

Abstract
Aims
A radioreceptor assay has been developed for α1-adrenoceptor subtypes and applied to a pharmacokinetic analysis of tamsulosin and terazosin.
Methods
Young, male, healthy volunteers received 0.4 mg tamsulosin (as Omnic® modified release capsules) or 5 mg terazosin (as Flotrin® tablets) in a randomized, cross-over design. Before and after 1, 3, 5, 7, 10 and 23.5 h plasma was analyzed by radioreceptor assay using cloned human α1A-, α1B- and α1D-adrenoceptors and specific h.p.l.c. analysis.
Results
Following ingestion of tamsulosin median peak plasma levels of 16 ng ml−1 were reached after 5 h and declined to 2 ng ml−1 at 23.5 h. The time course in the radioreceptor assay was similar, and at most time points binding to α1A-adrenoceptors was significantly greater than to α1B- and α1D-adrenoceptors. Following ingestion of terazosin median peak plasma levels of 91 ng ml−1 were reached after 1 h and declined to 11 ng ml−1 at 23.5 h. In the radioreceptor assay binding also peaked at 1 h and declined thereafter, but even after 23.5 h considerable binding activity remained detectable at all three subtypes. At most time points binding to the α1A- and α1D-adrenoceptor was significantly greater than to the α1B-adrenoceptor.
Conclusions
We conclude that α1-adrenoceptor antagonist pharmacokinetics can be monitored by radioreceptor assays in a subtype-selective manner. Tamsulosin and terazosin exhibit subtype selective receptor binding ex vivo. The discordance between terazosin blood levels as determined by h.p.l.c. and radioreceptor assay at late time points indicates the possible involvement of metabolites in in vivo terazosin effects.
Keywords: radioreceptor assay, tamsulosin, terazosin, α1-adrenoceptor subtypes
Introduction
α1-Adrenoceptor antagonists have long been used in the treatment of arterial hypertension [1, 2]. Recently, they have also been introduced for the symptomatic treatment of benign prostatic hyperplasia [3]. Receptor cloning and pharmacological analysis have identified the existence of at least three α1-adrenoceptor subtypes which are designated α1A (formerly α1c), α1B and α1D (formerly α1a/d) [4]. Since human prostate predominantly expresses α1A-adrenoceptors at the mRNA [5] and protein level [6], it has been postulated that selective binding to α1A-adrenoceptors may be sufficient to yield full therapeutic efficacy in benign prostatic hyperplasia with perhaps less side effects related to vasodilation [7]. On the other hand, it has been reported that some α1A-selective antagonists only poorly inhibit contraction of human prostate in vitro; it was proposed that an additional subtype designated α1L may be involved in prostatic contraction [8, 9]. More recent studies, however, have found that several compounds which have higher affinity for α1A- than for the putative α1L-adrenoceptors in standard binding assays using membrane preparations at room temperature also have low affinity for α1A-adrenoceptors when assayed in intact cells at 37° C; they have suggested that the α1L subtype may be a conformational state of the α1A-adrenoceptor [10]. Thus, the overall evidence suggests that indeed the α1A-adrenoceptor may be the main mediator of human prostatic contraction.
Most clinically used α1-adrenoceptor antagonists, e.g. terazosin, were originally developed for the treatment of arterial hypertension and do not discriminate α1-adrenoceptor subtypes [6, 11–14]. In contrast, the novel antagonist, tamsulosin, was specifically developed for the symptomatic treatment of benign prostatic hyperplasia and has about 15-fold selectivity for α1A- over α1B-adrenoceptors with intermediate affinity for α1D-adrenoceptors [6, 8, 13–16]. Interestingly placebo-controlled clinical trials with terazosin [17–19] have reported larger incidences of unwanted side effects than trials with tamsulosin [20, 21] but a direct comparison has not been performed.
The present study was designed to gain further insight into a potential role of α1-adrenoceptor subtype selectivity in the action of tamsulosin and terazosin. This was done by a pharmacokinetic analysis of receptor binding following a single, oral dose of tamsulosin (0.4 mg) and terazosin (5 mg) in a placebo-controlled, single-blind, randomized, three-way cross-over study. For this purpose we have developed a new radioreceptor assay [22] using cloned human α1-adrenoceptor subtypes stably expressed in rat-1 fibroblasts [23]. With this assay we have assessed binding to each of the three subtypes at 1, 3, 5, 7, 10 and 23.5 h following drug intake. Specific analysis of tamsulosin and terazosin plasma concentrations by specific h.p.l.c. analysis was performed in comparison.
Methods
Subjects
The study protocol was approved by the ethics committee at the University of Essen Medical School. Ten healthy male subjects (median age: 26.5 years, range: 22–36 years) participated after having given informed written consent. Each subject completed 3 study days during which they received in a single-blinded, randomized cross-over manner 1 tablet of 5 mg terazosin (purchased as Flotrin® from a German pharmacy), 1 capsule of 0.4 mg tamsulosin modified release formulation (Omnic®, provided by Yamanouchi Europe B.V., Leiderdorp, Netherlands), or 1 capsule of placebo matching the tamsulosin capsule. Study days were at least 7 days apart to secure sufficient drug washout.
