinogens [84, 85]. They inhibit the farnesylation of small G-proteins, which suggests that this type of molecule prevents oncogene activation by inhibiting post-translational farnesylation of the p21 ras protein [86].
NSAIDs might inhibit proliferation in carcinogenesis by inhibition of expression of the oncogene c-myc [39]. Expression of c-myc occurs early in EGF-induced cell proliferation. PGs are required, but not sufficient, for c-myc expression and DNA synthesis stimulated by EGF in BALB/c 3T3 cells [87]. Indomethacin inhibits both DNA synthesis and oncogene expression in this system, and this inhibition is reversed by addition of PGG2 [39, 88, 89]. Phorbol ester-induced transcription of protooncogenes activates the AP-1 family of transcription factors, these in turn increase the expression of a variety of response genes which regulate proliferation and differentiation and are thus germane to tumour promotion. That chemopreventive agents can suppress tumour promotor-induced AP-1 activation has been shown for curcumin in mouse fibroblasts [90], even though the mechanism is unclear. Our own work has demonstrated that genistein and the related isoflavan equol inhibit TPA-induced expression of the c-fos gene [91], but not TPA-induced activation of AP-1 in human breast cancer cells (Wrigley, Munks, Gescher, unpublished), which reflects a multitude of complex effects on early response genes and transcription factors impacting on downstream gene transcriptional activity.
Role of understanding mechanisms in chemopreventive drug discovery
The accumulating knowledge of the mechanisms by which agents may cause anticarcinogenesis has recently been harnessed in screens for novel chemopreventive substances [92, 93]. In these screens, which consist of panels of biochemical and cell physiological assays, fractions of plant extracts or pure agents are investigated for their abilities to suppress promotion-related events like the ones outlined above, among them AA metabolism, TPA-induced free radical formation, EGFR PTK activity, activation of transcription factors and terminal differentiation, in addition to properties germane to cancer initiation. In the absence of a definitive understanding of the importance of these processes in chemoprevention, the criteria which define ‘positive hits’ in the screens are somewhat ill-defined, but it seems important that ‘positive’ compounds are active in more than one of the individual tests which constitute the screen. The adequacy and usefulness of these screens remains to be established, but some interesting new compounds with chemopreventive efficacy in rodent tumour intervention studies have already been identified. Among them are the rotenoids, terpenoids from an African Leguminosa [94], and the polyphenolic stilbene analogue resveratrol, a constituent of grape skin [95] (see Figure 1). These substances were initially identified on the basis of their high abilities in one specific test, in the case of the rotenoids inhibition of ODC, and for resveratrol inhibition of COX.
Tumour suppressive mechanisms and design of chemoprevention trials
Cancer chemopreventive substances, such as genistein and curcumin, are found in the diet of humans, and it is feasible that their presence already affects the incidence of human cancer. Recommendations for dietary modification, including reduction of meat and fat consumption, as well as increasing consumption of fruits and vegetables, is clearly sound advice. The simpler concept of reducing caloric intake, should also be emphasised. But is it enough? On the basis of results in animals, some of which have been outlined above, we can expect with a high degree of confidence that some cancer chemopreventive agents reduce the risk of cancer also in humans. Dietary suggestions are very important, but it is not sufficient to rely on ‘An apple a day keeping the doctor away’ when faced with a disease as severe as cancer. In the light of the potential benefits offered by effective cancer chemoprevention, the implementation of therapeutic intervention trials using diet constituents with suspected chemopreventive activity or synthetic analogues thereof seems to be timely and sagacious. These trials will furnish the urgently needed clinical data which will ultimately confirm or refute claims of chemopreventive potency, and help identify subpopulations of individuals who would derive maximal benefit. The design of clinical chemoprevention trials is not yet clearly defined and continues to evolve [96], but a few generalities about each phase can be stated (Table 2). Phase I trials determine the dose-related safety of drugs and frequently include pharmacokinetic studies. Phase II and III trials serve to test drug efficacy. If absence of toxicity has been demonstrated in a phase I trial, a phase II trial using a placebo-controlled, double-blind design follows. A major goal of the phase II trial is the evaluation of toxicity observed during a prolonged administration period. In the phase II study multiple dosage levels should be evaluated. In most cases, phase II trials are conducted in populations at increased risk of succumbing to a particular malignancy. These trials are small-scale, short-term evaluations of the biological activities of new potential chemopreventive agents, determined by intermediate endpoints. If safety and efficacy are judged to be satisfactory, plans will be made for a definitive phase III intervention trial, the type of trial mentioned above. The ultimate evaluation in a phase III trial involves the recruitment of a large cohort of subjects at risk for the disease of interest who will be randomized to the agent of choice at one or multiple dose levels compared with a group receiving placebo. The long duration of large-scale trials allows for confirmation of the limited toxicity and efficacy data determined in early-stage clinical studies and for the possible appearance of adverse effects or agent efficacy not previously detected. Treatment of a large cohort is often preceeded by that of a vanguard group studied for 2 or more years to evaluate toxicity before the remainder of the cohort is recruited. In these large trials in which the efficacy of chemopreventive agents is tested, incidence of new primary tumours is the main endpoint.
