

Abstract
Cytochrome P450 CYP2D6 is the most extensively characterized polymorphic drug-metabolizing enzyme. A deficiency of the CYP2D6 enzyme is inherited as an autosomal recessive trait; these subjects (7% of Caucasians, about 1% of Orientals) are classified as poor metabolizers. Among the rest (extensive metabolizers), enzyme activity is highly variable, from extremely high in ultrarapid metabolizers, to markedly reduced in intermediate metabolizers. The CYP2D6 gene is highly polymorphic, with more than 70 allelic variants described so far. Of these, more than 15 encode an inactive or no enzyme at all. Others encode enzyme with reduced, ‘normal’ or increased enzyme activity. The CYP2D6 gene shows marked interethnic variability, with interpopulation differences in allele frequency and existence of ‘population-specific’ allelic variants, for instance among Orientals and Black Africans. The CYP2D6 enzyme catalyses the metabolism of a large number of clinically important drugs including antidepressants, neuroleptics, some antiarrhythmics, lipophilic β-adrenoceptor blockers and opioids. The present-day knowledge on the influence of the genetic variability in CYP2D6 on the clinical pharmacokinetics and therapeutic effects/adverse effects of psychotropic drugs is reviewed.
Keywords: antidepressants, CYP2D6, debrisoquine, neuroleptics, polymorphism
Introduction
Many drugs, especially lipophilic compounds such as psychotropics need to be metabolized prior to excretion in urine. Oxidative phase I catalysed metabolism by cytochrome P450 (CYP) enzymes plays a major role in this respect. In the 1960s it was shown that the 30-to 40-fold variability in plasma concentrations of the tricyclic antidepressant nortriptyline in patients treated with the same dose is due to a pronounced variation in the rate of metabolism of the drug [1,2]. Twin studies further showed that the rate of metabolism had a strong genetic component [2] and in 1980 the 10-hydroxylation of nortriptyline was shown to be catalysed by the polymorphic debrisoquine/sparteine hydroxylase (CYP2D6) [3].
This short review deals with the molecular genetics of CYP2D6 and its clinical relevance. Many recent reviews and books related to various aspects of this are available for further reading [4–8].
The CYP2D6 polymorphism
The discovery of the debrisoquine/sparteine hydroxylation polymorphism
In 1977, the hydroxylation of the antihypertensive drug debrisoquine was shown to be polymorphic in nature [9,10]. Independently, Eichelbaum et al.[11] showed that the oxidation of sparteine was also polymorphic. The metabolic ratios (MR; parent drug/metabolite) of the two drugs were closely correlated [12], showing that the same enzyme, now termed CYP2D6, was responsible for the two metabolic reactions.
The incidence of poor metabolizers (PM) of debrisoquine/sparteine with deficient CYP2D6 activity has been investigated in many populations, in most of them with a fairly small number of subjects [13,14]. Among 1011 Swedish Caucasians we found 69 (6.3%) PM of debrisoquine [15]. This incidence is very similar to that in other European Caucasian populations (7–10%) [13,14]. In collaboration with Lou and associates in Beijing it was shown that the incidence of PM among 695 Chinese was only 1.0% using the antimode established in Caucasian populations [15]. A similar low incidence of PM has been shown in Japanese [16] and Koreans [17].
Molecular genetics of the poor metabolizers
The gene encoding the CYP2D6 enzyme is localized on chromosome 22 [18]. Three major mutant alleles, now termed CYP2D6*3,*4 and *5[23] (Table 1), associated with the PM phenotype, were found early on in Caucasians [19–22]. In Swedish Caucasians, the CYP2D6*4 allele occurs with a frequency of 22% and accounts for more than 75% of the mutant alleles in this population [23]. The CYP2D6*4 allele is almost absent in Chinese and this is the reason for the low incidence (1%) of PM in this population compared with 6% in Caucasians [15]. The frequency of the gene deletion (CYP2D6*5) on the other hand is very similar, i.e. 4–6% in different populations (Table 1). The CYP2D6 gene has turned out to be extremely polymorphic with more than 70 allelic variants described so far (http://www.imm.ki.se/CYPalleles/cyp2d6.htm). In addition to the CYP2D6*3, *4 and *5, alleles, a large number of low-frequency alleles associated with the PM phenotype have been identified. Usually a few variants account for most of the mutant alleles in a population. The alleles of importance may, however, vary between populations (see below), which needs to be taken into consideration when applying genotyping methodology in clinical research or patient care.
Table 1.
Frequency of CYP2D6*1 or*2 alleles (causing ‘normal’ enzyme activity) and some alleles causing no or deficient CYP2D6 activity in three different ethnic populations.
| CYP2D6 alleles | Functional mutation | Consequence | Swedish | Allele frequency (%) Chinese | Zimbabwean |
|---|---|---|---|---|---|
| *1 or *2 | 69 | 43 | 54 | ||
| *3 | A2637 del | Frame shift | 2 | 0 | 0 |
| *4 | G1934A | Splicing defect | 22 | 0–1 | 2 |
| *5 | Gene deletion | No enzyme | 4 | 6 | 4 |
| *10 | C188T | Unstable enzyme | n.d. | 51 | 6 |
| *17 | C1111T | Reduced affinity | n.d. | n.d. | 34 |
Alleles in Orientals and Africans encoding CYP2D6 with decreased activity
Our early studies comparing CYP2D6 activity between Swedish and Chinese subjects revealed that the distribution of the MR of Chinese extensive metabolizers (EM) was shifted to the right compared with Swedish EM (P < 0.01) [15]. This showed that the mean rate of hydroxylation of debrisoquine was lower in Chinese EM compared with Caucasian EM. This right shift in MR in Orientals is due to the high frequency of a mutant CYP2D6*10 allele [24,25] with the SNP C188T causing a Pro34Ser amino acid substitution and an unstable enzyme with decreased catalytic activity [25] (Figure 1). The frequency of this CYP2D6*10 allele is similar, about 50%, in Chinese, Japanese and Koreans, but extremely low among Caucasians (Table 1).
Figure 1.
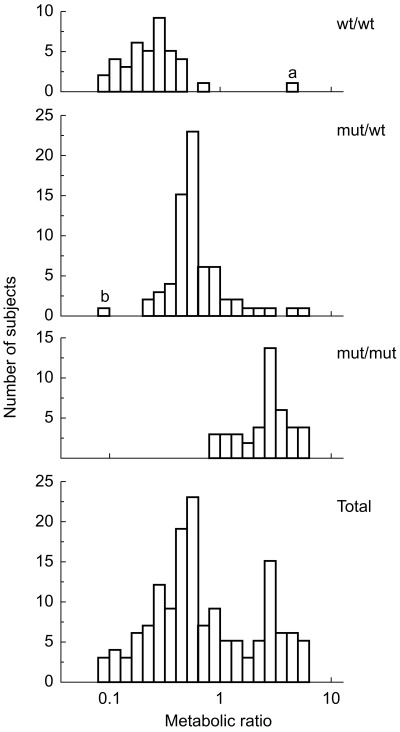
Distribution of the debrisoquine MR (parent drug/4-hydroxy metabolite) in three genotype groups related to the CYP2D6*10 allele in 152 Korean subjects. wt = CYP2D6*1(or*2) and mut = CYP2D6*10. Reproduced with permission from Roh et al.[26].
Masimirembwa et al.[27] found a right shift of the MR in black Zimbabweans, similar to that found in Orientals. A mutated allele encoding an enzyme with decreased activity was subsequently identified and named CYP2D6*17. The frequency of this allele was found to be 34% in Zimbabweans [27] (Table 1), 17% in Tanzanians [28], 28% in Ghanaians [29] and 9% in Ethiopians [30]. This and many other studies demonstrate the genetic heterogeneity of different black populations of Africa. There are thus three fairly population specific alleles with CYP2D6*4 in Caucasians, *10 in Asians and *17 in Africans. These mutations must have occurred after the separation of the respective populations from each other.