On each study day the subjects reported to the laboratory at 07.00 h after an overnight fast. An indwelling catheter was placed into a forearm vein for blood withdrawals. The subjects remained in the supine position until after the last blood withdrawal 24 h after drug ingestion. Blood samples were taken 2 h before and 1, 3, 5, 7, 10 and 23.5 h after drug intake. The blood samples were collected into EDTA-coated tubes to prevent coagulation. They were stored on ice immediately. Plasma samples were generated by centrifugation and stored at −20° C until analysis, i.e. up to 4 months. Each blood sample was used for h.p.l.c. and radioreceptor analysis. A light snack was allowed after the 7 h blood withdrawal and a pizza or pasta dish after the 10 h withdrawal. While no other food was given until after the last blood withdrawal, drinking of water was allowed ad libitum.
H.p.l.c. analysis
H.p.l.c. analysis for tamsulosin content was performed by the biopharmaceutical department of Yamanouchi Europe B.V. (Leiderdorp, Netherlands). The plasma samples were analysed by reversed phase h.p.l.c. with fluorescence detection. Tamsulosin and the internal standard AB289 were extracted from plasma by liquid-liquid extraction. The extracts were redissolved into the mobile phase (1.85 g KH2PO4, 0.14 g Na2HPO4, 254 g H2O, 56 g acetonitrile, 8 ml ethyl acetate) and injected on a 125×4 mm C18 column. Detection was performed fluorimetrically at an excitation wavelength of 275 nm with emission being read at 325 nm. This method is suitable for quantification of tamsulosin in human plasma between 0.5 and 50 ng ml−1.
H.p.l.c. analysis for terazosin content was performed by the National Poisons Unit, Guy's and St Thomas’ NHS Trust (London, UK). The samples were analysed by h.p.l.c. with fluorescence detection. The internal standard solution, aqueous dimethothiazine (5 mg l−1, 20 μl), NaOH solution (4 m, 50 μl) and saturated NaCl solution (100 μl) were added to plasma (50 μl). Terazosin and the internal standard were extracted into methyl tertiary-butyl ether (200 μl) by mixing for 30 s and centrifugation at 11 000 rev min−1 for 4 min. A portion (40 μl) of the extract was injected onto the h.p.l.c. column (mobile phase: 40 mm ammonium perchlorate in methanol:deionised water (9+1) adjusted to pH 6.8 with 1% (v/v) methanolic perchloric acid). Detection was by fluorescence monitoring with excitation at 250 nm. The limit of accurate measurement of the assay was 5 ng ml−1.
Radioreceptor assays
Radioreceptor assays were performed using membrane preparations from rat-1 fibroblasts which had been transfected with cDNAs encoding the human α1A-, α1B- or α1D-adrenoceptors [23]. These cell lines had kindly been provided by Dr Geoff Johnston at Pfizer Central Research (Sandwich, Kent, UK). They were cultured in Dulbecco's modified Eagle's medium supplemented with 4.5 g l−1 glucose, 1 mm l-glutamine and 5% heat-inactivated fetal calf serum. A membrane preparation from these cells was prepared as previously described [24].
Radioligand binding assays were performed in a total volume of 300 μl consisting of 200 μl plasma sample and 100 μl buffer (50 mm Tris, 0.5 mm EDTA, pH 7.5) including [3H]-prazosin and aliquots of the membrane preparation from the transfected rat-1 cells (≈25–35, 3–6 and 40–80 μg protein for α1A-, α1B- and α1D-adrenoceptors, respectively, depending on the respective receptor expression densities). The mixtures were incubated for 60 min at 25° C. Incubations were terminated by rapid vacuum filtration over Whatman GF/C filters using a Brandell cell harvester. Non-specific binding was defined as binding in the presence of 50 μm phentolamine. Samples from each time point of the tamsulosin and terazosin study days were assayed in quadruplicate in competition binding assays. Samples from various time points of the placebo study day were pooled to perform a [3H]-prazosin saturation binding experiment using eight radioligand concentrations; the results of this saturation experiment were used to calculate apparent Kd values for [3H]-prazosin for the plasma of each subject. For each α1-adrenoceptor subtype and subject samples from the placebo day (saturation binding) and the tamsulosin and terazosin day (competition binding) were run side-by-side in the same assay. While interference by endogenously released catecholamines cannot fully be excluded, this is quite unlikely because the subjects remained resting in the supine position throughout the study and because the drugs had no major effects on resting supine blood pressure.