Table 2.
Considerations governing clinical chemoprevention trials.
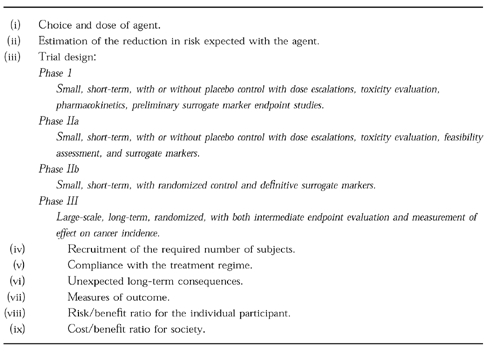
In the light of the enormous difficulties and effort associated with the design and conduct of large scale phase II and III chemoprevention studies, it seems pertinent to carry out specifically designed, small-scale, controlled studies in order to test and validate potential mechanism-based biomarkers. In order that such markers can be used systematically, they have to be proven to be specific for the process of carcinogenesis under study, correlate quantitatively or qualitatively with the degree of tumour progression, and be modulatable by the selected preventive agent. They should also be easily measurable in small specimens of body fluids or tissues, and sampling should be tolerable when performed repeatedly. Such studies will explore for example whether products of the interaction of DNA with reactive oxygen species are suitable surrogate markers for the chemopreventive efficacy of antioxidants, if downregulation of COX2 is a marker for colon cancer preventive agents such as curcumin, or whether alterations in levels of signal transduction molecules downstream of EGFR PTK or of transcription factors such as AP-1 in the target tissue are related to the efficacy of genistein.
To increase the cost/benefit ratio of intervention trials, specific subpopulations of very high risk individuals should be identified on the basis of constitutive or acquired abnormalities detectable in the target tissues. Recently a whole issue of the Journal of Cellular Biology has been devoted to cancer risk markers in chemoprevention trials [97]. The estimated sample size for the trial will be based on the estimated magnitude of the risk reduction associated with the agent, the baseline risk of the subjects enrolled, and the proposed duration of both the administration of the agent and the length of the observation time following administration of the agent. The ability of a prospective chemoprevention trial to deliver the desired outcome is dependent on the ability to recruit an adequate number of subjects in the appropriate risk category, the willingness of the subjects to remain in the trial and to take the chemopreventive agent as prescribed, and the level of risk reduction associated with long-term administration of the agent. Either loss of subjects from the trial before the planned duration is reached, or failure of the subjects to consume a sufficient proportion of the prescribed dose of the agent can have an enormous detrimental effect on the statistical power of the trial. Considerable efforts are required to recruit willing and committed subjects of adequate risk and to ensure as many of them as possible take the agent as prescribed for the entire planned duration of the trial. The major cause of attrition from a chemoprevention trial is clinical toxicity or the fear that it will occur. Therefore chemopreventive agents must be given with high therapeutic indices which maximise efficacy whilst minimising toxicity. Ethical considerations also dictate that agents designed to prevent malignancy in high risk subjects, who are nonetheless healthy individuals, must impose little or no serious toxicity and minimal symptomatic side effects.