In Caucasians and Orientals a close geno-and phenotype relationship has been demonstrated [23, 25, 26]. However, in studies in Ethiopia [30], Ghana [29] and Tanzania [28] a lower CYP2D6 activity in relation to genotype has been demonstrated, indicating that in Africa, environmental factors, e.g. infections or food constituents are probably of importance in addition to genetic factors.
Gene duplication, multiduplication and amplification as causes of increased CYP2D6 activity
The problems in treating PM of debrisoquine have been extensively discussed over the years since the discovery of the CYP2D6 polymorphism [14]. Much less attention has been given to patients at the other extreme, i.e. ultrarapid metabolizers. In 1993, we described a Swedish family with the father and his daughter and son having 12 extra copies of a functional CYP2D6*2 gene in the CYP2D locus [31]. These subjects were ultrarapid metabolizers of debrisoquine with MR 0.01–0.02 (Figure 2). In another family, duplication of the CYP2D6*2 gene was also associated with extremely high CYP2D6 activity [31]. This was the first demonstration of an inherited duplication/amplification of an active gene encoding a drug metabolizing enzyme. We also described two patients, who had to be treated with extremely high doses of antidepressants [32,33]. One of the patients is further described below. The CYP2D locus of these patients was found to contain a duplication of the CYP2D6*2 gene. A population study confirmed the association between the duplication/multiduplication of the CYP2D6*2 gene and low debrisoquine MR [34].
Figure 2.
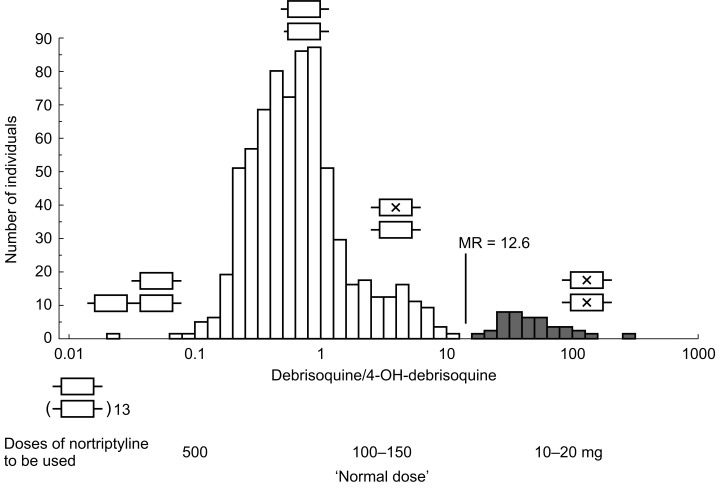
Distribution of the urinary debrisoquine MR in 757 healthy Swedish subjects with schematic presentation of CYP2D6 genotypes, where a cross in an allele indicates a detrimental mutation. Also tentative doses of nortriptyline to be used in different genotypes are indicated. From [64].
In Swedish Caucasians the frequency of subjects having duplicated/multiduplicated genes is about 1–2% [34]. Going south in Europe, the reported frequency increases to 3.6% in Germany [35], 7–10% in Spain [36,37] and 10% on Sicily in Italy [38]. The frequency is as high as 29% in black Ethiopians [30] and 20% in Saudi Arabians [39]. There is thus a European-African north–south gradient in the incidence of CYP2D6 gene duplication. It has been speculated that the high incidence in Spain and Italy may have an ancestry in the Arabian conquest in the Mediterranean area [36]. Caucasian subjects with a CYP2D6 gene duplication have been shown to be ultrarapid metabolizers of debrisoquine with MR usually between 0.01 and 0.15 [31, 34–36]. In the study of Aklillu et al.[30], black Ethiopians with multiple CYP2D6 genes had higher MR, usually between 0.1 and 1. These subjects do thus not have the ultrarapid metabolism of debrisoquine demonstrated for Caucasians with multiple genes. This might be due to environmental factors in Africa causing a decreased activity.
Clinical relevance of the CYP2D6 polymorphism
Since the discovery of the CYP2D6 polymorphism almost 100 drugs have been shown to be substrates for this enzyme. Some of these drugs are shown in Table 2. The clinical importance of the polymorphism depends on a number of factors including whether the parent compound, metabolite(s) or both are metabolized or formed by CYP2D6; whether the parent compound, the metabolite(s) or both are active; the potency of the active species; and the overall contribution of the CYP2D6-dependent pathway to the clearance of the drug. Furthermore, the therapeutic index of the drug (narrow-broad), possible saturation of the CYP2D6-dependent pathway, and the contribution of other pathways of elimination need to be considered. Thus, the clinical impact of CYP2D6 dependent metabolism needs to be carefully investigated for each substrate. So far, the majority of in vivo data on the role of CYP2D6 are from single-dose pharmacokinetic studies. The increasing availability of genotyping methods has made clinical studies in patients receiving therapeutic doses possible. We will here highlight these aspects with some examples, mainly from the field of psychopharmacology.
Table 2.
Some drugs whose metabolism is catalysed by CYP2D6.
| β-Adrenoceptor blockers | Antidepressants | Neuroleptics |
| Metoprolol | Amitriptyline | Haloperidol |
| Propranolol | Clomipramine | Perphenazine |
| Timolol | Desipramine | Risperidone |
| Fluoxetine | Thioridazine | |
| Antiarrhythmic drugs | Fluvoxamine | Zuclopenthixol |
| Encainide | Imipramine | |
| Flecainide | Mianserin | Miscellaneous |
| Perhexilene | Nortriptyline | Codeine |
| Propafenone | Paroxetine | Debrisoquine |
| Sparteine | Venlafaxine | Dextromethorphan |
| Phenformin | ||
| Tolterodine | ||
| Tramadol |
Nortriptyline
Nortriptyline was one of the first clinically important drugs to be shown to be metabolized by CYP2D6 [3,40]. These early studies (prior to the era of genotyping) were performed in phenotyped panels of healthy subjects and the results have been confirmed in vivo in patients as well as in vitro using human liver microsomes and expressed enzymes. In a recent study by Dalén et al.[41], nortriptyline was given as a single oral dose to 21 healthy Swedish Caucasian subjects with different CYP2D6 genotypes. As seen in Figure 3, there was a decrease in the plasma concentrations of nortriptyline from subjects with 0 functional genes (CYP2D6*4/*4 genotype) to those with 1, 2 and 3 (gene duplication) functional genes. The plasma concentrations of the parent drug were extremely low in one subject with 13 CYP2D6*2 genes. The plasma concentrations of the main metabolite 10-hydroxynortriptyline showed the opposite pattern, i.e. highest concentrations in the subject with 13 functional genes and lowest in the PM (Figure 3). This study clearly shows the impact of the detrimental CYP2D6*4 allele as well as of the duplication/amplification of the CYP2D6*2 gene on the metabolism of nortriptyline [41]. A relationship between the CYP2D6 genotype and steady-state plasma concentrations of nortriptyline has also been demonstrated in Swedish depressed patients treated with nortriptyline [42].
Figure 3.
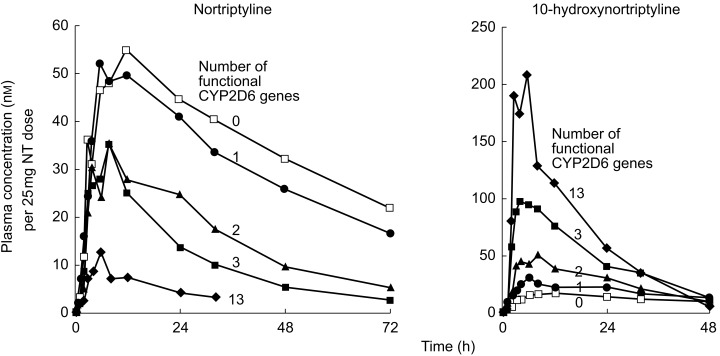
Mean plasma concentrations of nortriptyline (left) and 10-hydroxynortriptyline (right) in different CYP2D6 genotype groups after a single oral dose of nortriptyline. The numerals close to the curves represent the number of functional CYP2D6 genes in each genotype group. In groups with 0–3 functional genes, there were five subjects in each group while there was only one subject with 13 functional genes. Reproduced with permission from Dalén et al.[41].