Data analysis
Analysis of the radioreceptor assay data was performed according to procedures published for a β-adrenoceptor subtype radioreceptor assay [22]. Active drug concentration i was calculated as multiples of apparent affinity (Ki) equivalents at the α1-adrenoceptor subtypes, i.e. as ‘i/Ki’ according to equation [1]
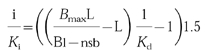 |
In this equation L is the concentration of the radioligand [3H]-prazosin, Kd is the apparent affinity of the radioligand when assayed in the presence of plasma as determined for each subject and α1-adrenoceptor subtype in the saturation binding experiments, Bl is the amount of the radioligand bound in the presence of plasma at the radioligand concentration L, and nsb is the amount of non-specific binding in the presence of plasma at the radioligand concentration L; the factor 1.5 is introduced to correct for dilution of the 200 μl sample in the 300 μl assay volume. Similar data were obtained when the pooled Kd of all subjects (α1A: 4349 [3263, 7722] pm, α1B: 335 [266, 394] pm, α1D: 698 [391, 858] pm; n=10 each for median values with upper and lower quartile in squared brackets) rather than the individually determined Kd values were used in the calculations (data not shown). Bmax is calculated individually for each data set based upon the law of mass action according to equation [2]
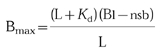 |
Analysis of saturation binding experiments was performed by fitting rectangular hyperbolic functions to the experimental data using non-linear regression analysis with the Inplot programme (Graphpad Software, San Diego, CA). Analysis of the competition binding data was performed with a spreadsheet to implement the above function using the Microsoft Excel programme.
All data are given as median with upper and lower quartiles of 10 subjects. Statistical significance of differences in drug binding to the α1-adrenoceptor subtypes was assessed by Wilcoxon's signed rank test with P<0.05 being considered significant.
Results
H.p.l.c. analysis
Median plasma concentrations of tamsulosin peaked 5 h following drug intake at 16.1 (12.4; 17.4) ng ml−1; thereafter they gradually declined to 2.1 (1.9; 2.8) ng ml−1, i.e. 13% of peak values after 23.5 h (Figure 1). Plasma concentrations of terazosin had peaked 1 h following drug intake and median Cmax was 91 (71; 97) ng ml−1 declining gradually to 11 (10; 14) ng ml−1, i.e. 12% of peak values, after 23.5 h (Figure 2).
Figure 1.
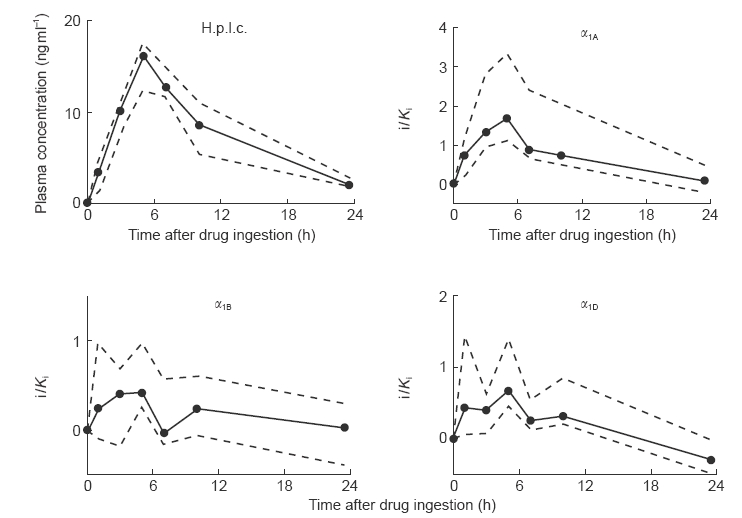
Pharmacokinetic profile of a single oral dose of tamsulosin (0.4 mg) in h.p.l.c. and radioreceptor analysis. Ten young, healthy, male subjects received a single oral dose of 0.4 mg tamsulosin. Before and 1, 3, 5, 7, 10 and 23.5 h after drug intake a blood sample was taken. Plasma derived from those samples was analyzed by h.p.l.c. and used in the radioreceptor assay. Drug levels in the radioreceptor assay are expressed as multiples of the concentration which occupies 50% of the human α1A-, α1B- or α1D-adrenoceptor (i/Ki) as calculated according to equation (1). Data are median values from 10 subjects (filled circles) with upper and lower quartiles (dashed lines).
Figure 2.
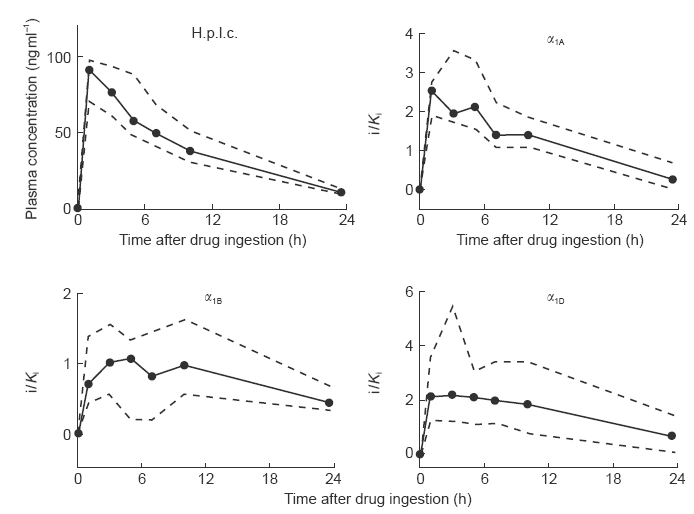
Pharmacokinetic profile of a single oral dose of terazosin (5 mg) in h.p.l.c. and radioreceptor analysis. Ten young, healthy, male subjects received a single oral dose of 5 mg terazosin. Before and 1, 3, 5, 7, 10 and 23.5 h after drug intake a blood sample was taken. Plasma derived from those samples was analyzed by h.p.l.c. and used in the radioreceptor assay. Drug levels are expressed as multiples of the concentration which occupies 50% of the human α1A-, α1B- or α1D-adrenoceptor (i/Ki) as calculated according to equation (1). Data are median values from 10 subjects (filled circles) with upper and lower quartiles (dashed lines).
