

Abstract
Aims
Short-term disulfiram administration has been shown to selectively inhibit CYP2E1 activity but the effects of chronic disulfiram administration on the activities of drug metabolizing enzymes is unclear. The purpose of this study was to evaluate the effects of disulfiram given for 11 days on selected drug metabolizing enzyme activities.
Methods
Seven healthy volunteers were given disulfiram 250 mg daily for 11 days. Activities of the drug metabolizing enzymes CYP1A2, CYP2C19, CYP2D6, CYP2E1 and N-acetyltransferase were determined using the probe drugs caffeine, mephenytoin, debrisoquine, chlorzoxazone, and dapsone, respectively. Chlorzoxazone was administered before disulfiram administration and after the second and eleventh doses of disulfiram, while the other probe drugs were given before disulfiram administration and after the eleventh disulfiram dose.
Results
Disulfiram administration markedly inhibited chlorzoxazone 6-hydroxylation by more than 95%, but did not affect metabolism of debrisoquine or mephenytoin. Caffeine N3-demethylation was decreased by 34% (P < 0.05). Monoacetyldapsone concentrations were markedly elevated by disulfiram administration resulting in a nearly 16-fold increase in the dapsone acetylation index, calculated as the plasma concentration ratio of monoacetyldapsone to dapsone. CYP-mediated dapsone N-hydroxylation was not significantly altered.
Conclusions
These data suggest that disulfiram-mediated inhibition is predominantly selective for CYP2E1. The magnitude of CYP2E1 inhibition was similar after both acute and chronic disulfiram administration. The effects on caffeine N3-demethylation (CYP1A2) and dapsone metabolism suggest that chronic disulfiram administration may affect multiple drug metabolizing enzymes, which could potentially complicate the use of chronically administered disulfiram as a diagnostic inhibitor of CYP2E1.
Keywords: acetylation, caffeine, chlorzoxazone, cytochrome P450, dapsone, deacetylation, debrisoquine, disulfiram, mephenytoin
Introduction
Disulfiram and its primary metabolite, diethyldithiocarbamate, have been identified as selective mechanism-based inhibitors of human cytochrome P450 (CYP) 2E1 in vitro[1,2]. Kharasch et al.[3,4] demonstrated in normal healthy volunteers that a single 500 mg dose of disulfiram markedly reduced (by 93%) the 6-hydroxylation of chlorzoxazone, a putative index of CYP2E1 activity. They subsequently showed, using probe drugs selectively metabolized by individual CYP enzymes, that disulfiram 500 mg had no effect on the metabolism of coumarin (CYP2A6), tolbutamide (CYP2C9), mephenytoin (CYP2C19), dextromethorphan (CYP2D6) or intravenously administered midazolam (CYP3A) [5,6]. Selective effects were further demonstrated by Damkier et al.[7], who showed that short-term treatment (for 5 days) with disulfiram 200 mg had no effect on the metabolism of caffeine (CYP1A2), tolbutamide (CYP2C9), mephenytoin (CYP2C19), sparteine (CYP2D6) or quinidine (CYP3A). Collectively, these observations support the suggestion that single-dose disulfiram administration could be used to assess the in vivo role of CYP2E1 in the biotransformation of a drug [3,6].
While evidence supports that acute administration of disulfiram results in selective CYP2E1 inhibition, the effect of chronic dosing is unclear. Chronic, long-term disulfiram administration has been shown to inhibit the metabolism of antipyrine, theophylline, caffeine, phenytoin, and warfarin [8–13], drugs for which CYP2E1 is not the predominant pathway of elimination. These observations suggest that with long-term administration (> 5 days) the effect of disulfiram becomes nonselective, which would severely limit its use as a diagnostic CYP2E1 inhibitor for cases in which disulfiram must be administered for longer periods of time, such as when evaluating the in vivo role of CYP2E1 in the metabolism of drugs with long elimination half-lives [14]. Thus, the purpose of this study was to evaluate the effects of disulfiram given for 11 days on the activities of the drug metabolizing enzymes CYP1A2, CYP2C19, CYP2D6, CYP2E1 and N-acetyltransferase using the probe drugs caffeine, mephenytoin, debrisoquine, chlorzoxazone and dapsone, respectively.
Methods
Eight normal healthy male volunteers agreed to participate in this study after providing written informed consent. This study was approved by the local Institutional Review Board. All subjects were nonsmokers (self-reported) and healthy as confirmed by medical history, physical examination, blood chemistries and urinalysis. Subjects were instructed to abstain from caffeine or alcohol-containing products for at least two days before each study visit and none of the subjects was receiving any over the counter or prescription medications.