Conclusions
Even though the literature on cancer chemopreventive agents and diet constituents is rapidly expanding, the development of such compounds for clinical use is still in its infancy. For the prevention of human cancer, agents which suppress rather than block tumour development are of particular importance. Many agents, among them several retinoids, are under clinical evaluation, and genistein, curcumin and other substances are about to be investigated in the clinic. Some of the mechanisms of tumour suppression currently under study are outlined above. Their overall role in the mediation of the effects of these agents is far from clear, but it is probable that the most effective agents exert their activity via more than one mechanism. A better understanding of these mechanisms will help confirm the validity of screens for the discovery of new chemoprotective agents, suggest new ones, and help identification of surrogate markers to assess the trial outcome. It will also contribute to the knowledge base which will ultimately help identify subjects or patients who are most likely to benefit from chemopreventive treatment, and who are least likely to experience detrimental effects, such as those observed in the β-carotene studies. Finally a cautionary note. It is pertinent to realise that the epidemiological, migration and diet studies which are the basis of much of the knowledge we have of diet-derived chemoprevention, have identified particular dietary components (foods), not the substances in the foods. This review shows that individual compounds have been isolated and shown to possess biological activity in certain in vitro systems and in vivo models indicative of cancer chemoprevention. This approach may miss the real value of the food matrix in toto. Synergism between biologically active dietary components may be the real basis of dietary cancer prevention. It has been suggested that synergism between weak environmental oestrogens could be detrimental and responsible for several toxicities observed in animals and humans [98]. Likewise, synergism might just as well occur in a beneficial direction. A combination of ‘anticancer factors’ provided by a food, rather than an isolated food component, may be needed to be biologically effective. In this way the food matrix would be analogous to combination chemotherapy. Undoubtedly the study of specific substances is crucial to furnish information which will ultimately constitute the scientific basis for rationally-guided and precise nutritional advice. Yet it is tantalising to envisage that the rapid expansion of our abilities to alter gene expression in plants may ultimately enable the tailoring of foods to specific human health needs including the chemoprevention of certain cancers.
References
- 1.Weinstein IB. Cancer prevention: recent progress and future opportunities. Cancer Res. 1991;51:5080s–5085s. [PubMed] [Google Scholar]
- 2.Wattenberg LW. Chemoprophylaxis of carcinogenesis: A review. Cancer Res. 1966;26:1520–1526. [PubMed] [Google Scholar]
- 3.Ruddon RW. Cancer Biology. Oxford University Press; 1995. [Google Scholar]
- 4.Wattenberg LW. Chemoprevention of cancer. Cancer Res. 1985;45:1–8. [PubMed] [Google Scholar]
- 5.Kelloff GJ, Boone CW, Crowell JA, et al. Chemopreventive drug development: perspectives and progress. Cancer Epidemiol Biomarkers Prev. 1994;3:85–98. [PubMed] [Google Scholar]
- 6.Greenwald P. Cancer risk factors for selecting cohorts for large-scale chemoprevention trials. J Cell Biochem. 25S:29–36. [PubMed] [Google Scholar]
- 7.The alpha-tocopherol; beta-carotene cancer prevention study group. The effects of vitamin E and beta-carotene on the incidence of lung cancer and other cancers in male smokers. N Engl J Med. 1994;330:1029–1035. doi: 10.1056/NEJM199404143301501. [DOI] [PubMed] [Google Scholar]
- 8.Omenn GS, Goodman GE, Thornquist MD, et al. Effects of a combination of beta-carotene and vitamin A on lung cancer and cardiovascular disease. N Engl J Med. 1996;334:1150–1155. doi: 10.1056/NEJM199605023341802. [DOI] [PubMed] [Google Scholar]