We used the single dose results of Dalén et al.[41] (Figure 3) to simulate steady-state concentrations of nortriptyline in the different genotype groups after different daily doses of the drug assuming linear kinetics (Figure 4). A dose of 25 mg three times daily, which is usually recommended as a starting dose, resulted in concentrations near the upper limit of the recommended therapeutic interval (200–600 nm) in subjects with 0 (PM) and 1 (heterozygotes) functional CYP2D6 genes (Figure 4, upper curves). Subjects with 2 or more functional genes fall below 200 nm. At the usually recommended daily dose of nortriptyline 150 mg, subjects with 0 or 1 functional genes would attain levels above 600 nm and might therefore be at higher risk of developing adverse drug reactions. Subjects with 2 functional genes, who constitute about half of Caucasian populations, are in the middle of the therapeutic interval (Figure 4, middle curves). Subjects with gene duplication or amplification might require increased doses of nortriptyline, e.g. 75 mg three times daily (Figure 4, bottom curves) or higher. One out of 10 patients in Italy and Spain, where gene duplication is common, might require increased doses of CYP2D6 substrates. It should be underscored that the curves presented in Figure 4 are simulated from the single dose data of Figure 3 assuming linear kinetics. Early studies by Alexanderson [43] showed that single dose data of nortriptyline can be used to predict steady state concentrations. However, dose-dependent kinetics of this drug seem to occur in extensive metabolizers when high doses of nortriptyline saturate the capacity for hydroxylation [44,45]. It should also be remembered that the 10-hydroxy metabolite of nortriptyline, formed by CYP2D6, is pharmacologically active although its relative contribution to the clinical effect and toxicity of nortriptyline has not been clearly elucidated [46]. The contribution of this metabolite is probably more important in ultrarapid metabolizers than in other patients.
Figure 4.
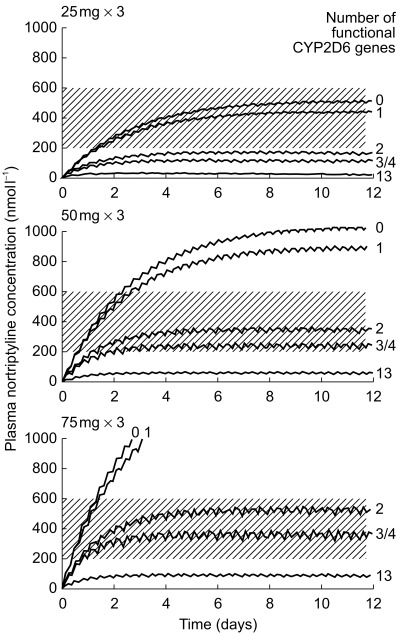
Steady-state levels of nortriptyline simulated from the single dose results of Dalén et al.[41] in different CYP2D6 genotypes. The shaded area indicates the commonly recommended ‘therapeutic’ range of 200–600 nm.
Using the same protocol as in the study of Dalén et al.[41], we investigated the influence of the Oriental-specific CYP2D6*10 allele on the disposition of nortriptyline in Chinese subjects living in Sweden [47]. Recently, Morita et al.[48] showed the influence of the CYP2D6*10 allele on the steady-state plasma levels of nortriptyline and its 10-hydroxy metabolite in Japanese depressed patients. From these two studies it may be concluded that CYP2D6*10 encodes an enzyme with decreased activity to metabolize nortriptyline. This effect is less pronounced than that of the Caucasian-specific CYP2D6*4 allele, which encodes no enzyme at all.
Genotyping or phenotyping for CYP2D6 may be a tool to predict proper initial dosing of drugs such as nortriptyline in individual patients, especially those with extremely low (PM) or high (UM) CYP2D6 activity. This can be demonstrated with our experience with two patients for whom the dosage of nortriptyline and other antidepressants needed to be individualized (Figure 2) [32, 33, 49, 50].
Patient 1. A 69 year old woman was hospitalized for moderate to severe depression and treated with nortriptyline in a modest dose of 25 mg three times daily. Two days after the start of treatment she complained of dizziness [49]. After a further 6 days of treatment, she complained of increasing tiredness and vertigo and appeared slightly confused. Low clearance of nortriptyline was suspected, blood was taken for nortriptyline analysis and the dosage was decreased to 25 mg once daily. The plasma concentration of nortriptyline after 8 days of treatment with the 75 mg daily dose was 1300 nm (recommended plasma concentration range 200–600 nm). The concentration on 25 mg daily for 12 days was 742 nm. When the dosage was further reduced to 20 mg at night, the patient had no side-effects and made an excellent recovery [49]. Thirty days later, after she had stopped taking nortriptyline, the patient was phenotyped with debrisoquine. She had a high metabolic ratio of 43 and was thus classified as a PM of debrisoquine.
Patient 2. A 41 year old woman had been treated for long periods with high doses of nortriptyline (300–500 mg day−1) to achieve ‘therapeutic’ plasma levels (200–600 nm) [32]. The mean (± s.d.) plasma level in seven samples drawn at a dose of 300 mg day−1 was 291 ± 56 nm. The plasma concentration of unconjugated 10-hydroxynortriptyline was about 10 times higher than that of the parent drug, which is much higher than usual. A debrisoquine phenotyping test confirmed that this patient is an ultrarapid metabolizer of debrisoquine (and nortriptyline) with a metabolic ratio of 0.07 [32]. This metabolic ratio was one of the lowest seen in a Swedish population (Figure 2). As amitriptyline is also partly metabolized by CYP2D6, it was not surprising that this patient also had low plasma concentrations of amitriptyline and its metabolite nortriptyline when treated with amitriptyline. In 1993, 8 years after we first described this patient, we were able to demonstrate that she had a duplication of the CYP2D6 gene and thus 3 functional genes, leading to increased activity of the enzyme [33].
Other antidepressants
CYP2D6 has been shown to catalyse the metabolism of a number of other antidepressants including the serotonin-reuptake inhibitors paroxetine, fluvoxamine and fluoxetine as well as venlafaxine and mianserin (Table 2). As an example, Sindrup et al.[51] found 25-fold differences in plasma concentrations of paroxetine between PM and EM after a single oral dose of the drug. The interphenotype difference was, however, only two-fold at steady-state, due to paroxetine's saturable metabolism catalysed by CYP2D6 in EM. Özdemir et al.[52] also found two-fold higher median steady-state plasma concentrations of paroxetine in heterozygous EM than in homozygous EM subjects, with a considerable overlap in the distribution of paroxetine concentrations between the two genotypes. Unfortunately, no PM or UM subjects were included in that study. The clinical significance of the CYP2D6 genotype on the steady state plasma levels of fluoxetine and fluvoxamine is not known. Venlafaxine undergoes CYP2D6 dependent metabolism to the active major metabolite O-desmethylvenlafaxine, while its N-desmethylation is catalysed by CYP3A4, and possibly by CYP2C19 and CYP2C9 [53,54]. PM of CYP2D6 had a more than 4-fold lower oral clearance of venlafaxine compared to EM, mainly due to a decreased capacity to form the O-desmethylated metabolites [55]. In a study on 33 patients with depression treated with 225 mg venlafaxine/day, a significant relationship between the CYP2D6 genotype and the ratio O-desmethylvenlafaxine/venlafaxine was found, PM having extremely low ratios and UM high ratios compared with homo-and heterozygous EM [56]. A significant relationship between the CYP2D6 genotype (i.e. the presence of the CYP2D6*10 allele) and the plasma kinetics of venlafaxine and O-desmethylvenlafaxine has also been shown in Japanese subjects [57]. A relationship between the PM phenotype and cardiovascular toxicity of venlafaxine has been suggested, based on four patients with PM phenotype which was either genetically determined or due to inhibition of CYP2D6 activity by concomitant drugs [55]. The side-effects reported included palpitations, shortness of breath and proarrhythmias due to the heterogeneity in cardiac repolarization.