Subjects received the probe drug chlorzoxazone (250 mg) on three occasions: prior to disulfiram administration, after the second daily disulfiram dose and after the eleventh daily disulfiram dose. Subjects also received, in combination with chlorzoxazone, the probe drugs caffeine (100 mg), dapsone (100 mg), debrisoquine (10 mg), and mephenytoin (100 mg), prior to disulfiram administration and after the eleventh daily dose. Subjects received disulfiram (250 mg orally) each morning and all doses were administered by clinic personnel. All probe drugs were given orally with eight ounces of water, the morning after an overnight fast. The five probe drugs were administered simultaneously as a cocktail, which we have previously shown to be devoid of any interaction at the doses used [15]. In each session, heparinized plasma samples were collected prior to drug administration and at 0.5, 1, 2, 4, 6, 8, and 10 h after probe administration. Urine was collected from 0-8 h into a container with ascorbic acid as a preservative for the unstable dapsone hydroxylamine metabolite. Plasma harvested by centrifugation, and urine aliquots were stored frozen at −20 ° C until analysed.
Analytical techniques
The following drugs and metabolites were measured by high performance liquid chromatographic techniques described previously: caffeine and paraxanthine in plasma [16]; chlorzoxazone and 6-hydroxychlorzoxazone in plasma and 6-hydroxychlorzoxazone in urine [17]; dapsone (DDS) and dapsone hydroxylamine (HDA) in urine and dapsone and monoacetyldapsone in plasma [18]; debrisoquine (DB) and 4-hydroxydebrisoquine (HDB) in urine [19]; and 4′-hydroxymephenytoin (HMP) in urine [15]. The within and between-day coefficients of variation for each of these assays was ≤10%. All of the assay procedures utilized in this study were tested with the other probe drugs and metabolites to ensure that no analytical interference would occur with simultaneous administration.
Data analysis
Chlorzoxazone and 6-hydroxychlorzoxazone pharmacokinetic data were presented in detail previously [14]. In this report, the chlorzoxazone metabolic ratio, calculated as the concentration ratio of 6-hydroxychlorzoxazone to chlorzoxazone in a 4 h plasma sample, was used as an index of CYP2E1 activity [20,21]. The concentration of paraxanthine (1,7 dimethylxanthine) divided by the concentration of caffeine in the 8 h plasma sample was used to assess CYP1A2 activity [22,23]. The ability to N-hydroxylate dapsone (CYP-mediated) was estimated by the urinary recovery ratio [24]:
in which HDA is the urinary recovery of dapsone hydroxylamine in an 8 h urine sample and DDS is the 8 h urinary recovery of dapsone. Acetylator phenotype was defined as the ratio of monoacetyldapsone to dapsone in the 8 h plasma sample; subjects having an acetylation ratio of 0.35 or greater were classified as rapid acetylators [24]. The area under the concentration time curve (AUC) for monoacetyldapsone and dapsone from 0 to 10 h after dapsone administration was calculated by the trapezoidal rule. The activity of CYP2D6 was estimated using the debrisoquine recovery ratio [25]:
where HDB and DB are the urinary recoveries of 4-hydroxydebrisoquine and debrisoquine in 8 h, respectively. The total urinary recovery of 4′-hydroxymephenytoin (µmol) was used as the phenotypic measure of CYP2C19 activity [26].
Statistical analysis
Data are presented as median and range. Statistical test values are Hodges-Lehmann estimates of median differences with exact 95% confidence intervals. Computations were performed using StatXact 4 (Cytel Software Corporation, USA). Differences were considered statistically significant when the 95% confidence intervals excluded zero.
Results
Seven healthy volunteers completed the study. One subject withdrew consent for reasons unrelated to study participation. The four African-American and three Caucasian subjects averaged 29 ± 6 years of age and weighed 77 ± 12 kg. Disulfiram treatment and single-dose probe drug administrations were well tolerated by all study participants.
