

Abstract
Aims
To study the effects of rifampicin on the pharmacokinetics and pharmacodynamics of nilvadipine.
Methods
Five healthy adult volunteers received nilvadipine (4 mg) orally before and after a 6 day treatment with rifampicin. Blood and urine were collected and assayed for plasma nilvadipine and urinary 6β-hydroxycortisol and cortisol.
Results
The treatment with rifampicin reduced the mean (± s.d.) AUC of nilvadipine from 17.4 ± 8.4 to 0.6 ± 0.4 µg l−1 h (mean difference −16.8 µg l−1 h, 95% CI −9.4, 24.2 µg l−1 h). While the administration of nilvadipine alone elicited a significant (P < 0.05) hypotensive (mean difference for diastolic blood pressure –8 mmHg, 95% CI −4, −12 mmHg) and reflex tachycardia (mean difference 5 beats min−1, 95% CI 1, 9 beats min−1), the treatment with rifampicin abolished these responses. The urinary 6β-hydroxycortisol/cortisol ratio showed a significant (P < 0.05) increase from 10.3 ± 4.0 to 50.3 ± 24.6 by rifampicin: mean difference 40.1, 95% CI 20.4, 59.8.
Conclusions
Because rifampicin may greatly decrease the oral bioavailability of nilvadipine, caution is needed when these two drugs are to be coadministered.
Keywords: calcium channel antagonist, CYP3A4, drug interaction, enzyme induction, nilvadipine, rifampicin
Introduction
Rifampicin may substantially decrease the oral bioavailability of different types of prototype calcium channel antagonists (e.g. verapamil [1] and nifedipine [2]) to the extent that the therapeutic effects of these drugs are attenuated or almost abolished. Previous studies have revealed that these drug interactions can be attributable to induction of hepatic and intestinal CYP3A4 activity by rifampicin [1, 2]. Recently, we reported that rifampicin can also attenuate the hypotensive effects of the newer dihydropyridine calcium channel antagonists (i.e. nisoldipine, nifedipine, barnidipine or manidipine) [3]. In this case report four elderly hypertensive patients, whose blood pressure had been well-controlled by one of the above drugs, developed sustained hypertension after rifampicin was coadministered for the treatment of tuberculosis. These data indicate that rifampicin may affect the disposition not only of nifedipine but also of the newer dihydropyridine calcium channel antagonists.
Nilvadipine is one of the second-generation dihydropyridine calcium channel antagonists widely used for the treatment of essential hypertension in Japan and Europe [4]. Because nilvadipine possesses pharmacokinetic properties similar to those of nifedipine [4], coadministration of rifampicin may be expected to affect the disposition and pharmacological effects of nilvadipine. In the present study, this hypothesis was tested in healthy subjects.
Methods
Five healthy volunteers (one female and four males; age, 32 ± 7 years; body weight, 58 ± 6 kg [mean±s.d.]) participated in the study. All subjects were non-smokers and received no concurrent medication. Physical and laboratory tests including complete blood cell counts, blood biochemistry and urinalysis, showed no abnormalities. The study protocol was approved by the institutional ethics committee and written informed consent was obtained from each subject.
The pharmacokinetics and cardiovascular effects of nilvadipine were studied before and after a 6 day treatment with rifampicin (450 mg day−1, Rifadin®, Daiichi Pharmaceutical Co. Ltd, Japan). The volunteers received nilvadipine (4 mg) orally (Nivadil®, Fujisawa Pharmaceutical Co Ltd, Japan) after an overnight fast and blood samples were withdrawn at 0, 1, 2, 4, 8, 12 and 24 h postdose. The samples were immediately centrifuged and separated plasma was kept frozen at −20 ° C until analysed. In addition, urine was collected over 24 h postdose for assay of 6β-hydroxycortisol and cortisol. Supine and standing blood pressure and pulse rates were measured by sphygmomanometer at baseline and 1 h postdose of nilvadipine.