Haloperidol
Llerena et al.[58] gave low single oral doses (2–4 mg) of haloperidol to panels of six EM and six PM of debrisoquine. PM eliminated haloperidol more slowly than EM, the mean plasma half-life being longer (29.4 and 16.3 h, respectively; P < 0.01) and the mean clearance lower (1.16 and 2.49 l h−1 kg−1, respectively; P < 0.05) [58]. The plasma level of reduced haloperidol was also higher in PM than in EM [59]. In a clinical study involving eight Caucasian patients with schizophrenia treated with depot haloperidol (decanoate), the dopamine D2 receptor occupancy was determined by positron emission tomography 1 and 4 weeks after intramuscular injection of the drug [60]. One of the patients was genotypically a PM of debrisoquine. Of the group, he had the highest plasma concentration of haloperidol and also the highest D2 receptor occupancy.
Two studies from Japan [61,62] have shown a relationship between increased steady state haloperidol plasma concentrations and the presence of CYP2D6*10 (and *5) alleles in Japanese patients treated with 12 mg daily oral doses of haloperidol. In a recent study in Korea [63], a relationship between haloperidol and reduced haloperidol concentrations and CYP2D6 genotype was established in patients receiving less than 20 mg haloperidol daily, but not in patients receiving higher doses. We believe that the high affinity-low capacity CYP2D6 plays an important role at low concentrations/doses of haloperidol, while the low affinity-high capacity CYP3A4 becomes more important at higher doses. Thus, the importance of the CYP2D6 genotype depends on the dose range used. The metabolic pathway of haloperidol catalysed by CYP2D6 is presently not known.
Other neuroleptics
Also many other neuroleptics such as perphenazine, zuclopenthizol, thioridazine and risperidone have been shown to be metabolized by CYP2D6 (Table 2) [64]. At steady state, the median oral clearance of perphenazine was three-fold lower in PM than in homozygous EM of debrisoquine [65]. Similarly, there was a two-fold difference in the clearance of zuclopenthixol in the two genotype groups [65]. That PM reach higher average plasma concentrations per dose unit of perphenazine and zuclopenthixol than EM has been confirmed by others [66,67]. All these studies have only included patients on oral treatment. We have recently demonstrated that the difference between PM and EM in steady-state plasma levels of zuclopenthixol is also seen during treatment with intramuscular zuclopenthixol decanoate [68].
The metabolism of risperidone to its active metabolite 9-hydroxyrisperidone is mainly dependent on CYP2D6, leading to significantly higher dose-corrected steady-state plasma levels of the parent drug in PM than in EM [38] (Figure 5). However, the concentrations of the ‘active moiety’, i.e. the sum of risperidone and 9-hydroxyrisperidone, did not differ between the genotype groups (Figure 5). Thus, as the parent compound and metabolite are considered pharmacologically equipotent, the polymorphic metabolism is in this case not expected to be of clinical significance. It is to be pointed out that in all the published studies, there was a large overlap in the steady state plasma concentrations of neuroleptics between the different genotype groups, indicating that other factors in addition to the CYP2D6 genotype are of major importance for the interindividual variability in pharmacokinetics.
Figure 5.
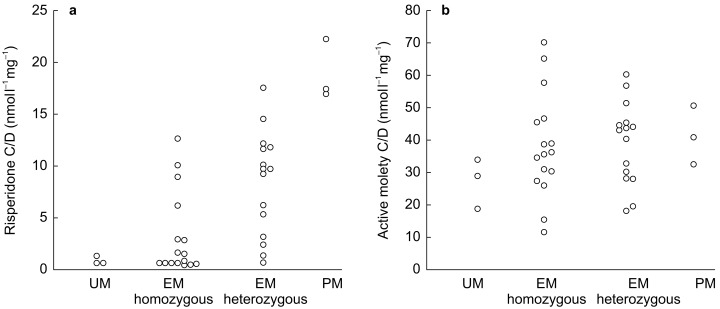
Relationship between the CYP2D6 genotype and the plasma concentration-to-dose (C/D) ratios of risperidone (left) and the active moiety of risperidone (risperidone plus 9-hydroxyrisperidone, right). UM=ultrarapid metabolizers, defined as a carrier of gene duplication; EM homozygous=extensive metabolizers homozygous for the functional CYP2D6*1 allele; EM heterozygous=EM heterozygous for a defective CYP2D6 gene; PM=poor metabolizers carrying two defect genes. Reproduced with permission from Scordo et al.[38].
Therapeutic effects/side-effects and CYP2D6 genotype
Although the pharmacokinetic consequences of polymorphic metabolism are relatively well documented for a number of CYP2D6 substrates, its clinical impact with respect to therapeutic response and dosing remains scanty and mainly based on case reports (see above). The clearest example of clinical significance relates to the antianginal drug perhexilene. Patients with perhexilene-induced peripheral neuropathy achieved higher blood concentrations than patients without this adverse effect [69]. In a retrospective study it was shown that of 20 patients developing peripheral neuropathy while taking this drug, 11 were PM (55%) and of the 9 EM, 6 had a debrisoquine MR higher than 1 [70]. The polymorphism apparently contributed to the withdrawal of perhexilene from the market in many countries in 1980s, but in patients with ‘normal’ debrisoquine metabolism, other factors probably contributed to the development of the neuropathy.
A number of studies have explored the relationship between adverse effects of psychotropic drugs and CYP2D6 genotype. For example, Chen & coworkers [71] found a doubling of the frequency of defective CYP2D6 alleles in depressed patients with adverse reactions compared with those without such reactions. The possible association between the CYP2D6 genotype and neuroleptic-induced movement disorders has been addressed in several studies [72–79] (Table 3). The results have been inconsistent but tend to show a slight overrepresentation of mutated CYP2D6 alleles in patients with tardive dyskinesia and parkinsonism during neuroleptic treatment. Possible explanations for the partly inconsistent or negative results are that in some studies the patients were taking various neuroleptics including those not metabolized by CYP2D6, or on polytherapy with drugs that are potent inhibitors of CYP2D6. In a recent 1 year follow-up pilot study of 100 consecutive psychiatric in-patients genotyped for CYP2D6 on admission, Chou et al.[80] found a trend towards greater number of adverse drug effects from drugs primarily metabolized by CYP2D6, when one moves from UM to PM. The costs of treating the patients with extremes of CYP2D6 activity (i.e. PM and UM) was on average 4000–6000 dollars greater per year than the costs of treating patients with the EM or intermediate metabolizer (heterozygous EM) genotypes. The total duration of hospital stay was also longer for patients in the PM group. This pilot study needs a large clinical trial to confirm these preliminary and interesting issues.
Table 3.
Studies on the relationship between CYP2D6 genotype and neuroleptic-induced movement disorders.
| Study | Side-effect | Relationship with CYP2D6 genotype |
|---|---|---|
| Arthur et al.[72] | Tardive dyskinesia | No |
| Armstrong et al.[73] | Acute dystonia | No |
| Chronic movement disorders | No (non-sign tendency) | |
| Andreasson et al.[74] | Tardive dyskinesia | No (non-sign tendency) |
| Parkinsonism | No | |
| Akathisia | No | |
| Kapitany et al.[75] | Tardive dyskinesia | Yes |
| Ohmori et al.[76] | Tardive dyskinesia | No |
| Vandel et al.[77] | Extrapyramidal side- effects | Yes |
| Hamelin et al.[78] | Not defined | No |
| Scordo et al.[79] | Extrapyramidal side- effects | No |
Antiarrhythmic drugs have a narrow therapeutic index and many of them (flecainide, encainide, propafenone, mexiletine, N-propyl-ajmaline) are CYP2D6 substrates. PM might thus be expected to have an increased risk of concentration-dependent adverse effects. Encainide is an exception as its O-desmetyl metabolite, the formation of which is catalysed by CYP2D6, is 6–10 times more potent than the parent drug in blocking sodium channels [81]. Although PM treated with encainide develop less QRS and QT prolongation than EM, both phenotype groups showed a similar antiarrhythmic response at comparable doses [82]. An increased frequency of fatal nervous system adverse effects has been reported in PM during propafenone treatment [83]. It has also been suggested that determination of the phenotype (or genotype) of patients before therapy with N-propyl-ajmaline might be useful to select the individual optimum dose of the drug [84]. No prospective studies have, however, tested this hypothesis.