As expected, disulfiram administration profoundly decreased chlorzoxazone 6-hydroxylation (Table 1; Figure 1). The chlorzoxazone metabolic ratio (Figure 1) was decreased from a median value of 0.86 to 0.04 after acute and 0.04 after chronic disulfiram administration (median difference: −0.66; 95% confidence interval of the difference: [−1.27, −0.40]). The effects of disulfiram on the metabolism of the other CYP probe drugs were far less dramatic (Table 1). There was no significant difference in the 8 h urinary recovery of 4′-hydroxymephenytoin or the debrisoquine recovery ratio, indices of CYP2C19 and CYP2D6 activity, respectively (Table 1). CYP1A2 activity, as measured by the ratio of plasma paraxanthine to caffeine concentrations (caffeine metabolic ratio) at 8 h, was decreased after disulfiram administration in all seven subjects by an average of 34% (P < 0.05) (Tables 1). The dapsone recovery ratio, an index of CYP-mediated dapsone N-hydroxylation, was decreased in five of the six evaluable subjects by an average of 22%, which was not statistically significant (median difference: −0.14; 95% confidence interval of the difference: −0.32, 0.11). One subject was not included in the analysis due to unmeasureable dapsone hydroxylamine in the baseline assessment.
Table 1.
Phenotypic indexes of drug metabolizing enzyme activities before and during concomitant administration of disulfiram (data are median (range)). The difference is the Hodges-Lehmann point estimate of the median difference with the exact 95% confidence intervals (CI).
| Index | Pathway | Baseline | Disulfiram | Difference (95% CI) |
|---|---|---|---|---|
| Caffeine metabolic ratio | CYP1A2 | 0.92 (0.50–2.78) | 0.52 (0.32–2.23) | −0.41 (−0.70, −0.12)* |
| Chlorzoxazone metabolic ratio | CYP2E1 | 0.86 (0.34–1.74) | 0.04 (0.02–0.07) | −0.66 (−1.27, −0.40)* |
| Dapsone recovery ratio | 0.61 (0.51–0.67) | 0.47 (0.30–0.71) | −0.14 (−0.32, 0.11) | |
| Dapsone acetylation ratio | NAT | 0.18 (0.13–0.67) | 2.87 (1.43–16.24) | 6.27 (1.37, 12.25)* |
| Debrisoquine recovery ratioa | CYP2D6 | 0.53 (0.02–0.74) | 0.61 (0.01–0.76) | 0.005 (−0.11, 0.10) |
| 4′-Hydroxymephenytoin recovery (µmol)b | CYP2C19 | 122 (14–192) | 117 (7–203) | −3.5 (−18.5, 43.0) |
The 95% confidence limit excludes zero;
n = 6, one poor metabolizer excluded from analysis;
n = 6, one poor metabolizer excluded from analysis.
Figure 1.
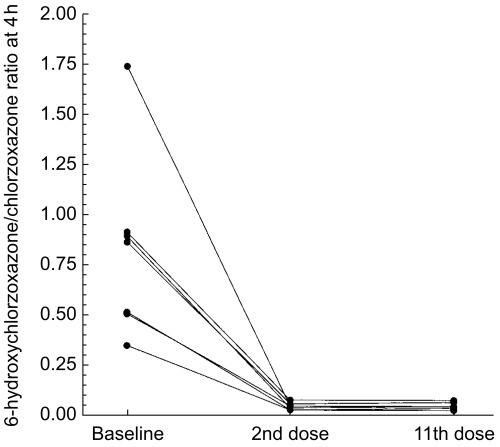
Ratio of 6-hydroxychlorzoxazone to chlorzoxazone in plasma, before disulfiram administration and after the second and eleventh daily disulfiram doses.
Disulfiram administration altered plasma concentrations of both monoacetyldapsone and dapsone as shown in Figure 2. The monoacetyldapsone AUC(0,10 h) increased from 3.5 to 24.0 mg h l−1 (median difference: 20.6; 95% confidence interval of the difference 6.1, 43.2), while the dapsone AUC(0,10 h) decreased from 9.9 to 7.0 mg h l−1 (median difference: −2.9; 95% confidence interval of the difference: −4.7, −1.6). Accordingly, the acetylation ratio of monacetyldapsone to dapsone at 8 h was increased by more than 16-fold in these subjects during disulfiram administration (P < 0.05) (Table 1). The increase in the acetylation ratio was primarily due to the large increase (900%) in monoacetyldapsone concentrations with a concomitant 35% reduction in dapsone plasma concentrations at 8 h (Figure 2). The change in the acetylation ratio was greater in the three phenotypic rapid acetylators as compared with the four phenotypic slow acetylators (Figure 3).
Figure 2.