Plasma nilvadipine concentrations were assayed by capillary column gas chromatography with electron-capture detection [5]. Within-and between-day assay variability was less than 5% and the lower limit of detection with a signal to noise ratio of 2 was 0.1 µg l−1. Urinary concentrations of 6β-hydroxycortisol and cortisol were measured by h.p.l.c. with ultraviolet absorption according to the method of Bienvenu et al.[6]
The area under the plasma concentration-time curves (AUC) of nilvadipine was calculated by use of the trapezoidal rule. Plasma concentrations below the limit of quantification were omitted from the analysis. Comparisons of mean blood pressure and heart rates between the baseline and 1 h postdose of nilvadipine before and after the treatment with rifampicin were made by the Student's t-test for paired data and those for AUC were made by the Wilcoxon signed rank test. Data are expressed as mean±s.d. A P value of < 0.05 was considered statistically significant.
Results
All volunteers completed the study without suffering appreciable adverse drug reactions. Individual plasma nilvadipine concentration-time curves before and after the treatment with rifampicin are shown in Figure 1. In all subjects the AUC of the drug after the treatment with rifampicin were substantially less than that before treatment. The mean peak plasma concentrations for nilvadipine before treatment with rifampicin (6.4 ± 2.5 µg l−1) was approximately 20 times greater than that after treatment (0.3 ± 0.2 µg l−1) (P < 0.05, mean difference −5.6 µg l−1, 95% CI −3.4, −7.8 µg l−1). As shown in Figure 1 plasma nilvadipine concentrations were measurable only around the peak plasma concentrations for most of the subjects after treatment with rifampicin. The mean AUC for nilvadipine before rifampicin treatment (17.4 ± 8.4 µg l−1 h) was approximately 30 times greater than that after treatment (0.6 ± 0.4 µg l−1 h) (P < 0.05, mean difference −16.8 µg l−1 h, 95% CI −9.4, −24.2 µg l−1 h). Thus, mean apparent oral clearance (CL/F) for nilvadipine was increased significantly (P < 0.05) by treatment with rifampicin from 5.2 ± 2.9 l h−1 kg−1 to 155.7 ± 62.8 l h−1 kg−1 (mean difference 150.5 l h−1 kg−1, 95% CI 87.3, 213.7 l h−1 kg−1).
Figure 1.
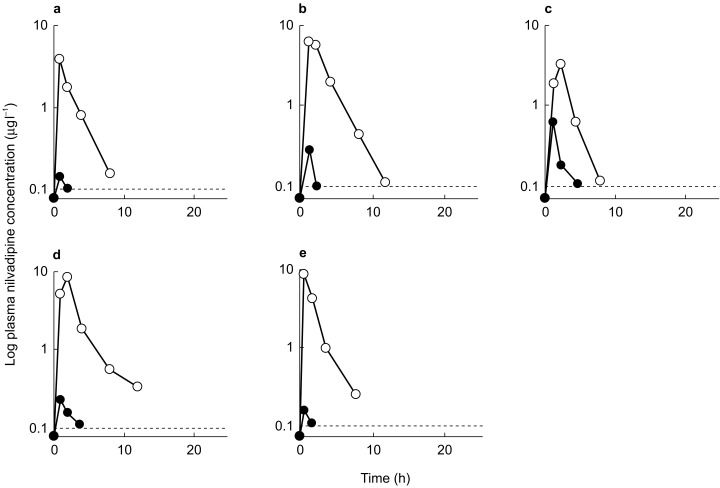
Plasma concentration-time curves of orally administered nilvadipine (4 mg) before (○) and after a 6 day treatment with rifampicin (•) in five healthy subjects designated a to e. The dotted, horizontal lines represents detection limit of the assay for nilvadipine.