Future perspectives
The pronounced interindividual variation in the rate of drug metabolism has been known for many years. It was initially only of academic interest, but today the pharmaceutical industry has to document the metabolism of a new drug in development before registration. The knowledge of how a drug is metabolized and which enzymes are involved helps to predict drug–drug interactions and how fast an individual patient may metabolize a specific drug.
With the high-throughput screening techniques available today, pharmaceutical industries may screen for drug candidates which are metabolized by and/or inhibitors of e.g. CYP2D6. Such drugs are often not developed further. If this had been the case in the past, we would not have many of the important drugs available today (see Table 2). The novel antimuscarinic drug tolterodine may serve as an example of a drug metabolized by CYP2D6, but not discontinued for development. In early Phase I studies, it was discovered that tolterodine is hydroxylated by CYP2D6, but the surrogate antimuscarinic effect on salivation was the same in EM and PM [85]. The reason is that 5-hydroxytolterodine is an active metabolite and responsible for all or some of the effect in EM.
Phenotype analysis, with for example, debrisoquine or sparteine should be avoided during on-going drug treatment as many drugs, especially psychotropics may inhibit CYP2D6 activity, leading to increased MRs. Genotyping may of course be performed independent of ongoing drug treatment. As pointed out above, interethnic differences must be considered when choosing which alleles are to be identified and when interpreting the genotype-phenotype relationship. With the development of new, less expensive and more rapid genotyping methods, the use of such techniques in drug development, academic research and clinical practice is expected to increase in the future. Today, genotyping methods are available for the identification of PM as well as of UM with CYP2D6 gene duplication/amplification. However, only about 40% of subjects with debrisoquine MRs of 0.1 or lower carry duplicated/multiduplicated genes, leaving about 60% of very rapid metabolizers unidentified by genotyping [23]. Efforts to find other molecular genetic explanations to ultrarapid metabolism in these subjects have so far been unsuccessful. The clinical usefulness of genotyping would be greatly increased if it would allow more accurate prediction of the catalytic activity (MR) within the EM phenotype.
The subgroup of so called intermediate metabolizers (IM) with severely impaired yet residual CYP2D6 activity and accounting for 10–15% of EM is a group of interest in this respect. It has been shown that the metabolic clearance of IM for sparteine oxidation is five-fold lower on average compared with ‘normal’ EM [86]. Interestingly, Raimundo et al.[87] have recently identified a mutation in the CYP2D6 5′-flanking region which was associated with a functional bimodality of the frequent CYP2D6*2 allele, but it accounts for only a small proportion of the total EM subjects. Analysis of such functionally important allelic variants of CYP2D6 would substantially improve the relatively poor genotype-phenotype correlations among EMs and might be clinically valuable for drugs with a narrow therapeutic range in order to identify patients at risk of concentration-dependent adverse effects or therapeutic failure. Further studies are required to establish whether the dosage of some drugs metabolized by CYP2D6 can be prospectively individualized based on the genotype and whether this would improve treatment outcome and be cost-effective.
The studies performed in the authors' laboratory have been supported by the Swedish Medical Research Council (3902), National Institutes of Health, USA (GM 60548–01A2) and Karolinska Institutet.
References
- 1.Hammer W, Sjöqvist F. Plasma levels of monomethylated tricyclic antidepressants during treatment with imipramine-like compounds. Life Sci. 1967;6:1895–1903. doi: 10.1016/0024-3205(67)90218-4. [DOI] [PubMed] [Google Scholar]
- 2.Alexanderson B, Evans DAP, Sjöqvist F. Steady-state plasma levels of nortriptyline in twins: influence of genetic factors and drug therapy. Br Med J. 1969;4:764–768. doi: 10.1136/bmj.4.5686.764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bertilsson L, Eichelbaum M, Mellström B, Säwe J, Schulz HU, Sjöqvist F. Nortriptyline and antipyrine clearance in relation to debrisoquine hydroxylation in man. Life Sci. 1980;27:1673–1677. doi: 10.1016/0024-3205(80)90642-6. [DOI] [PubMed] [Google Scholar]
- 4.Bertilsson L, Kalow W. Interethnic differences is drug disposition and effects. In: Pacifici GM, Pelkonen O, editors. Interindividual Variability in Drug Metabolism in Humans. London: Taylor & Francis; 2001. pp. 15–74. [Google Scholar]
- 5.Ingelman-Sundberg M, Oscarsson M, McLellan RA. Polymorphic human cytochrome P450 enzymes: an opportunity for individualized drug treatment. Trends Pharmacol Sci. 1999;20:324–349. doi: 10.1016/s0165-6147(99)01363-2. [DOI] [PubMed] [Google Scholar]
- 6.Evans WE, Relling MV. Pharmacogenomics: Translating functional genomics into rational therapeutics. Science. 1999;286:487–491. doi: 10.1126/science.286.5439.487. DOI: 10.1126/science.286.5439.487. [DOI] [PubMed] [Google Scholar]
- 7.Wolf CR, Smith G, Smith RL. Clinical Review. Pharmacogenetics. Br Med J. 2000;320:987–990. doi: 10.1136/bmj.320.7240.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meyer UA. Pharmacogenetics and adverse drug reactions. Lancet. 2000;356:1667–1671. doi: 10.1016/S0140-6736(00)03167-6. [DOI] [PubMed] [Google Scholar]
- 9.Mahgoub A, Idle JR, Dring DG, Lancaster R, Smith RL. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2:584–586. doi: 10.1016/s0140-6736(77)91430-1. [DOI] [PubMed] [Google Scholar]
- 10.Tucker GT, Silas JH, Iyun AO, Lennard MS, Smith AJ. Polymorphic hydroxylation of debrisoquine in man. Lancet. 1977;2:718. doi: 10.1016/s0140-6736(77)90527-x. [DOI] [PubMed] [Google Scholar]
- 11.Eichelbaum M, Spannbrucker N, Steinke B, Dengler HJ. Defective N-oxidation of sparteine in man: a new pharmacogenetic defect. Eur J Clin Pharmacol. 1979;16:183–187. doi: 10.1007/BF00562059. [DOI] [PubMed] [Google Scholar]
- 12.Eichelbaum M, Bertilsson L, Säwe J, Zekorn C. Polymorphic oxidation of sparteine and debrisoquine. Related pharmacogenetic entities. Clin Pharmacol Ther. 1982;31:184–186. doi: 10.1038/clpt.1982.29. [DOI] [PubMed] [Google Scholar]