Ex vivo radioreceptor assay
The median intraassay coefficient of variation of the binding assay was calculated from the quadruplicate determination of the −2 h time point in each experiment and was 4.3% (2.7; 5.8), 3.9% (2.9; 4.7) and 5.9% (4.0; 7.6) for the assay with α1A-, α1B- and α1D-adrenoceptors, respectively (n=24 each). Our study design did not allow a formal calculation of the interassay coefficient of variation of the radioreceptor assay. However, the interassay variance can be estimated from the replicate determination of the −2 h time point from the tamsulosin and terazosin study days; the mean deviation of each replicate from the mean of both measurements was expressed as percentage of the mean and was 5.3% (4.1; 8.9), 3.0% (1.0; 5.8) and 3.2% (2.0; 5.4) for the assay with α1A-, α1B-and α1D-adrenoceptors, respectively (n=12 each). The intersubject coefficient of variation was determined at the time of peak plasma concentrations of parent compound, i.e. after 5 h for tamsulosin and after 1 h for terazosin. For both compounds the median intersubject coefficient of variation was smaller in the h.p.l.c. assay (25% and 35%, respectively) than in the radioreceptor assays for α1A-adrenoceptors (61% and 42%, respectively), α1B-adrenoceptors (104% and 74%, respectively) or α1D-adrenoceptors (131% and 109%, respectively) when assayed on parallel samples. Similarly, the interquartile ranges in the radioreceptor assays for all three subtypes were consistently greater than those for the h.p.l.c. measurements (Figures 1–2). Thus, the radioreceptor assay appears to yield greater data scatter than the specific h.p.l.c. methods.
Following tamsulosin ingestion, binding to α1A-, α1B-and α1D-adrenoceptors in the radioreceptor assay peaked after 5 h and was 1.70 (1.10; 3.33), 0.43 (0.26; 0.97) and 0.69 (0.46; 1.40) times Ki, respectively (Figure 1). From there blood levels declined gradually and were no longer significantly different from 0 after 23.5 h for all three subtypes (Figure 1). At most time points binding to α1A-adrenoceptors was significantly greater than that to α1B-adrenoceptors, and after 5 and 7 h also significantly greater than binding to α1D-adrenoceptors (Figure 3).
Figure 3.
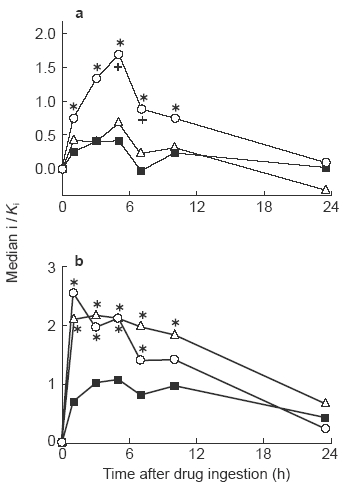
Analysis of α1-adrenoceptor subtype-selectivity of plasma obtained following tamsulosin (0.4 mg) and terazosin (5 mg) intake. Data for tamsulosin (a) and terazosin (b) are taken from Figures 1 and 2, respectively, and replotted to allow a direct comparison of plasma binding to the α1-adrenoceptor subtypes (○α1A; ▪α1B; ▵α1D). * and +: P<0.05 vsα1B- and α1-adrenoceptors, respectively, in a Wilcoxon's signed rank test.
The behaviour of terazosin in the radioreceptor assay was more complex. Binding to the α1A-adrenoceptor (similar to the h.p.l.c. data) peaked after 1 h and was 2.56 (1.93; 2.75) times Ki; from there it declined gradually to 0.27 (0.03; 0.71) times Ki after 23.5 h (Figure 2). Binding to the α1B- and α1D- adrenoceptors did not exhibit a clear time-dependency between 1 and 10 h after terazosin intake. At the 1 h time point binding to the α1B- and α1D-adrenoceptor was 0.70 (0.42; 1.37) and 2.14 (1.26; 3.57) times Ki, respectively. Even after 23.5 h considerable binding was maintained, i.e. 0.45 (0.34; 0.69) and 0.70 (0.09; 1.49) at the α1B-and and α1D-adrenoceptor, respectively (Figure 2), which corresponds to 64% and 33% of peak values, respectively. Binding to α1A- and α1D-adrenoceptors at most time points was significantly greater than that to α1B-adrenoceptors (Figure 3).