- 9.Albanes D, Heinonen OP, Taylor PR, et al. Alpha-Tocopherol and beta-carotene supplements and lung cancer incidence in the Alpha-Tocopherol, Beta-Carotene Cancer Prevention Study: effects of base line characteristics and study compliance. J Natl Cancer Inst. 1996;88:1560–1570. doi: 10.1093/jnci/88.21.1560. [DOI] [PubMed] [Google Scholar]
- 10.Omenn GS, Goodman GE, Thornquist MD, et al. Risk factors for lung cancer and for intervention effects in CARET, the Beta-Carotene and retinol Efficacy Trial. J Natl Cancer Inst. 1996;88:1550–1559. doi: 10.1093/jnci/88.21.1550. [DOI] [PubMed] [Google Scholar]
- 11.Lee JS, Lippman SM, Hong WK, et al. Determination of biomarkers for intermediate end points in chemoprevention trials. Cancer Res. 1992;52:2707s–2710s. [PubMed] [Google Scholar]
- 12.Lotan R. Retinoids in cancer chemoprevention. FASEB J. 1996;10:1031–1039. doi: 10.1096/fasebj.10.9.8801164. [DOI] [PubMed] [Google Scholar]
- 13.Hong WK, Itri LM. Retinoids and human cancer. In: Sporn MB, Roberts AB, Goodman DS, editors. The retinoids. New York: Raven Press; 1994. pp. 597–658. [Google Scholar]
- 14.Moon RC, Mehta RG, Rao KJVN. Retinoids and cancer in experimental animals. In: Sporn MB, Roberts AB, Goodman DS, editors. The retinoids. New York: Raven Press; 1994. pp. 573–595. [Google Scholar]
- 15.Goodman DS. Vitamin A and retinoids in health and disease. N Engl J Med. 1984;310:1023–1031. doi: 10.1056/NEJM198404193101605. [DOI] [PubMed] [Google Scholar]
- 16.Dowling JE, Wald J. The biological function of vitamin A acid. Proc Natl Acad Sci USA. 1960;46:587–608. doi: 10.1073/pnas.46.5.587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thompson JN, Howell JM, Pitt GAJ. Vitamin A and reproduction in rats. Proc R Soc Lond. 1964;159:510–535. doi: 10.1098/rspb.1964.0017. [DOI] [PubMed] [Google Scholar]
- 18.Wolbach SB, Howe PR. Tissue changes following deprivation of fat soluble A vitamin. J Exp Med. 1925;42:753–777. doi: 10.1084/jem.42.6.753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saffiotti U, Montesano R, Sellakumar AR, Bork SA. Experimental cancer of the lung. Inhibition by vitamin A of the induction of tracheobronchial metaplasia and squamous cell tumor. Cancer. 1967;20:857–864. doi: 10.1002/1097-0142(1967)20:5<857::aid-cncr2820200545>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 20.Peck GL, DiGiovanna JJ. Synthetic retinoids in dermatology. In: Sporn MB, Roberts AB, Goodman DS, editors. The retinoids. New York: Raven Press; 1994. pp. 631–658. [Google Scholar]
- 21.Morriss-Kay G. Retinoids and embryos. In: Degos L, Parkinson DR, editors. Retinoids in Oncology. New York: Springer, Berlin, Heidelberg; 1995. pp. 43–54. [Google Scholar]
- 22.Messina MJ, Persky V, Detchell KD, Barnes S. Soy intake and cancer risk: a review of the in vitro and in vivo data. Nutr Cancer. 1994;21:113–131. doi: 10.1080/01635589409514310. [DOI] [PubMed] [Google Scholar]
- 23.Coward L, Barnes NC, Setchell KDR, Barnes S. Genistein and daidzein, and their beta-glycoside conjugates: antitumor isoflavones in soybean foods of the American and Asian diets. J Agric Food Sci. 1993;41:1961–1967. [Google Scholar]
- 24.Jones AE, Price KR, Fenwick GR. Development and application of a high-performance liquid chromatographic method for the analysis of phytoestrogens. J Sci Food Agric. 1989;46:357–364. [Google Scholar]
- 25.Adlercreutz H, Mazur W. Phyto-oestrogens and Western diseases. Ann Med. 1997;29:95–120. doi: 10.3109/07853899709113696. [DOI] [PubMed] [Google Scholar]
- 26.Barnes S. Effect of genistein on in vitro and in vivo models of cancer. J Nutr. 1995;125:777s–783s. doi: 10.1093/jn/125.3_Suppl.777S. [DOI] [PubMed] [Google Scholar]
- 27.Lamartiniere CA, Moore JB, Brown NM, Thompson R, Hardin MY, Barnes S. Genistein suppresses mammary cancer in rats. Carcinogenesis. 1995;16:2833–2840. doi: 10.1093/carcin/16.11.2833. [DOI] [PubMed] [Google Scholar]
- 28.Govindarajan VS. Turmeric chemistry, technology and quality. CRC Rev Food Sci Nutr. 1980;12:199–301. doi: 10.1080/10408398009527278. [DOI] [PubMed] [Google Scholar]
- 29.Stoner GD, Mukhtar H. Polyphenols as cancer chemopreventive agents. J Cell Biochem. 1995;22:169–180. doi: 10.1002/jcb.240590822. [DOI] [PubMed] [Google Scholar]
- 30.Huang MT, Smart RC, Wong CQ, Conney AH. Inhibitory effect of curcumin, chlorogenic acid and ferulic acid on tumor promotion in mouse skin by 12-O-tetradecanoylphorbol-13-acetate. Cancer Res. 1988;48:5941–5946. [PubMed] [Google Scholar]