- 13.Alván G, Bechtel P, Iselius L, Gundert-Remy U. Hydroxylation polymorphisms of debrisoquine and mephenytoin in European populations. Eur J Clin Pharmacol. 1990;39:533–537. doi: 10.1007/BF00316090. [DOI] [PubMed] [Google Scholar]
- 14.Eichelbaum M, Gross AS. The genetic polymorphism of debrisoquine/sparteine metabolism – clinical aspects. In: Kalow W, editor. Pharmacogenetics of Drug Metabolism. New York: Pergamon Press Inc; 1992. pp. 625–648. [Google Scholar]
- 15.Bertilsson L, Lou YQ, Du YL, et al. Pronounced differences between native Chinese and Swedish populations in the polymorphic hydroxylations of debrisoquine and S-mephenytoin. Clin Pharmacol Ther. 1992;51:388–397. doi: 10.1038/clpt.1992.38. [DOI] [PubMed] [Google Scholar]
- 16.Nakamura K, Goto F, Ray WA, et al. Interethnic differences in genetic polymorphism of debrisoquin and mephenytoin hydroxylation between Japanese and Caucasian populations. Clin Pharmacol. 1985;38:402–408. doi: 10.1038/clpt.1985.194. [DOI] [PubMed] [Google Scholar]
- 17.Sohn D-R, Shin S-G, Park C-W, Kusaka M, Chiba K, Ishizaki T. Metoprolol oxidation polymorphism in a Korean population. comparison with native Japanese and Chinese populations. Br J Clin Pharmacol. 1991;32:504–507. doi: 10.1111/j.1365-2125.1991.tb03939.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Eichelbaum M, Baur MP, Dengler HJ, et al. Chromosomal assignment of human cytochrome P450 (debrisoquine/sparteine type) to chromosome 22. Br J Clin Pharmacol. 1987;23:455–458. doi: 10.1111/j.1365-2125.1987.tb03075.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Skoda RC, Gonzalez FJ, Demierre A, Meyer UA. Two mutant alleles of the human cytochrome P450 db1 gene (P450, II, D1) associated with genetically deficient metabolism of debrisoquine and other drugs. Proc Natl Acad Sci USA. 1988;85:5240–5243. doi: 10.1073/pnas.85.14.5240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Gaedigk A, Blum M, Gaedigk R, Eichelbaum M, Meyer UA. Deletion of the entire cytochrome P450 CYP2D6 gene as a cause of impaired drug metabolism in poor metabolizers of the debrisoquine/sparteinepolymorphism. Am J Human Genet. 1991;48:943–950. [PMC free article] [PubMed] [Google Scholar]
- 21.Heim HM, Meyer UA. Genotyping of poor metabolisers of debrisoquine by allele-specific PCR amplification. Lancet. 1990;336:529–532. doi: 10.1016/0140-6736(90)92086-w. [DOI] [PubMed] [Google Scholar]
- 22.Heim HM, Meyer UA. Evolution of a highly polymorphic human cytochrome P450 gene cluster: CYP2D6. Genomics. 1992;14:49–58. doi: 10.1016/s0888-7543(05)80282-4. [DOI] [PubMed] [Google Scholar]
- 23.Dahl M-L, Johanssen I, Porsmyr Psalmertz M, Ingelman-Sundberg M, Sjoqvist F. Analysis ofthe CYP2D6 gene in relation to debrisoquin and desipramine hydroxylation in a Swedish population. Clin Pharmacol Ther. 1992;51:12–17. doi: 10.1038/clpt.1992.2. [DOI] [PubMed] [Google Scholar]
- 24.Wang SL, Huang JD, Lai MD, Liu B-H, Lai M-L. Molecular basis of genetic variation in debrisoquine hydroxylation in Chinese subjects: Polymorhism in RFLP and DNA sequence of CYP2D6. Clin Pharmacol Ther. 1993;53:410–418. doi: 10.1038/clpt.1993.44. [DOI] [PubMed] [Google Scholar]
- 25.Johansson I, Oscarsson M, Yue Q-Y, Bertilsson L, Sjöqvist F, Ingelman-Sundberg M. Genetic analysis of the Chinese CYP2D locus. Characterization of variant CYP2D6 genes present in subjects with diminished capacity for debrisoquine hydroxylation. Mol Pharmacol. 1994;46:452–459. [PubMed] [Google Scholar]
- 26.Roh H-K, Dahl M-L, Johansson I, Ingelman-Sundberg M, Cha Y-N, Bertilsson L. Debrisoquine and S-mephenytoin hydroxylation phenotypes and genotype in a Korean population. Pharmacogenetics. 1996;6:441–447. doi: 10.1097/00008571-199610000-00008. [DOI] [PubMed] [Google Scholar]
- 27.Masimirembwa C, Persson I. Bertilsson l, Hasler J, Ingelman-Sundberg M. A novel mutant variant of the CYP2D6 gene (CYP2D6*17) common in a black African population: association with diminished debrisoquine hydroxylase activity. Br J Clin Pharmacol. 1996;42:713–719. doi: 10.1046/j.1365-2125.1996.00489.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wennerholm A, Johansson I, Massele AY, Jande M, Alm C, Aden Abdi Y, Dahl M-L, Ingelman-Sundberg M, Bertilsson L, Gustafsson LL. Decreased capacity for debrisoquine metabolism among black Tanzanians. Analyses of the CYP2D6 genotype and phenotype. Pharmacogenetics. 1999;9:707–714. [PubMed] [Google Scholar]
- 29.Griese E-U, Asante-Poku S, Ofori-Adjei D, Mikus G, Eichelbaum M. Analysis of the CYP2D6 gene mutations and their consequences for enzyme function in a West African population. Pharmacogenetics. 1999;9:715–723. [PubMed] [Google Scholar]
- 30.Aklillu E, Persson I, Bertilsson L, Johansson I, Rodriguez F, Ingelman-Sundberg M. Frequent distribution of ultrarapid metabolizers of debrisoquine in an Ethiopian population carrying duplicated and multiduplicated functional CYP2D6 alleles. J Pharmacol Exp Ther. 1996;278:441–446. [PubMed] [Google Scholar]
- 31.Johansson I, Lundqvist E, Bertilsson L, Dahl M-L, Sjöqvist F, Ingelman-Sundberg M. Inherited amplification of an active gene in the cytochrome P450 CYP2D-locus as a cause of ultrarapid metabolism of debrisoquine. Proc Natl Acad Sci (USA) 1993;90:11825–11829. doi: 10.1073/pnas.90.24.11825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bertilsson L, Åberg-Wistedt A, Gustafsson LL, Nordin C. Extremely rapid hydroxylation of debrisoquine – A case report with implication for treatment with nortriptyline and other tricyclic antidepressants. Ther Drug Monit. 1985;7:478–480. [PubMed] [Google Scholar]
- 33.Bertilsson L, Dahl M-L, Sjöqvist F, et al. Molecular basis for rational megaprescribing in ultrarapid hydroxylators of debrisoquine. Lancet. 1993;341:63. doi: 10.1016/0140-6736(93)92546-6. [DOI] [PubMed] [Google Scholar]
- 34.Dahl M-L, Johansson I, Bertilsson L, Ingelman-Sundberg M, Sjöqvist F. Ultrarapid hydroxylation of debrisoquine in a Swedish population. Analysis of the molecular genetic basis. J Pharmacol Exp Ther. 1995;274:516–520. [PubMed] [Google Scholar]
- 35.Sachse C, Brockmöller J, Bauer S, Roots I. Cytochrome P450 2D6 variants in a Caucasian population. allele frequencies and phenotypic consequences. Am J Hum Genet. 1997;60:284–295. [PMC free article] [PubMed] [Google Scholar]
- 36.Bernal M-L, Sinues B, Johansson I, et al. Ten percent of North Spanish individuals carry duplicated or triplicated CYP2D6 genes associated with ultrarapid metabolism of debrisoquine. Pharmacogenetics. 1999;9:657–660. [PubMed] [Google Scholar]