Discussion
Classical pharmacokinetic analysis is based on quantification of drug levels in blood or plasma by specific h.p.l.c. or antibody-based techniques, e.g. radioimmunoassay. Using these techniques, the pharmacokinetic profiles of tamsulosin and terazosin have been documented. Thus, tamsulosin in its modified release formulation reaches its maximum plasma concentration at 5–6 h and has a half-life (t1/2) of 11–13 h [25]. While terazosin similarly has a t1/2 of 12 h, it is absorbed more quickly and reaches its maximum concentrations after 1–2 h [25, 26]. In the present study maximum plasma concentrations for tamsulosin and terazosin in plasma occured at time points which are consistent with the previously reported values. Reliable formal calculations of plasma half-life and tmax values were not feasible from the present data, particularly for tamsulosin, since the number and spacing of time points in the present study were not designed for such analysis. Nevertheless our h.p.l.c. data are compatible with the above estimates of plasma half-life values for tamsulosin and terazosin.
While such specific measurements yield sensitive and reliable estimates of drug levels in blood or plasma, they also have several disadvantages. Firstly, measurement of drug plasma levels does not account for a possible contribution of drug metabolites to the observed effects, particularly when such metabolites have not yet been fully identified. Secondly, active drug levels are frequently lower than plasma concentrations due to plasma protein binding, which may be different for a parent compound and its metabolites. Thirdly, drug metabolites may have a pharmacodynamic profile which is distinct from that of their parent compound, e.g. with regard to receptor subtype affinity and specificity. These disadvantages become important when a comparison of pharmacokinetic and pharmacodynamic parameters is intended, particularly when subtype-selective drugs are involved.
To circumvent these problems radioreceptor assays have been introduced into the quantitative analysis of drug effects in man [22]. They determine the ability of plasma of a drug-treated subject to compete for radioligand binding to the receptor of interest relative to the ability of plasma from the non-treated subject. This technique is effectively independent of knowledge of the chemical identity of drug metabolites and evaluates the parent compound and all of its metabolites according to their relative contribution to receptor binding. The presence of plasma in the radioreceptor assay automatically corrects for plasma protein binding. To secure autocorrection for plasma protein binding, it is key that a major fraction of the assay volume is plasma in order to maintain plasma protein binding and not to dilute the drugs present in the sample too much. In the present study this was achieved using 200 μl of plasma in a total assay volume of 300 μl. When [3H]-prazosin saturation binding experiments were performed in the presence of plasma, the apparent Kd values were higher than those typically observed at cloned subtypes [8, 24]. While this may be partially explained by the known plasma protein binding of prazosin, it was noted that the apparent affinity at the α1A-adrenoceptor was much lower than with the other subtypes. While we have no good explanation for this aberrant behaviour of the α1A-adrenoceptor in the presence of plasma, it should be noted that the shift from membrane binding at 25° C to whole cell binding at 37° C also reduces the apparent affinity of [3H]-prazosin at α1A- but not at α1B- or α1D-adrenoceptors [10].
Radioreceptor assay techniques have originally been used for studies on β-adrenoceptor antagonists [22]. Thereafter, they have also been applied to studies of muscarinic acetylcholine receptors [27] and α1-adrenoceptors [28]. When the assay is carried out in parallel on subtypes of a given receptor, e.g. on β1- and β2-adrenoceptors [22, 29] or M1 and M2 muscarinic acetylcholine receptors [27], the assay allows a receptor subtype-specific analysis of receptor binding. A simultaneous radioreceptor assay for subtypes of β-adrenoceptors [29] or muscarinic acetylcholine receptors [27] has allowed to assign distinct functions to a specific subtype in vivo in man.
In a radioreceptor assay it is possible to quantitate blood concentrations relative to known standards, which have also been evaluated in the presence of plasma [28]. However, a quantitative analysis of this type implies that drug metabolites behave very similar to the parent compound. An alternative method of analysis of radioreceptor assay data has been developed by Wellstein et al. [22]. This procedure does not require knowledge of the absolute affinities of all chemical entities participating in receptor occupancy. Therefore, drug concentrations are not given in molar units but rather as multiples of the concentration occupying 50% of the receptor, i.e. i/Ki equivalents. This procedure was used in the present study. Thus, the aim of the present study was to establish a radioreceptor assay for human α1-adrenoceptor subtypes and use it to test whether tamsulosin and terazosin behave as subtype-selective drugs in man in vivo.