- 31.Nagabhushan M, Bhide SV. Curcumin as an inhibitor of cancer. J Am Coll Nutr. 1992;11:192–198. [PubMed] [Google Scholar]
- 32.Mehta RG, Moon RC. Characterization of effective chemopreventive agents in mammary gland in vitro using an initiation-promotion protocol. Anticancer Res. 1991;11:593–596. [PubMed] [Google Scholar]
- 33.Rao CV, Simi B, Reddy BS. Inhibitory effect by dietary curcumin of azoxymethane-induced ornithine decarboxylase, tysosine protein kinase, arachidonic acid metabolism and aberrant crypt foci formation in the rat colon. Carcinogenesis. 1993;14:2219–2225. doi: 10.1093/carcin/14.11.2219. [DOI] [PubMed] [Google Scholar]
- 34.Kensler TW, Trush MA, Guyton KZ. Free radicals as targets for cancer chemoprevention: Prospects and problems. In: Steel VE, Stoner GDM, Boon CW, Kelloff GJ, editors. Cellular and Molecular Targets for Chemoprevention. Boca Raton, FL: CRC Press; 1992. pp. 173–191. [Google Scholar]
- 35.Irani K, Xia Y, Zweier JL, et al. Mitogenic signaling mediated by oxidants in ras-transformed fibroblasts. Science. 1997;275:1649–1652. doi: 10.1126/science.275.5306.1649. [DOI] [PubMed] [Google Scholar]
- 36.Hartman PE, Shankel DM. Antimutagens and anticarcinogens: A survey of putative interceptor molecules. Environ Mol Mutagen. 1990;15:145–182. doi: 10.1002/em.2850150305. [DOI] [PubMed] [Google Scholar]
- 37.Twentyman PR, Fox NE, White DJ. Cyclosporin A and its analogues as modifiers of adriamycin and vincristine resistance in multi-drug resistant human lung cancer cell line. Br J Cancer. 1987;56:55–57. doi: 10.1038/bjc.1987.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marnett LJ. Asprin and the potential role of prostaglandins in colon cancer. Cancer Res. 1992;52:5575–5589. [PubMed] [Google Scholar]
- 39.Zenser TV, Davis BB. Arachidonic acid metablolism. In: Steel VE, Stoner GDM, Boon CW, Kelloff GJ, editors. Cellular and Molecular Targets for Chemoprevention. Boca Raton, FL: CRC Press; pp. 225–243. [Google Scholar]
- 40.Plummer S. M., Hall, M., Faux, S.P. Oxygenation and genotoxicity of fecapentaene-12 are potentiated by prostaglandin H synthase. Carcinogenesis. 1995;16:1023–1028. doi: 10.1093/carcin/16.5.1023. [DOI] [PubMed] [Google Scholar]
- 41.Smith WL. The eicosanoids and their biochemical mechanisms of action. Biochem J. 1989;259:315–324. doi: 10.1042/bj2590315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Earnest DL, Hixson LJ, Finley PR, et al. Arachidonic acid cascade inhibitors in chemoprevention of human colon cancer: Preliminary studies. In: Wattenberg L, Lipkin M, Boone CW, Kelloff GJ, editors. Cancer Chemoprevention. Boca Raton, FL: CRC Press; 1992. pp. 165–180. [Google Scholar]
- 43.Ristimaki A, Honkanen N, Jankala H, Sipponen P, Harkonen M. Expression of cyclooxygenase-2 in human gastric carcinoma. Cancer Res. 1997;57:1276–1280. [PubMed] [Google Scholar]
- 44.Parrett ML, Harris RE, Joarder FS, Ross MS, Clausen KP, Robertson FM. Cyclooxygenase gene expression in human breast cancer. Int J Oncol. 1997;10:530–507. doi: 10.3892/ijo.10.3.503. [DOI] [PubMed] [Google Scholar]
- 45.Oshima M, Dinchuk JE, Kargman SL, et al. Suppression of intestinal polyposis in APC knockout mice by inhibition of cyclooxygenase 2 (COX-2) Cell. 1996;87:803–809. doi: 10.1016/s0092-8674(00)81988-1. [DOI] [PubMed] [Google Scholar]
- 46.Tsujii M, Dubois RN. Alterations in cellular adhesion and apoptosis in epithelial cells overexpressing prostaglandin endoperoxide synthase 2. Cell. 1995;83:493–501. doi: 10.1016/0092-8674(95)90127-2. [DOI] [PubMed] [Google Scholar]
- 47.Thun MJ, Namboodiri MM, Calle EE, Flanders WD, Heath CW. Aspirin use and risk of fatal cancer. Cancer Res. 1993;53:1322–1327. [PubMed] [Google Scholar]
- 48.Boolbol SK, Dannenberg AJ, Chadburn A, et al. Cyclooxygenase-2 overexpression and tumor formation are blocked by sulindac in a murine model of familial adenomatous polyposis. Cancer Res. 1996;56:2556–2560. [PubMed] [Google Scholar]
- 49.Holloway K, Plummer S. Induction of COX2 mRNA and NFkB DNA binding activity by TNF-alpha and fecapentaene-12 is inhibited by curcumin. Eur J Cancer Prev. (in press).