- 37.Agúndez JAG, Ledesma MC, Ladero JM, Benitez J. Prevalence of CYP2D6 gene duplication and its repercussion on the oxidative phenotype in a white population. Clin Pharmacol Ther. 1995;57:265–269. doi: 10.1016/0009-9236(95)90151-5. [DOI] [PubMed] [Google Scholar]
- 38.Scordo MG, Spina E, Facciolá G, Avenoso A, Johansson I, Dahl M-L. Cytochrome P450 2D6 genotype and steady state plasma levels of risperidone and 9-hydroxy risperidone. Psychopharmacology. 1999;147:300–305. doi: 10.1007/s002130051171. [DOI] [PubMed] [Google Scholar]
- 39.McLellan RA, Oscarson M, Seidegård JE, Evans DAP, Ingelman-Sundberg M. Frequent occurrence of CYP2D6 gene duplication in Saudi Arabians. Pharmacogenetics. 1997;7:187–191. doi: 10.1097/00008571-199706000-00003. [DOI] [PubMed] [Google Scholar]
- 40.Mellström B, Bertilsson L, Säwe J, Schulz HU, Sjöqvist F. E-and Z-hydroxylation of nortriptyline in man – relationship to polymorphic hydroxylation of debrisoquine. Clin Pharmacol Ther. 1981;30:189–193. doi: 10.1038/clpt.1981.147. [DOI] [PubMed] [Google Scholar]
- 41.Dalén P, Dahl M-L, Bernal Ruiz ML, Nordin J, Bertilsson L. 1998;63:444–452. doi: 10.1016/S0009-9236(98)90040-6. 10-Hydroxylation of nortriptyline in Caucasians with 0, 1, 2, 3 and 13 functional CYP2D6 genesClin Pharmacol Ther. [DOI] [PubMed] [Google Scholar]
- 42.Dahl M-L, Bertilsson L, Nordin C. Steady-state plasma levels of nortriptyline and its 10-hydroxy metabolite: relationship to the CYP2D6 genotype. Psychopharmacology. 1996;123:315–319. doi: 10.1007/BF02246640. [DOI] [PubMed] [Google Scholar]
- 43.Alexanderson B. Pharmacokinetics of nortriptyline in man after single and multiple oral doses: The predictability of steady-state plasma concentration from single-dose plasma level. Eur J Clin Pharmacol. 1972;4:82–91. doi: 10.1007/BF00562502. [DOI] [PubMed] [Google Scholar]
- 44.Vandel S, Bertschy G, Vandel B, Allers G, Volmat R. Nonlinear kinetics of nortriptyline in every day practice. Eur J Clin Pharmacol. 1990;39:97–98. doi: 10.1007/BF02657069. [DOI] [PubMed] [Google Scholar]
- 45.Jerling M, Alván G. Nonlinear kinetics of nortriptyline in relation to nortriptyline clearance as observed during therapeutic drug monitoring. Eur J Clin Pharmacol. 1994;46:67–70. doi: 10.1007/BF00195918. [DOI] [PubMed] [Google Scholar]
- 46.Nordin C, Bertilsson L. Active hydroxymetabolites of antidepressants. Emphasis on E-10-hydroxy-nortriptyline. Clin Pharmacokinet. 1995;28:26–40. doi: 10.2165/00003088-199528010-00004. [DOI] [PubMed] [Google Scholar]
- 47.Yue Q-Y, Zhong Z-H, Tybring G, et al. Pharmacokinetics of nortriptyline and its 10-hydroxy metabolite in Chinese subjects of different CYP2D6 genotypes. Clin Pharmacol Ther. 1998;64:384–390. doi: 10.1016/S0009-9236(98)90069-8. [DOI] [PubMed] [Google Scholar]
- 48.Morita S, Shimoda K, Someya T, Yoshimura Y, Kamijima K, Kato N. Steady-state plasma levels of nortriptyline and its hydroxylated metabolites in Japanese patients: Impact of CYP2D6 genotype on the hydroxylation of nortriptyline. J Clin Psychopharmacol. 2000;20:141–149. doi: 10.1097/00004714-200004000-00005. [DOI] [PubMed] [Google Scholar]
- 49.Bertilsson L, Mellström B, Sjöqvist F, et al. Slow hydroxylation of nortriptyline and concomitant poor debrisoquine hydroxylation: clinical implication. Lancet. 1982;i:560–561. doi: 10.1016/s0140-6736(81)92894-4. [DOI] [PubMed] [Google Scholar]
- 50.Sjöqvist F, Bertilsson L. Clinical pharmacology of antidepressant drugs: pharmacogenetics. In: Usdin E, Åsberg M, Bertilsson L, editors. Frontiers in Biochemical and Pharmacological Research in Depression. New York: Raven Press; 1984. pp. 359–372. [PubMed] [Google Scholar]
- 51.Sindrup SH, Brøsen K, Gram LF, et al. The relationship between paroxetine and the sparteine oxidation polymorphism. Clin Pharmacol Ther. 1992;51:278–287. doi: 10.1038/clpt.1992.23. [DOI] [PubMed] [Google Scholar]
- 52.Özdemir V, Tyndale RF, Reed K, et al. Paroxetine steady-state plasma concentration in relation to CYP2D6 genotype in extensive metabolizers. J Clin Psychopharmacol. 1999;19:472–475. doi: 10.1097/00004714-199910000-00014. DOI: 10.1097/00004714-199910000-00014. [DOI] [PubMed] [Google Scholar]
- 53.Otton SV, Ball SE, Cheung SW, Inaba T, Rudolf RL, Sellers EM. Venlafaxine oxidation in vitro is catalyzed by CYP2D6. Br J Clin Pharmacol. 1996;41:149–156. doi: 10.1111/j.1365-2125.1996.tb00173.x. [DOI] [PubMed] [Google Scholar]
- 54.Fogelman SM, Schmider J, Venkatakrishnan K, et al. O-and N-desmethylation of venlafaxine in vitro by human liver microsomes and by microsomes from cDNA-transfected cells: effects of metabolic inhibitors and SSRI antidepressants. Neuropsychopharmacology. 1999;20:480–490. doi: 10.1016/S0893-133X(98)00113-4. DOI: 10.1016/s0893-133x(98)00113-4. [DOI] [PubMed] [Google Scholar]
- 55.Lessard E, Yessine MA, Hamelin BA, O'Hara G, LeBlanc J, Turgeon J. Influence of CYP2D6 activity on the disposition and cardiovascular toxicity of the antidepressant agent venlafaxine in humans. Pharmacogenetics. 1999;9:435–443. [PubMed] [Google Scholar]
- 56.Veefkind AH, Haffmans PMJ, Hoencamp E. Venlafaxine serum levels and CYP2D6 genotype. Ther Drug Monit. 2000;22:202–208. doi: 10.1097/00007691-200004000-00011. DOI: 10.1097/00007691-200004000-00011. [DOI] [PubMed] [Google Scholar]
- 57.Fukuda T, Nishida Y, Zhou Q, Yamamoto I, Kondo S, Azuma J. The impact of the CYP2D6 and CYP2C19 genotypes on venlafaxine pharmacokinetics in a Japanese population. Eur J Clin Pharmacol. 2000;56:175–180. doi: 10.1007/s002280050737. [DOI] [PubMed] [Google Scholar]
- 58.Llerena A, Alm C, Dahl M-L, Ekqvist B, Bertilsson L. Haloperidol disposition is dependent of debrisoquine hydroxylation phenotype. Ther Drug Monit. 1992;14:92–97. doi: 10.1097/00007691-199204000-00003. [DOI] [PubMed] [Google Scholar]
- 59.Llerena A, Dahl ML, Ekqvist B, Bertilsson L. Haloperidol disposition is dependent on the debrisoquine hydroxylation phenotype: increased plasma levels of the reduced metabolite in poor metabolizers. Ther Drug Monit. 1992;14:261–264. doi: 10.1097/00007691-199206000-00014. [DOI] [PubMed] [Google Scholar]
- 60.Nyberg S, Farde L, Halldin C, Dahl M-L, Bertilsson L. D2 dopamine receptor occupancy during low-dose treatment with haloperidol decanoate. Am J Psychiatry. 1995;152:173–178. doi: 10.1176/ajp.152.2.173. [DOI] [PubMed] [Google Scholar]
- 61.Suzuki A, Otani K, Mihara K, et al. Effects of the CYP2D6 genotype on the steady-state plasma concentrations of haloperidol and reduced haloperidol in Japanese schizophrenic patients. Pharmacogenetics. 1997;7:415–418. doi: 10.1097/00008571-199710000-00013. [DOI] [PubMed] [Google Scholar]