The time course of tamsulosin levels following single oral administration as determined in the radioreceptor assay with α1A-adrenoceptors was similar to that observed by h.p.l.c. analysis of the parent compound. Thus, maximum tamsulosin concentrations in the h.p.l.c. assay and maximum binding to the α1A-adrenoceptor were seen 5 h following drug intake and from there plasma concentrations gradually declined to levels of 13% (h.p.l.c.) or even less (radioreceptor assays) of the peak concentrations after 23.5 h. The time dependency of binding to the α1B- and α1D-adrenoceptor was less clear than for the α1A-adrenoceptor. The most likely explanation of this difference is the smaller signal/noise ratios since the binding to the α1B- and α1D-adrenoceptors was less than with α1A-adrenoceptors at most time points. The order of binding in the radioreceptor assay, i.e. α1A>α1D α1B, corresponds well to various previous in vitro studies demonstrating an order of potency for tamsulosin of α1A≥α1D>α1B [6, 8, 13–16]. Taken together these data validate our approach of the radioreceptor assay.
The behaviour of terazosin in the radioreceptor assay was more complex. Binding to the α1A-adrenoceptor and terazosin concentrations in the h.p.l.c. analysis peaked after 1 h and declined to 11% and 12%, respectively, of peak levels after 23.5 h. In contrast binding to α1D- and α1B-adrenoceptors did not exhibit a clear peak or a clear time dependency between 1 and 10 h following terazosin intake. Moreover, after 23.5 h median binding to α1B- and α1D-adrenoceptors was still at 64% and 33%, respectively, of the median 1 h values while concentrations of parent compound in the h.p.l.c. analysis were only 12% of 1 h values. In contrast to the situation with tamsulosin, this cannot be explained by small signal/noise ratios. Moreover, our data suggest that terazosin may be somewhat selective for α1A- and α1D-adrenoceptors relative to α1B-adrenoceptors in vivo whereas terazosin has repeatedly been demonstrated to have similar affinity for all α1-adrenoceptor subtypes in vitro [6, 11–14]. The considerable binding activity in plasma of terazosin-treated subjects after 23.5 h and the apparent subtype-selectivity in vivo indicate the possibility that metabolites may contribute to α1-adrenoceptor binding activity in terazosin-treated subjects, particularly at late time points. Indeed terazosin has been demonstrated to undergo extensive metabolism in humans [26]. The α1-adrenoceptor subtype-selectivity of terazosin metabolites is not known. However, it is noteworthy that two of the three major terazosin metabolites are 6-O- and 7-O-demethyl-terazosin [26]. Demethylation of the corresponding moiety in the tamsulosin molecule (tamsulosin metabolite M4) interestingly yields compounds with selectivity for α1D- and α1A- relative to α1B-adrenoceptors [16], similar to what we observed in vivo with terazosin. Evaluation of this possibility appears intriguing, but unfortunately the terazosin metabolites were not available to us for investigation. Thus, confirmation of an involvement of metabolites in functional in vivo effects of terazosin has to await further studies.
In contrast after 23.5 h, when tamsulosin levels in the h.p.l.c. assay had declined to 13% of peak values, α1-adrenoceptor binding activity in plasma of tamsulosin-treated subjects was no longer significantly different from 0 with all three subtypes; moreover, the observed profile of α1-adrenoceptor subtype-selectivity ex vivo was similar to that reported in vitro. These data support the view that metabolites do not play a major role in the therapeutic effects of tamsulosin. Thus, tamsulosin appears to undergo less metabolism in man [30] than in rats or dogs [31], and the metabolites being formed appear to be rapidly eliminated from plasma and thus contribute only little to overall plasma levels [30]. Moreover, the limited contribution of metabolites is not likely to alter the qualitative pharmacological profile of tamsulosin in vivo since we have previously demonstrated that most of the tamsulosin metabolites which do occur have an affinity and α1-adrenoceptor subtype-selectivity similar to tamsulosin itself [16].
In summary our study demonstrates that the radioreceptor assay technique can be applied to human α1-adrenoceptor subtypes. Our data with tamsulosin suggest that the radioreceptor assay technique yields data which are compatible with the pharmacokinetic profile according to h.p.l.c. analysis and with known in vitro data regarding subtype-selectivity. Our data with terazosin, which does not discriminate α1-adrenoceptor subtypes in vitro, surprisingly indicate a possible in vivoα1-adrenoceptor subtype-selectivity. This unexpected feature and the discrepancies between the pharmacokinetic profile in the radioreceptor assay and of the parent compound in the h.p.l.c. assay indicate that metabolites may contribute to the in vivo profile of terazosin with regard to duration of action and α1-adrenoceptor subtype-selectivity. Thus, radioreceptor assays based on plasma may not fully reflect receptor occupancies at tissue sites of interest, but seem to provide considerable additional information relative to classical h.p.l.c. analysis. The application of radioreceptor assays based on α1-adrenoceptor subtypes may allow the association of distinct physiological effects with specific subtypes. However, such applications may be limited by the fact that data scatter is larger in the radioreceptor assay than with h.p.l.c. analysis.