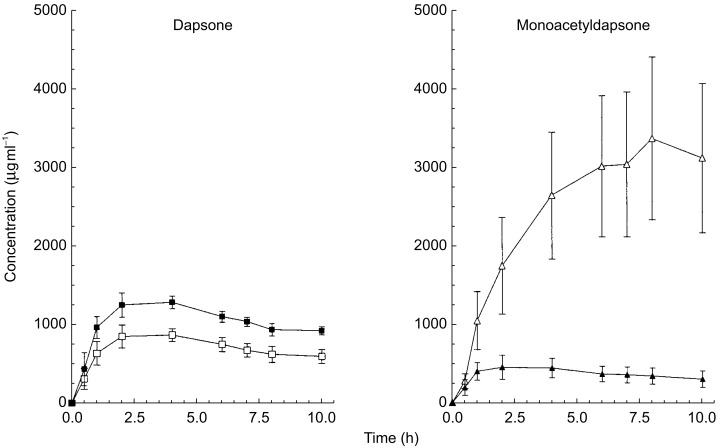
Concentration-time profiles of dapsone (left panel) and monoacetyldapsone (right panel) before (closed symbols) and during (open symbols) disulfiram administration.
Figure 3.
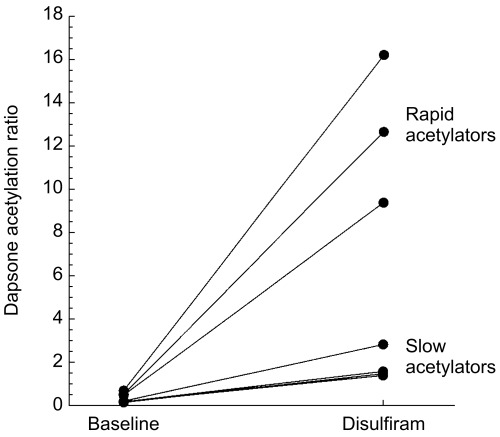
Dapsone acetylation ratio before and during disulfiram administration. The ratio increased 20-fold in phenotypic rapid acetylators and 12-fold in phenotypic slow acetylators.
Discussion
The results of this study confirm previous reports that disulfiram acutely inhibits chlorzoxazone metabolism and extends these observations by showing that the inhibition of chlorzoxazone metabolism was sustained at the same magnitude after 11 days of disulfiram administration [3,4]. The results from the other CYP probe drugs indicate that chronic disulfiram administration has no effect on CYP2D6 or CYP2C19 mediated metabolism, as indicated by the probe drugs debrisoquine and mephenytoin, respectively, and does not alter CYP-mediated dapsone hydroxylation, as shown by the dapsone recovery ratio. In contrast to previous reports with acute disulfiram administration, chronic disulfiram administration modestly decreased caffeine N3-demethylation. In addition, disulfiram administration markedly influenced dapsone pharmacokinetics as evidenced by a substantial increase in monoacetyladapsone plasma concentrations and a slight decrease in dapsone plasma concentrations, resulting in an increase in the dapsone acetylation ratio. Thus, the effects of disulfiram on drug metabolizing enzymes are not confined to CYP2E1.
Disulfiram and diethyldithiocarbamate are potent mechanism-based inhibitors of CYP2E1 in vitro[1,2] and single-dose disulfiram administration has been shown to profoundly inhibit CYP2E1-mediated chlorzoxazone metabolism in vivo[3,4]. In our study, both the formation clearance of 6-hydroxy-chlorzoxazone [14] and the single-point chlorzoxazone metabolic ratio were reduced by approximately 95% after both acute and chronic disulfiram administration. The magnitude of inhibition was similar after acute and chronic inhibition, which is anticipated since the effect is mediated through mechanism-based inhibition. That disulfiram-mediated inhibition was constant throughout the 11 day evaluation period is important for supporting the use of disulfiram to identify the role of CYP2E1 in the in vivo metabolism of a drug. This strategy would require the use of multiple-dose disulfiram to maintain inhibition for drugs with long-elimination half-lives, since it was shown that chlorzoxazone metabolism returns to 50% and 100% of baseline values three and eight days after a single 500 mg dose of disulfiram, respectively [4]. Thus, disulfiram administration should be continued throughout the sampling period for a drug with a long elimination half-life in order to avoid the confounding effect that CYP2E1 enzyme resynthesis would have on activity.
Disulfiram administration did not affect the debrisoquine recovery ratio or the urinary recovery of 4′-hydroxymephenytoin, indices of CYP2D6 and CYP2C19 activity, respectively. A lack of effect on CYP2D6 and CYP2C19 activities with chronic dosing is consistent with the observations following single-dose (500 mg) or five daily doses (200 mg) of disulfiram and the probe drugs mephenytoin (CYP2C19) and dextromethorphan or sparteine (CYP2D6) [6,7]. Thus, these data suggest that administration of disulfiram does not affect CYP2D6 or CYP2C19 activities in vivo.