- 62.Mihara K, Suzuki A, Kondo T, et al. Effects of the CYP2D6*10 allele on the steady-state plasma concentrations of haloperidol and reduced haloperidol in Japanese patients with schizophrenia. Clin Pharmacol Ther. 1999;65:291–294. doi: 10.1016/S0009-9236(99)70108-6. [DOI] [PubMed] [Google Scholar]
- 63.Roh H-K, Chung J-Y, Oh DY, Park CS, Svensson J-O, Dahl M-L, Bertilsson L. 2001;52:265–271. doi: 10.1046/j.0306-5251.2001.01437.x. Plasma concentrations of haloperidol are related to the CYP2D6 genotype at low, but not high doses of haloperidol in Korean schizophrenic patientsBr J Clin Pharmacol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bertilsson L, Dahl M-L. Polymorphic drug oxidation. Relevance to the treatment of psychiatric disorders. CNS Drugs. 1996;5:200–223. [Google Scholar]
- 65.Jerling M, Dahl M-L, Åberg-Wistedt A, et al. The CYP2D6 genotype predicts the oral clearance of the neuroleptic agents perphenazine and zuclopenthixol. Clin Pharmacol Ther. 1996;59:423–428. doi: 10.1016/S0009-9236(96)90111-3. [DOI] [PubMed] [Google Scholar]
- 66.Linnet K, Wiborg O. Steady-state serum concentrations of the neuroleptic perphenazine in relation to CYP2D6 genetic polymorphism. Clin Pharmacol Ther. 1996;60:41–47. doi: 10.1016/S0009-9236(96)90165-4. [DOI] [PubMed] [Google Scholar]
- 67.Linnet K, Wiborg O. Influence of CYP2D6 genetic polymorphism on ratios of steady-state serum concentrations to dose of the neuroleptic zuclopenthixol. Ther Drug Monit. 1996;18:629–634. doi: 10.1097/00007691-199612000-00001. [DOI] [PubMed] [Google Scholar]
- 68.Jaanson P, Marandi T, Kiivet RA, et al. 2001. Maintenance therapy of schizophrenic outpatients with zuclopenthixol decanoate: Associations between plasma zuclopenthixol concentrations, extrapyramidal side-effects, tardive dyskinesia and CYP2D6 genotype. Psychopharmacol (in revision)
- 69.Singlas E, Goujet MA, Simmon P. Pharmacokinetics of perhexilene maleate in anginal patients with and without peripheral neuropathy. Eur J Clin Pharmacol. 1978;14:195–201. doi: 10.1007/BF02089960. [DOI] [PubMed] [Google Scholar]
- 70.Shah RR, Oates NS, Idle JR, Smith RL, Lockhart JDF. Impaired oxidation of debrisoquine in patients with perhexilene neuropathy. Br Med J. 1982;284:295–299. doi: 10.1136/bmj.284.6312.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chen S, Chou W-H, Blouin RA, et al. The cytochrome P450 2D6 (CYP2D6) enzyme polymorphism. Screening costs and influence on clinical outcomes in psychiatry. Clin Pharmacol Ther. 1996;60:522–534. doi: 10.1016/S0009-9236(96)90148-4. [DOI] [PubMed] [Google Scholar]
- 72.Arthur H, Dahl M-L, Siwers B, Sjöqvist F. Polymorphic drug metabolism in schizophrenic patients with tardive dyskinesia. J Clin Psychopharmacol. 1995;15:211–216. doi: 10.1097/00004714-199506000-00010. DOI: 10.1097/00004714-199506000-00010. [DOI] [PubMed] [Google Scholar]
- 73.Armstrong M, Daly AK, Blennerhassett R, Ferrier N, Idle JR. Antipsychotic drug-induced movement disorders in schizophrenics in relation to CYP2D6 genotype. Br J Psychiatry. 1997;170:23–26. doi: 10.1192/bjp.170.1.23. [DOI] [PubMed] [Google Scholar]
- 74.Andreassen OA, MacEwan T, Guldbrandsen A-K, McCreadie RG, Steen VM. Non-functional CYP2D6 alleles and risk for neuroleptic-induced movement disorders in schizophrenic patients. Psychopharmacology. 1997;131:174–179. doi: 10.1007/s002130050281. DOI: 10.1007/s002130050281. [DOI] [PubMed] [Google Scholar]
- 75.Kapitany T, Meszaros K, Lenzinger E, et al. Genetic polymorphisms for drug metabolism (CYP2D6) and tardive dyskinesia in schizophrenia. Schizophrenia Res. 1998;32:101–106. doi: 10.1016/s0920-9964(98)00038-3. [DOI] [PubMed] [Google Scholar]
- 76.Ohmori O, Kojima H, Shinkai T, Terao T, Suzuki T, Abe K. Genetic association analysis between CYP2D6*2 allele and tardive dyskinesia in schizophrenic patients. Psychiatry Res. 1999;87:239–244. doi: 10.1016/s0165-1781(99)00065-7. [DOI] [PubMed] [Google Scholar]
- 77.Vandel P, Haffen E, Vandel S, et al. Drug extrapyramidal side effects. CYP2D6 genotypes and phenotypes. Eur J Clin Pharmacol. 1999;55:659–665. doi: 10.1007/s002280050689. [DOI] [PubMed] [Google Scholar]
- 78.Hamelin BA, Dorson PG, Pabis D, et al. CYP2D6 mutations and therapeutic outcome in schizophrenic patients. Pharmacotherapy. 1999;19:1057–1063. doi: 10.1592/phco.19.13.1057.31593. [DOI] [PubMed] [Google Scholar]
- 79.Scordo MG, Spina E, Romeo P, et al. CYP2D6 genotype and antipsychotic-induced extrapyramidal side effects in schizophrenic patients. Eur J Clin Pharmacol. 2000;56:679–683. doi: 10.1007/s002280000222. [DOI] [PubMed] [Google Scholar]
- 80.Chou WH, Yan F-X, de Leon J, et al. Extension of a pilot study: impact from the cytochrome P450 2D6 polymorphism on outcome and costs associated with severe mental illness. J Clin Psychopharmacol. 2000;20:246–251. doi: 10.1097/00004714-200004000-00019. DOI: 10.1097/00004714-200004000-00019. [DOI] [PubMed] [Google Scholar]
- 81.Roder DM, Duff HS, Alternbern DC, Woosley RL. Antiarrythmic activity of the O-desmethyl metabolite of encaidine. J Pharmacol Exp Ther. 1982;221:552–557. [PubMed] [Google Scholar]
- 82.Buchert E, Woosley RL. Clinical implications of variable antiarrythmic drug metabolism. Pharmacogenetics. 1992;20:2–11. doi: 10.1097/00008571-199202000-00002. [DOI] [PubMed] [Google Scholar]
- 83.Siddoway LA, Thompson KA, McAllister CB, et al. Polymorphism of propafenone metabolism and disposition in man: clinical and pharmacokinetic consequences. Circulation. 1987;75:785–791. doi: 10.1161/01.cir.75.4.785. [DOI] [PubMed] [Google Scholar]
- 84.Eichelbaum M, Gross AS. The genetic polymorphism of debrisoquine/sparteine metabolism – clinical aspects. Pharmacol Ther. 1990;46:377–394. doi: 10.1016/0163-7258(90)90025-w. [DOI] [PubMed] [Google Scholar]
- 85.Brynne N, Dalén P, Alván G, Bertilsson L, Gabrielsson J. Influence of CYP2D6 polymorphism on the pharmacokinetics and pharmacodynamics of tolterodine. Clin Pharmacol Ther. 1998;63:529–539. doi: 10.1016/S0009-9236(98)90104-7. [DOI] [PubMed] [Google Scholar]
- 86.Griese V, Zanger UM, Brudermanns U, et al. Assessment of the predictive power of genotypes for the in-vivo catalytic function of CYP2D6 in a German population. Pharmacogenetics. 1998;8:15–26. doi: 10.1097/00008571-199802000-00003. [DOI] [PubMed] [Google Scholar]
- 87.Raimundo S, Fischer J, Eichelbaum M, Griese E-U, Schwab M, Zanger UM. Elucidation of the genetic basis of the common ‘intermediate metabolizer’ phenotype for drug oxidation by CYP2D6. Pharmacogenetics. 2000;10:577–581. doi: 10.1097/00008571-200010000-00001. DOI: 10.1097/00008571-200010000-00001. [DOI] [PubMed] [Google Scholar]