- 50.Mestre JR, Subbaramaiah K, Sacks PG, et al. Retinoids suppress phorbol ester-mediated induction of cyclooxygenase-2. Cancer Res. 1997;57:1081–1085. [PubMed] [Google Scholar]
- 51.Tsai MJ, O'Malley BW. Molecular mechanisms of actions of steroid/thyroid receptor super-family members. Ann Rev Biochem. 1994;63:451–486. doi: 10.1146/annurev.bi.63.070194.002315. [DOI] [PubMed] [Google Scholar]
- 52.Gronemeyer H, Benhamou B, Berry M, et al. Mechanisms of antihormone action. J Steroid Biochem Mol Biol. 1992;41:217–221. doi: 10.1016/0960-0760(92)90347-l. [DOI] [PubMed] [Google Scholar]
- 53.Jordan VC. The strategic use of antiestrogens to control the development and growth of breast cancer. Cancer. 1992;70:977–982. [PubMed] [Google Scholar]
- 54.Martin PM, Horwitz KB, Ryan DS, McGuire WI. Phytoestrogen interaction with estrogen receptors in human breast cancer cells. Endocrinology. 1978;103:1860–1867. doi: 10.1210/endo-103-5-1860. [DOI] [PubMed] [Google Scholar]
- 55.Roberts AB, Sporn Sporn, MB MB. Tranforming growth factor β. Adv Cancer Res. 1988;51:107–145. [PubMed] [Google Scholar]
- 56.Sibrestein GB, Daniel CW. Reversible inhibition of mammary gland growth by tranforming growth factor-β. Science. 1987;237:291–293. doi: 10.1126/science.3474783. [DOI] [PubMed] [Google Scholar]
- 57.Sporn MB, Roberts AB, Glick AB, Luckert PH, Pollard M. Interactions of retinoids and transforming growth factor β in the chemoprevention of cancer. In: Sporn MB, editor. Control of growth factors and prevention of cancer. New York: Springer Verlag; 1992. pp. 37–46. [Google Scholar]
- 58.Russell WE, Coffey RJ, Jr, Ouellette AJ, Moses HL. Type β transforming growth factor reversibly inhibits the early proliferative response to partial hepatectomy in the rat. Proc Natl Acad Sci USA. 1988;85:5126–5130. doi: 10.1073/pnas.85.14.5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Glick AB, McCune BK, Abdulkarem N, et al. Complex regulation of TGFβ expression by retinoic acid in the vitamin A-deficient rat. Development. 1991;111:1081–1086. doi: 10.1242/dev.111.4.1081. [DOI] [PubMed] [Google Scholar]
- 60.Kumar R, Shoemaker AR, Verma AK. Retinoic acid nuclear receptors and tumor promotion: decreased expression of retinoic acid nuclear receptors by the tumor promotor 12-O-tetradecanoylphorbol-13-acetate. Carcinogenesis. 1994;15:701–705. doi: 10.1093/carcin/15.4.701. [DOI] [PubMed] [Google Scholar]
- 61.Xu XC, Sozzi G, Lee JS, et al. Suppression of retinoic acid receptor beta in non-small-cell lung cancer in vivo: Implications for lung cancer development. J Natl Cancer Inst. 1997;89:624–629. doi: 10.1093/jnci/89.9.624. [DOI] [PubMed] [Google Scholar]
- 62.Li JJ, Dong ZG, Dawson MI, Colburn NH. Inhibition of tumor promotor-induced transformation by retinoids that transrepress AP-1 without transactivating retinoic acid response element. Cancer Res. 1996;56:483–489. [PubMed] [Google Scholar]
- 63.Suganuma M, Okabe S, Suoka E, et al. A new process of cancer prevention through inhibition of tumor necrosis factor alpha expression. Cancer Res. 1996;56:3711–3715. [PubMed] [Google Scholar]
- 64.Cooper GM. Oncogenes. Boston: Jones and Bartlett Publishers; 1990. Oncogenes and growth factors; pp. 163–173. chapter 11. [Google Scholar]
- 65.Powis G. Signalling pathways as targets for anticancer drug development. Pharmacol Ther. 1994;62:57–95. doi: 10.1016/0163-7258(94)90005-1. [DOI] [PubMed] [Google Scholar]