Disulfiram administration had a modest effect on caffeine N3-demethylation causing a 34% decrease in the caffeine metabolic ratio, which has been validated as a surrogate measure of systemic caffeine clearance [22,23]. The magnitude of change in the caffeine metabolic ratio is consistent with the 30% and 29% decreases in systemic caffeine clearance observed in normal volunteers given disulfiram 250 mg or 500 mg day−1 for 4 days, respectively, and also the 30% decrease in theophylline clearance seen after 1 week of treatment with disulfiram 500 mg [9,10]. In contrast to these findings, Damkier et al.[7] did not observe a change in caffeine metabolic ratio in normal volunteers given five daily doses of disulfiram 200 mg. The smaller dose (200 mg vs 250 mg) and/or shorter duration of exposure (5 vs 11 days) relative to our study may have contributed to the disparate effect on CYP1A2 activity. The effect of single-dose disulfiram (500 mg) on CYP1A2 activity remains unknown. The decrease in caffeine metabolic ratio suggests that disulfiram inhibits CYP1A2 activity following chronic administration. However, it is also known that CYP2E1 contributes to caffeine 1-and 7-demethylations [27–29]. Thus, the magnitude of decrease may reflect decreases in both CYP1A2 and CYP2E1 activities. It is also possible in our study that caffeine metabolism may have been diminished during disulfiram administration by the substantially higher plasma concentrations of chlorzoxazone, since an interaction between these two probes (when both are given at higher doses than used in this study) has been reported [30]. Clearly, the magnitude of inhibition seen with disulfiram is modest compared with other known CYP1A2 inhibitors such as fluvoxamine [31].
The effect of disulfiram administration on dapsone disposition was marked, with the predominate effect observed being an increase in the plasma concentrations of monoacetyldapsone. As shown in Figure 4, the increase in monoacetyldapsone plasma concentrations that was observed could result from either inhibition of the metabolic breakdown of monoacetyldapsone, inhibition of monoacetyldapsone deacetylation, or a combination of both. Monoacetyldapsone hydroxylamine is not recovered in urine to any significant extent and no other urinary metabolites of dapsone have been identified [18,32]. An inhibition of deacetylase activity by disulfiram is plausible since disulfiram was shown to reduce the deacetylation of both diltiazem and cinobufagin in rat liver microsomes [33,34]. In addition, monoacetyldapsone is deacetylated by a carboxylesterase enzyme(s) and it is known that oral administration of disulfiram and its reduced metabolite, diethyldithiocarbamate, inhibits plasma and hepatic microsomal carboxylesterase in rats [35,36]. Concentration-time data for dapsone and monoacetyldapsone following oral administration of monoacetyldapsone indicate that deactylation is much slower in comparison with acetylation [32]. Thus, a decrease in monoacetyldapsone deacetylation would be expected to result in a decrease in dapsone concentrations, as was observed, since the equilibrium between monoacetyldapsone and dapsone would be altered. Characterization of the mechanism of the interaction between disulfiram, dapsone, and monoacetyldapsone will require further study.
Figure 4.
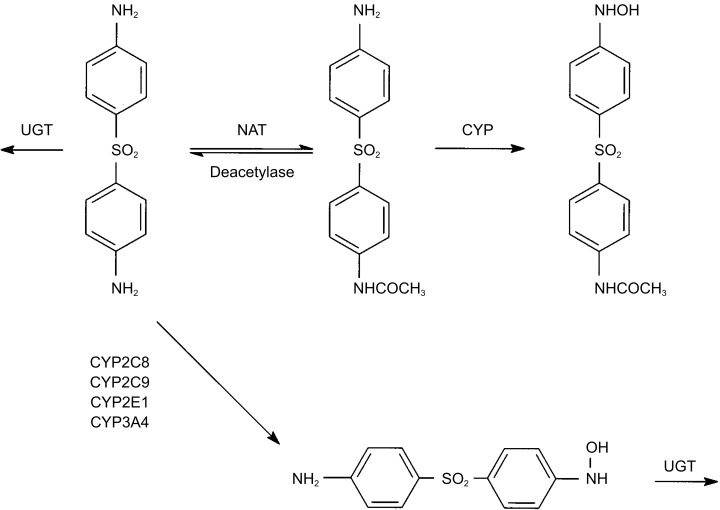
Dapsone metabolic pathways. NAT = N-acetyltransferase, CYP = Cytochrome P450, UGT = uridine 5′-diphosphate [UDP]-glucuronosyltransferase.