Acknowledgments
This study was funded in part by a grant from Boehringer Ingelheim (Ingelheim, Germany). We thank Dr H. Schumacher (Department of Biometrics, Boehringer Ingelheim) for help with the statistical analysis, Dr J. Schloos (Beiersdorf-Lilly GmbH, Hamburg, Germany) for advice on the principles of the radioreceptor assay, Dr G. Johnston (Pfizer, Sandwich, Kent, UK) for providing the cell lines, and Dr R. J. Flanagan (National Poisons Unit, London, UK) and Dr W. J. J. Krauwinkel (Yamanouchi Europe, Leiderdorp, The Netherlands) for performing the h.p.l.c. analysis.
References
- 1.Schäfers RF, Reid JL. Alpha Blockers. In: Kaplan NM, Brenner BM, Laragh JH, editors. New Therapeutic Strategies in Hypertension. New York: Raven Press; 1989. pp. 51–69. [Google Scholar]
- 2.Khoury AF, Kaplan NM. α-Blocker therapy of hypertension. An unfulfilled promise. J Am Med Ass. 1991;266:394–398. [PubMed] [Google Scholar]
- 3.Oesterling JE. Benign prostatic hyperplasia. Medical and minimally invasive treatment options. New Engl J Med. 1995;332:99–109. doi: 10.1056/NEJM199501123320207. [DOI] [PubMed] [Google Scholar]
- 4.Michel MC, Kenny BA, Schwinn DA. Classification of α1-adrenoceptor subtypes. Naunyn-Schmiedeberg's Arch Pharmacol. 1995;352:1–10. doi: 10.1007/BF00169183. [DOI] [PubMed] [Google Scholar]
- 5.Price DT, Schwinn DA, Lomasney JW, Allen LF, Caron MG, Lefkowitz RJ. Identification, quantification, and localization of mRNA for three distinct alpha1 adrenergic receptor subtypes in human prostate. J Urol. 1993;150:546–551. doi: 10.1016/s0022-5347(17)35544-1. [DOI] [PubMed] [Google Scholar]
- 6.Michel MC, Grübbel B, Taguchi K, Verfürth F, Otto T, Kröpfl D. Drugs for treatment of benign prostatic hyperplasia: affinity comparison at cloned α1-adrenoceptor subtypes and in human prostate. J Auton Pharmacol. 1996;16:21–28. doi: 10.1111/j.1474-8673.1996.tb00352.x. [DOI] [PubMed] [Google Scholar]
- 7.Chapple CR. α-Adrenergic blocking drugs in bladder outflow obstruction: what potential has α1-adrenoceptor selectivity? Br J Urol. 1995;76(Suppl. 1):47–55. [PubMed] [Google Scholar]
- 8.Ford APDW, Arredondo NF, Blue DR, et al. RS-17053 (N-[2-cyclopropylmethoxy-phenoxy)ethyl]-5-chloro-α,α-dimethyl-1H-indole-3-ethanamine hydrochloride), a selective α1A-adrenoceptor antagonist, displays low affinity for functional α1A-adrenoceptors in human prostate: implications for adrenoceptor classification. Mol Pharmacol. 1996;49:209–215. [PubMed] [Google Scholar]
- 9.Chess-Williams R, Chapple CR, Verfürth F, Noble AJ, Couldwell CJ, Michel MC. The effects of SB 216469, an antagonist which discriminates between the α1A-adrenoceptor and the human prostatic α1-adrenoceptor. Br J Pharmacol. 1996;119:1093–1100. doi: 10.1111/j.1476-5381.1996.tb16009.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Ford APDW, Daniels DV, Chang DJ, et al. Pharmacological pleiotropism of the human recombinant α1A-adrenoceptor: implications for α1-adrenoceptor classification. Br J Pharmacol. 1997;121:1127–1135. doi: 10.1038/sj.bjp.0701207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Goetz AS, Lutz MW, Rimele TJ, Saussy DLJ. Characterization of alpha-1 adrenoceptor subtypes in human and canine prostate membranes. J Pharmacol Exp Ther. 1994;271:1228–1233. [PubMed] [Google Scholar]
- 12.Weinberg DH, Trivedi P, Tan CP, et al. Cloning, expression and characterization of human α adrenergic receptors α1A, α1B and α1C. Biochem Biophys Res Commun. 1994;201:1296–1304. doi: 10.1006/bbrc.1994.1845. [DOI] [PubMed] [Google Scholar]
- 13.Foglar R, Shibata K, Horie K, Hirasawa A, Tsujimoto G. Use of recombinant α1-adrenoceptors to characterize subtype selectivity of drugs for the treatment of prostatic hypertrophy. Eur J Pharmacol. 1995;288:201–207. doi: 10.1016/0922-4106(95)90195-7. [DOI] [PubMed] [Google Scholar]
- 14.Testa R, Guarneri L, Taddei C, et al. Functional antagonistic activity of Rec 15/2739, a novel alpha-1 antagonist selective for the lower urinary tract, on noradrenaline-induced contraction of human prostate and mesenteric artery. J Pharmacol Exp Ther. 1996;277:1237–1246. [PubMed] [Google Scholar]