- 66.Shin DM, Ro JY, Hong WK, Hittelman WN. Dysregulation of epidermal growth factor receptor expression in premalignant lesion during head and neck tumorigenesis. Cancer Res. 1994;54:3153–3159. [PubMed] [Google Scholar]
- 67.Levitzky A. Signal-transduction therapy. A novel approach to disease management. Eur J Biochem. 1994;226:1–13. doi: 10.1111/j.1432-1033.1994.tb20020.x. [DOI] [PubMed] [Google Scholar]
- 68.Gullick WJ. Prevalence of aberrant expression of the epidermal growth factor receptors in human cancers. Br Med Bull. 1991;47:87–98. doi: 10.1093/oxfordjournals.bmb.a072464. [DOI] [PubMed] [Google Scholar]
- 69.Lee DC, Fenton SE, Berkowitz EA, Hissong MA. Transforming growth factor alpha: expression regulation, and biological activities. Pharmacol Rev. 1995;47:51–85. [PubMed] [Google Scholar]
- 70.Salomon DS, Brandt R, Ciardiello F, Normanno N. Epidermal growth factor-related peptides and their receptors in human malignancies. Crit Rev Onol Hematol. 1995;19:183–232. doi: 10.1016/1040-8428(94)00144-i. [DOI] [PubMed] [Google Scholar]
- 71.Kumar V, Bustin SA, McKay IA. Transforming growth factor alpha. Cell Biol Int. 1995;19:373–388. doi: 10.1006/cbir.1995.1083. [DOI] [PubMed] [Google Scholar]
- 72.Boonstra J, Rijken P, Humbel B, Cremers F, Verkleij A, van Bergen-Henegouwen P. The epidermal growth factor. Cell Biol Int. 1995;19:413–430. doi: 10.1006/cbir.1995.1086. [DOI] [PubMed] [Google Scholar]
- 73.Briscoe J, Guschin D, Muller M. Just another signalling pathway. Curr Biol. 1994;4:1033–1035. doi: 10.1016/s0960-9822(00)00236-0. [DOI] [PubMed] [Google Scholar]
- 74.Akiyama T, Ishida J, Nakagawa S, et al. Genistein, a specific inhibitor of tyrosine-specific protein kinases. J Biol Chem. 1987;262:5592–5595. [PubMed] [Google Scholar]
- 75.Barnes S. Effect of genistein on in vitro and in vivo models of cancer. J Nutr. 1995;125:777s–783s. doi: 10.1093/jn/125.3_Suppl.777S. [DOI] [PubMed] [Google Scholar]
- 76.Peterson TG, Barnes S. Isoflavones inhibit the growth of human prostate cancer cell lines without inhibiting epidermal growth factor receptor autophosphorylation. Prostate. 1993;22:335–345. doi: 10.1002/pros.2990220408. [DOI] [PubMed] [Google Scholar]
- 77.Peterson TG, Barnes S. Genistein inhibits both estrogen and growth factor-stimulated proliferation of human breast cancer cells. Cell Growth Diff. 1996;7:1345–1351. [PubMed] [Google Scholar]
- 78.Kikkawa U, Nishizuka Y. The role of protein kinase C in transmembrane signalling. Ann Rev Cell Biol. 1986;2:149–178. doi: 10.1146/annurev.cb.02.110186.001053. [DOI] [PubMed] [Google Scholar]
- 79.Liu JY, Lin SJ, Lin JK. Inhibitory effects of curcumin on protein kinase C activity induced by 12-O-tetradecanoylphorbol-13-acetate in NIH 3T3 cells. Carcinogenesis. 1993;14:857–861. doi: 10.1093/carcin/14.5.857. [DOI] [PubMed] [Google Scholar]
- 80.Korutla L, Cheung JY, Mendelsohn J, Kumar R. Inhibition of ligand-induced activation of epidermal growth factor receptor tyrosine phosphorylation by curcumin. Carcinogenesis. 1995;16:1741–1745. doi: 10.1093/carcin/16.8.1741. [DOI] [PubMed] [Google Scholar]
- 81.Tseng CP, Verma AK. Inhibition of 12-O-tetradecanoyl-13-phorbol-13-acetate-induced ornithine decarboxylase activity by genistein, a tyrosine kinase inhibitor. Mol Pharmacol. 1996;50:249–257. [PubMed] [Google Scholar]
- 82.Weinstein IB. Strategies for inhibiting multistage carcinogenesis based on signal transduction pathways. Mutat Res. 1988;202:413–420. doi: 10.1016/0027-5107(88)90202-3. [DOI] [PubMed] [Google Scholar]