Dapsone is commonly used as a probe of N-acetyltransferase activity with the acetylation ratio serving as a single-point index of N-acetyltransferase activity [24]. The acetylation ratio is determined using plasma concentrations of dapsone and monoacetyldapsone, which reach equilibrium approximately 4 h after dosing [24,32]. Disulfiram administration caused a 16-fold increase in the acetylation ratio (Figure 3), due largely to the substantial increase in monoacetyldapsone concentrations. As discussed, the acetylation ratio may have been increased due to apparent effects of disulfiram on monoacetyldapsone metabolism. Clearly factors that could independently affect monoacetyldapsone metabolism (e.g. deacetylation) would confound interpretation of the acetylation ratio and thereby severely limit the use of dapsone as a probe of acetylation in the context of drug–drug interaction studies.
CYP-mediated dapsone metabolism, as estimated using the dapsone recovery ratio, was not affected by disulfiram administration. There is controversy regarding which CYP enzyme(s) is/are most relevant to dapsone metabolism in vivo. While the dapsone recovery ratio was originally proposed as an index of CYP3A activity, recent data demonstrate that in addition to (and possibly more important than) CYP3A4, the enzymes CYP2E1 [37], CYP2C8 and CYP2C9 [38,39] can also hydroxylate dapsone. It has been suggested that at plasma dapsone concentrations typically observed following a single 100 mg dose (∼5 µm), CYP2E1 is primarily responsible for dapsone hydroxylation [37]. However, this is not consistent with the data from the present study, where the dapsone recovery ratio was decreased by only 22% (P > 0.05) in the presence of substantial CYP2E1 inhibition. It is interesting to note that inhibition of monoacetyldapsone deacetylation could result in reduction in the 8 h urinary recovery of dapsone hydroxylamine, since the altered equilibrium (Figure 4) would result in less dapsone being available for subsequent hydroxylation (or potentially, less monoacetyldapsone hydroxylamine being converted to dapsone hydroxylamine).
A limitation of the present study is that there was not an assessment of the effects of chronic disulfiram administration on CYP2C9 and CYP3A activities. Chronic disulfiram administration has previously been shown to decrease phenytoin clearance [12,40] and augment warfarin pharmacodynamics [13] suggesting that disulfiram may affect CYP2C9 activity, although Damkier et al.[7] did not observe a significant change in tolbutamide metabolism (CYP2C9) in five volunteers given five daily doses of disulfiram 200 mg and Svendsen et al.[40] did not observe a change in tolbutamide clearance after four daily doses of disulfiram in 10 volunteers. Data pertaining to the effect of disulfiram on CYP3A in vivo are limited. Single-dose disulfiram did not alter the clearance of intravenously administered midazolam [5] and multiple dose disulfiram given for 5 or 10 days did not alter quinidine 3-hydroxylation [7] or alprazolam clearance [41], respectively.
In conclusion, disulfiram administration substantially inhibited CYP2E1-mediated hydroxylation of chlorzoxazone. The extent of CYP2E1 inhibition observed with a dose of 250 mg was similar after both acute and chronic administration. Disulfiram modestly decreased caffeine N3-demethylation, and had no effect on debrisoquine or mephenytoin metabolism. An interaction between dapsone and disulfiram resulted in an increase in monoacetyldapsone concentrations and an associated increase in the dapsone acetylation ratio; dapsone hydroxylation was unchanged. These data suggest that disulfiram, when used as an inhibitor of CYP-mediated metabolism, is predominately selective for CYP2E1 when given at a daily dose of 250 mg. However, effects on CYP1A2 and deacetylation may potentially confound use of disulfiram as a diagnostic CYP2E1 inhibitor.