The effects of rifampicin treatment on the cardiovascular responses to an orally administered nilvadipine and on the urinary β-hydroxycortisol/cortisol ratio are shown in Table 1. While the administration of nilvadipine elicited a significant (P < 0.05) decrease in the diastolic blood pressure and a significant increase in heart rate in the control period, no significant responses to nilvadipine were observed after the treatment with rifampicin. In addition, rifampicin significantly (P < 0.01) increased the urinary 6β-hydroxycortisol/cortisol ratio (mean difference 40.1, 95% CI 20.4, 59.8).
Table 1.
The effects of a 6 day treatment with rifampicin on the pharmacodynamic responses to the oral administration of a single dose of nilvadipine and the mean urinary 6β-hydroxycortisol/cortisol ratios in healthy subjects.
| Parameter | Control | After rifampicin treatment | ||
|---|---|---|---|---|
| baseline | 1 h post-dose | baseline | 1 h post-dose | |
| Standing | ||||
| SBP (mmHg) | 120 ± 17 | 115 ± 17 | 123 ± 14 | 116 ± 9 |
| DBP (mmHg) | 69 ± 11 | 61 ± 8a | 68 ± 12 | 70 ± 10 |
| HR (beats min−1) | 74 ± 8 | 86 ± 13a | 87 ± 18 | 92 ± 10 |
| Supine | ||||
| SBP (mmHg) | 113 ± 18 | 114 ± 14 | 127 ± 4 | 117 ± 13 |
| DBP (mmHg) | 64 ± 10 | 62 ± 6 | 73 ± 7 | 64 ± 11 |
| HR (beats min−1) | 74 ± 9 | 81 ± 17 | 86 ± 6 | 74 ± 8 |
| 6β-hydroxycortisol/cortisol ratio | 10.3 ± 4.0 | 50.3 ± 24.6b | ||
SBP=systolic blood pressure, DBP=diastolic blood pressure, HR=heart rate.
Mean±s.d. data.
P < 0.05 compared with the values obtained before the administration of nilvadipine,
P < 0.01 compared with the value obtained from the control period.
Discussion
We have shown that a 6 day treatment with rifampicin may substantially reduce the AUC of orally administered nilvadipine to less than 5% of the control value and also attenuate its hypotensive effect and reflex tachycardia in normotensive healthy subjects. The results of the present study are consonant with our anecdotal findings [3] and those previously reported [1, 2] that the coadministration of rifampicin with nifedipine and other dihydropyridines as well as verapamil can abolish the hypotensive and other pharmacological effects of these cardiovascular drugs.
A possible mechanism(s) for the drug interaction between rifampicin and nilvadipine could be induction by the former of hepatic and intestinal CYP3A4 activity as has been shown for other calcium channel antagonists [1, 2]. CYP3A4 dominates the metabolism of most dihydropyridine calcium channel antagonists and it is likely that this CYP isoform plays a major role in the disposition of nilvadipine. Nilvadipine undergoes extensive presystemic elimination, because its oral bioavailability ranges from 14 to 19% [4] and its intestinal absorption is almost complete. The finding that the treatment with rifampicin elicited a 5-fold increase in the urinary 6β-hydroxycortisol/cortisol ratio, an index of hepatic CYP3A activity [6, 7], indicates that there was a substantial induction of this enzyme in the livers of our subjects. However, because rifampicin elicited a quantitatively much greater (30-fold) reduction in the AUC of nilvadipine than the decrease in the urinary 6β-hydroxycortisol/cortisol ratio, induction of CYP3A4 activity in the gut wall and or other mechanism(s) might have also been associated with the reduction in the oral bioavailability of nilvadipine. Westphal et al.[8] reported that rifampicin induces not only CYP3A4 but also a drug efflux pump, P-glycoprotein, in the gut wall in humans. Because there is a substantial overlap in substrate specificity between P-glycoprotein and CYP3A4 [9], rifampicin may diminish the oral bioavailability of substrate common to both proteins to a much greater extent than that expected by the induction of CYP3A4 activity in the liver. At present, it remains unclear whether nilvadipine is a substrate of P-glycoprotein, but Kim et al.[9] reported that nifedipine, a similar dihydropyridine calcium antagonist, is not a good substrate for P-glycoprotein.