- 83.Zarbl H, Sukumar S, Arther AV, Martin-Zanca Martin-Zanca, D D, Barbacid M. Direct mutagenesis of Ha-ras-1 oncogenes by N-nitroso-N-methylurea during initiation of mammary carcinogenesis in rats. Nature. 1985;315:382–385. doi: 10.1038/315382a0. [DOI] [PubMed] [Google Scholar]
- 84.Haag JD, Lindstrom MJ, Gould MN. Limonene-induced regression of mammary carcinomas. Cancer Res. 1992;52:4021–4026. [PubMed] [Google Scholar]
- 85.Haag JD, Crowell PL, Gould MN. Enhanced inhibition of protein isoprenylation and tumor growth by perillyl alcohol, an hydroxylated analog of D-limonene. Proc Am Assoc Cancer Res. 1992;33:3135. [Google Scholar]
- 86.Crowell PL, Chang RR, Ren Z, Elson CE, Gould MN. Selective inhibition of isoprenylation of 21–26-KDa proteins by the anticarcinogen d-limonene and its metabolites. J Biol Chem. 1991;266:17679–17685. [PubMed] [Google Scholar]
- 87.Sebti SM, Tkalcevic GT, Jani JP. Lovastatin, a cholestrol biostynthesis inhibitor, inhibits the growth of human H-ras oncogene transformed cells in nude mice. Cancer Commun. 1991;3:141–147. doi: 10.3727/095535491820873371. [DOI] [PubMed] [Google Scholar]
- 88.Nolan RD, Danilowicz RM, Eling TE. Role of arachidonic acid metabolism in the mitogenic response of BALB/c 3T3 fibroblasts to epidermal growth factor. Mol Pharmacol. 1988;33:650–656. [PubMed] [Google Scholar]
- 89.Handler JA, Danilowicz RM, Eling TE. Mitogenic signaling by epidermal growth factor (EGF), but not platelet-derived growth factor, requires arachidonic acid metabolism in BALB/c3T3 cells. J Biol Chem. 1990;265:3669–3673. [PubMed] [Google Scholar]
- 90.Huang TS, Lee SC, Lin JK. Suppression of c-Jun/AP-1 activation by an inhibitor of tumor promotion in mouse fibroblast cells. Proc Natl Acad Sci USA. 1991;88:5292–5296. doi: 10.1073/pnas.88.12.5292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schultze-Mosgau M, Gant TW, Gescher A. Regulation of c-fos transcription by chemopreventive isoflavonoids and lignans in MDA-MB-468 breast cancer cells. Int J Cancer. (in press). [DOI] [PubMed]
- 92.Pezzuto JM. Plant-derived anticancer agents. Biochem Pharmacol. 1997;53:121–133. doi: 10.1016/s0006-2952(96)00654-5. [DOI] [PubMed] [Google Scholar]
- 93.Sharma S, Stutzman JD, Kelloff GJ, Steele VE. Screening of potential chemopreventive agents using biochemical markers of carcinogenesis. Cancer Res. 1994;54:5848–5855. [PubMed] [Google Scholar]
- 94.Gerhäuser C, Mar W, Lee SK, et al. Rotenoids mediate potent cancer chemopreventive activity through transcriptional regulation of ornithine decarboxylase. Nature Med. 1995;1:260–266. doi: 10.1038/nm0395-260. [DOI] [PubMed] [Google Scholar]
- 95.Jang M, Cai L, Udeani GO, et al. Cancer chemopreventive activity of resveratrol, a natural product derived from grapes. Science. 1997;275:218–220. doi: 10.1126/science.275.5297.218. [DOI] [PubMed] [Google Scholar]
- 96.Goodman GE. The clinical evaluation of cancer chemoprevention agents: defining and contrasting phase I, II, and II objectives. Cancer Res. 1992;52:2752–2757. [PubMed] [Google Scholar]
- 97.J Cell Biol. 1996;25(Suppl.):1–206. [Google Scholar]
- 98.Soto AM, Fernandez MF, Luizzi MF, Oles Karasko AS, Sonnenschein C. Developing a marker of exposure to xenoestrogen mixtures in human serum. Environ Health Perspect. 1997;105:647–654. doi: 10.1289/ehp.97105s3647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Macara IG. Oncogenes and cellular signal transduction. Physiol Rev. 1989;69:797–820. doi: 10.1152/physrev.1989.69.3.797. [DOI] [PubMed] [Google Scholar]