References
- 1.Guengerich FP, Kim DH, Iwasaki M. Role of human cytochrome P-450 IIE1 in the oxidation of many low molecular weight cancer suspects. Chem Res Toxicol. 1991;4:168–179. doi: 10.1021/tx00020a008. [DOI] [PubMed] [Google Scholar]
- 2.Snyderwine EG, Kroll R, Rubin RJ. The possible role of the ethanol-inducible isozyme of cytochrome P450 in the metabolism and distribution of carbon disulfide. Toxicol Appl Pharmacol. 1988;93:11–21. doi: 10.1016/0041-008x(88)90021-x. [DOI] [PubMed] [Google Scholar]
- 3.Kharasch ED, Thummel KE, Mhyre J, Lillibridge JH. Single-dose disulfiram inhibition of chlorzoxazone metabolism: a clinical probe for P450 2E1. Clin Pharmacol Ther. 1993;53:643–650. doi: 10.1038/clpt.1993.85. [DOI] [PubMed] [Google Scholar]
- 4.Emery MG, Jubert C, Thummel KE, Kharasch ED. Duration of cytochrome P-450 2E1 (CYP2E1) inhibition and estimation of functional CYP2E1 enzyme half-life after single-dose disulfiram administration in humans. J Pharmacol Exp Ther. 1999;291:213–219. [PubMed] [Google Scholar]
- 5.Kharasch ED, Hankins DC, Baxter PJ, Thummel KE. Single-dose disulfiram does not inhibit CYP2A6 activity. Clin Pharmacol Ther. 1998;64:39–45. doi: 10.1016/S0009-9236(98)90020-0. [DOI] [PubMed] [Google Scholar]
- 6.Kharasch ED, Hankins DC, Jubert C, Thummel KE, Taraday JK. Lack of single-dose disulfiram effects on cytochrome P-450 2C9, 2C19, 2D6, and 3A4 activities: evidence for specificity toward P-450 2E1. Drug Metab Dispos. 1999;27:717–723. [PubMed] [Google Scholar]
- 7.Damkier P, Hansen LL, Brosen K. Effect of diclofenac, disulfiram, itraconazole, grapefruit juice and erythromycin on the pharmacokinetics of quinidine. Br J Clin Pharmacol. 1999;48:829–838. doi: 10.1046/j.1365-2125.1999.00099.x. DOI: 10.1046/j.1365-2125.1999.00099.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Loft S, Sonne J, Pilsgaard H, Dossing M, Poulsen E. Inhibition of antipyrine elimination by disulfiram and cimetidine: the effect of concomitant administration. Br J Clin Pharmacol. 1986;21:75–77. doi: 10.1111/j.1365-2125.1986.tb02825.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Loi CM, Day JD, Jue SG, et al. Dose-dependent inhibition of theophylline metabolism by disulfiram in recovering alcoholics. Clin Pharmacol Ther. 1989;45:476–486. doi: 10.1038/clpt.1989.61. [DOI] [PubMed] [Google Scholar]
- 10.Beach CA, Mays DC, Guiler RC, Jacober CH, Gerber N. Inhibition of elimination of caffeine by disulfiram in normal subjects and recovering alcoholics. Clin Pharmacol Ther. 1986;39:265–270. doi: 10.1038/clpt.1986.37. [DOI] [PubMed] [Google Scholar]
- 11.Poulsen HE, Ranek L, Jorgensen L. The influence of disulfiram on acetaminophen metabolism in man. Xenobiotica. 1991;21:243–249. doi: 10.3109/00498259109039466. [DOI] [PubMed] [Google Scholar]
- 12.Olesen OV. Disulfiram (Antabuse) as inhibitor of phenytoin metabolism. Acta Pharmacol Toxicol. 1966;24:317–322. doi: 10.1111/j.1600-0773.1966.tb00394.x. [DOI] [PubMed] [Google Scholar]
- 13.O'Reilly RA. Interaction of sodium warfarin and disulfiram (Antabuse) in man. Ann Intern Med. 1973;78:73–76. doi: 10.7326/0003-4819-78-1-73. [DOI] [PubMed] [Google Scholar]
- 14.Frye RF, Tammara B, Cowart TD, Bramer SL. Effect of disulfiram-mediated CYP2E1 inhibition on the disposition of vesnarinone. J Clin Pharmacol. 1999;39:1177–1183. [PubMed] [Google Scholar]
- 15.Frye RF, Matzke GR, Adedoyin A, Porter J, Branch RA. Validation of the Pittsburgh cocktail approach to assess selective regulation of drug metabolizing enzymes. Clin Pharmacol Ther. 1997;62:365–376. doi: 10.1016/S0009-9236(97)90114-4. [DOI] [PubMed] [Google Scholar]
- 16.Frye RF, Stiff DD, Branch RA. A sensitive method for the simultaneous determination of caffeine and its dimethylxanthine metabolites in human plasma: Application to CYP1A2 phenotyping. J Liq Chromatogr Rel Techn. 1998;21:1161–1171. [Google Scholar]