Because only a small number of subjects was studied, our statistical inference about the effects of coadministration of rifampicin on the cardiovascular responses to nilvadipine should be interpreted with caution. The reason why quantitatively smaller changes were observed in the pharmacodynamics of nilvadipine as compared with its pharmacokinetics was probably because we performed the study in healthy, normotensive subjects given a single oral dose. However, we consider that the substantial changes in the AUC of nilvadipine elicited by the coadministration of rifampicin would lead to a clinically relevant attenuation of the hypotensive effect of the drug in hypertensive patients. In this context, careful monitoring is recommended when rifampicin is coadministered with nilvadipine and other dihydropyridine calcium channel antagonists in hypertensive patients.
The present study was supported by a grant-in-aid from Meiji Pharmaceutical University.
References
- 1.Fromm MF, Dilger K, Busse D, Kroemer HK, Eichelbaum M, Klotz U. Gut metabolism of verapamil in older people: effect of rifampicin-mediated enzyme induction. Br J Clin Pharmacol. 1998;45:247–255. doi: 10.1046/j.1365-2125.1998.00669.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Holtbecker N, Fromm MF, Kroemer HK, Ohnhaus EE, Heidemann H. The nifedipine–rifampin interaction. Evidence of induction of gut wall metabolism. Drug Metab Dispos. 1996;24:1121–1123. [PubMed] [Google Scholar]
- 3.Yoshimoto H, Takahashi M, Saima S. Influence of rifampicin on antihypertensive effects of dihydropyridine calcium channel blockers in four elderly patients. Nippon Ronen Igakkai Zasshi. 1996;33:692–696. doi: 10.3143/geriatrics.33.692. (in Japanese) [DOI] [PubMed] [Google Scholar]
- 4.Brogden RN, McTavish D. Nilvadipine. A review of its pharmacodynamic and pharmacokinetic properties, therapeutic use in hypertension and potential in cerebrovascular disease and angina. Drugs Aging. 1995;6:150–171. doi: 10.2165/00002512-199506020-00007. [DOI] [PubMed] [Google Scholar]
- 5.Tokuma Y, Fujiwara T, Sekiguchi M, Noguchi H. Determination of nilvadipine in plasma and urine by capillary column gas chromatography with electron-capture detection. J Choromatogr. 1987;415:156–162. doi: 10.1016/s0378-4347(00)83204-1. [DOI] [PubMed] [Google Scholar]
- 6.Bienvenu T, Rey E, Pons G, D'Athis P, Olive G. A simple non-invasive procedure for the investigation of cytochrome P-450 IIIA dependent enzyme in humans. Int J Clin Pharmacol Ther Toxicol. 1991;29:441–445. [PubMed] [Google Scholar]
- 7.Ged C, Roulilllon JM, Pichard L, et al. The increase in urinary excretion of 6β-hydroxycortisol as a maker of human hepatic cytochrome P450IIIA induction. Br J Clin Pharmacol. 1989;28:373–387. doi: 10.1111/j.1365-2125.1989.tb03516.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Westphal K, Weinbrenner A, Zschiesche M, et al. Induction of P-glycoprotein by rifampin increases intestinal secretion of talinolol in human beings: a new type of drug/drug interaction. Clin Pharmacol Ther. 2000;68:345–355. doi: 10.1067/mcp.2000.109797. [DOI] [PubMed] [Google Scholar]
- 9.Kim RB, Wandel C, Leake B, et al. Interrelationship between substrates and inhibitors of human CYP3A and P-glycoprotein. Pharmaceut Res. 1999;16:408–414. doi: 10.1023/a:1018877803319. [DOI] [PubMed] [Google Scholar]