- 15.Shibata K, Foglar R, Horie K, et al. KMD-3213, a novel, potent, α1A-adrenoceptor-selective antagonist: characterization using recombinant human α1-adrenoceptors and native tissues. Mol Pharmacol. 1995;48:250–258. [PubMed] [Google Scholar]
- 16.Taguchi K, Saitoh M, Sato S, Asano M, Michel MC. Effects of tamsulosin metabolites at alpha-1 adrenoceptor subtypes. J Pharmacol Exp Ther. 1997;280:1–5. [PubMed] [Google Scholar]
- 17.Brawer MK, Adams G, Epstein H. Terazosin in the treatment of benign prostatic hyperplasia. Arch Fam Med. 1993;2:929–935. doi: 10.1001/archfami.2.9.929. [DOI] [PubMed] [Google Scholar]
- 18.Lepor H, Williford WO, Barry MJ, et al. The efficacy of terazosin, finasteride, or both in benign prostatic hyperplasia. New Engl J Med. 1996;335:533–539. doi: 10.1056/NEJM199608223350801. [DOI] [PubMed] [Google Scholar]
- 19.Roehrborn CG, Oesterling JE, Auerbach S, et al. The Hytrin community assessment trial study: a one-year study of terazosin versus placebo in the treatment of patients with symptomatic benign prostatic hyperplasia (BPH) Urology. 1996;47:159–168. doi: 10.1016/s0090-4295(99)80409-9. [DOI] [PubMed] [Google Scholar]
- 20.Lepor H. Clinical evaluation of tamsulosin, a prostate selective alpha 1c antagonist. J Urol. 1995;153(Suppl. 274A) [Google Scholar]
- 21.Chapple CR, Wyndaele JJ, Nordling J, Boeminghaus F, Ypma AFGVM, Abrams P. Tamsulosin, the first prostate-selective α1A-adrenoceptor antagonist. A meta-analysis of two randomized, placebo-controlled multicentre studies in patients with benign prostatic obstruction (symptomatic BPH) Eur Urol. 1996;29:155–167. [PubMed] [Google Scholar]
- 22.Wellstein A, Belz GG, Palm D. Beta adrenoceptor subtype binding activity in plasma and beta blockade by propranolol and beta-1 selective bisoprolol in humans. Evaluation with Schild-plots. J Pharmacol Exp Ther. 1988;246:328–337. [PubMed] [Google Scholar]
- 23.Schwinn DA, Johnston GL, Page SO, et al. Cloning and pharmacological characterization of human alpha-1 adrenergic receptors: sequence corrections and direct comparison with other species. J Pharmacol Exp Ther. 1995;272:134–142. [PubMed] [Google Scholar]
- 24.Michel MC, Insel PA. Comparison of cloned and pharmacologically defined rat tissue α1-adrenoceptor subtypes. Naunyn-Schmiedeberg's Arch Pharmacol. 1994;350:136–142. doi: 10.1007/BF00241087. [DOI] [PubMed] [Google Scholar]
- 25.Rabasseda X, Fitzpatrick JM. Tamsulosin: the first prostate-selective α1A-adrenoceptor antagonist for the treatment of symptomatic benign prostatic hyperplasia. Drugs of Today. 1996;32:259–268. [Google Scholar]
- 26.Sonders RC. Pharmacokinetics of terazosin. Am J Med. 1986;80(Suppl. 5b):20–24. doi: 10.1016/0002-9343(86)90847-8. [DOI] [PubMed] [Google Scholar]
- 27.Pitschner HF, Wellstein A. Dose-response curves of pirenzepine in man in relation to M1- and M2-cholinoceptor occupancy. Naunyn-Schmiedeberg's Arch Pharmacol. 1988;338:207–210. doi: 10.1007/BF00174872. [DOI] [PubMed] [Google Scholar]
- 28.Borbe HO, Peter G. Radioreceptor assay for the determination of α1-adrenoceptor-binding material in rat plasma following a single oral administration of naftopidil. Arzneim-Forsch/Drug Res. 1990;40:253–256. [PubMed] [Google Scholar]
- 29.Brodde O-E, Daul A, Wellstein A, Palm D, Michel MC, Beckeringh JJ. Differentiation of β1- and β2-adrenoceptor-mediated effects in humans. Am J Physiol. 1988;254:199–206. doi: 10.1152/ajpheart.1988.254.2.H199. [DOI] [PubMed] [Google Scholar]
- 30.Soeishi Y, Matsushima H, Watanabe T, Higuchi S, Cornelissen K, Ward J. Absorption, metabolism and excretion of tamsulosin hydrochloride in man. Xenobiotica. 1996;26:637–645. doi: 10.3109/00498259609046739. [DOI] [PubMed] [Google Scholar]
- 31.Soeishi Y, Matsushima H, Teraya Y, Watanabe T, Higuchi S, Kaniwa H. Metabolism of tamsulosin in the rat and dog. Xenobiotica. 1996;26:355–365. doi: 10.3109/00498259609046714. [DOI] [PubMed] [Google Scholar]