- 17.Frye RF, Stiff DD. Determination of chlorzoxazone and 6-hydroxychlorzoxazone in human plasma and urine by high-performance liquid chromatography. J Chromatogr B Biomed Appl. 1996;686:291–296. doi: 10.1016/s0378-4347(96)00227-7. [DOI] [PubMed] [Google Scholar]
- 18.May DG, Porter JA, Uetrecht JP, Wilkinson GR, Branch RA. The contribution of N-hydroxylation and acetylation to dapsone pharmacokinetics in normal subjects. Clin Pharmacol Ther. 1990;48:619–627. doi: 10.1038/clpt.1990.204. [DOI] [PubMed] [Google Scholar]
- 19.Frye RF, Branch RA. Improved high performance liquid chromatographic determination of debrisoquine and 4-hydroxydebrisoquine in human urine following direct injection. J Chromatogr B Biomed Appl. 1996;677:178–182. doi: 10.1016/0378-4347(95)00380-0. [DOI] [PubMed] [Google Scholar]
- 20.Girre C, Lucas D, Hispard E, Menez C, Dally S, Menez JF. Assessment of cytochrome P4502E1 induction in alcoholic patients by chlorzoxazone pharmacokinetics. Biochem Pharmacol. 1994;47:1503–1508. doi: 10.1016/0006-2952(94)90524-x. [DOI] [PubMed] [Google Scholar]
- 21.Frye RF, Adedoyin A, Mauro K, Matzke GR, Branch RA. Use of chlorzoxazone as an in vivo probe of cytochrome P450 2E1: choice of dose and phenotypic trait measure. J Clin Pharmacol. 1998;38:82–89. doi: 10.1002/j.1552-4604.1998.tb04381.x. [DOI] [PubMed] [Google Scholar]
- 22.Fuhr U, Rost KL. Simple and reliable CYP1A2 phenotyping by the paraxanthine/caffeine ratio in plasma and saliva. Pharmacogenetics. 1994;4:109–116. doi: 10.1097/00008571-199406000-00001. [DOI] [PubMed] [Google Scholar]
- 23.Tanaka E, Ishikawa A, Yamamoto Y, et al. A simple useful method for the determination of hepatic function in patients with liver cirrhosis using caffeine and its three major dimethylmetabolites. Int J Clin Pharmacol Ther Toxicol. 1992;30:336–341. [PubMed] [Google Scholar]
- 24.May DG, Arns PA, Richards WO, et al. The disposition of dapsone in cirrhosis. Clin Pharmacol Ther. 1992;51:689–700. doi: 10.1038/clpt.1992.81. [DOI] [PubMed] [Google Scholar]
- 25.Kaisary A, Smith P, Jacqz E, et al. Genetic predisposition to bladder cancer. ability to hydroxylate debrisoquine and mephenytoin as risk factors. Cancer Res. 1987;47:5488–5493. [PubMed] [Google Scholar]
- 26.Wedlund PJ, Aslanian WS, McAllister CB, Wilkinson GR, Branch RA. Mephenytoin hydroxylation deficiency in Caucasians. Frequency of a new oxidative drug metabolism polymorphism. Clin Pharmacol Ther. 1984;36:773–780. doi: 10.1038/clpt.1984.256. [DOI] [PubMed] [Google Scholar]
- 27.Berthou F, Flinois JP, Ratanasavanh D, Beaune P, Riche C, Guillouzo A. Evidence for the involvement of several cytochromes P-450 in the first steps of caffeine metabolism by human liver microsomes. Drug Metab Dispos. 1991;19:561–567. [PubMed] [Google Scholar]
- 28.Gu L, Gonzalez FJ, Kalow W, Tang BK. Biotransformation of caffeine, paraxanthine, theobromine and theophylline by cDNA-expressed human CYP1A2 and CYP2E1. Pharmacogenetics. 1992;2:73–77. doi: 10.1097/00008571-199204000-00004. [DOI] [PubMed] [Google Scholar]
- 29.Tassaneeyakul W, Birkett DJ, McManus ME, et al. Caffeine metabolism by human hepatic cytochromes P450. contributions of 1A2, 2E1 and 3A isoforms. Biochem Pharmacol. 1994;47:1767–1776. doi: 10.1016/0006-2952(94)90304-2. [DOI] [PubMed] [Google Scholar]
- 30.Berthou F, Goasduff T, Lucas D, Dreano Y, Le Bot MH, Menez JF. Interaction between two probes used for phenotyping cytochromes P4501A2 (caffeine) and P4502E1 (chlorzoxazone) in humans. Pharmacogenetics. 1995;5:72–79. doi: 10.1097/00008571-199504000-00003. [DOI] [PubMed] [Google Scholar]
- 31.Jeppesen U, Gram LF, Vistisen K, Loft S, Poulsen HE, Brosen K. Dose dependent inhibition of CYP1A2, CYP2C19 and CYP2D6 by citalopram, fluoxetine, fluvoxamine and paroxetine. Eur J Clin Pharmacol. 1996;51:73–78. doi: 10.1007/s002280050163. [DOI] [PubMed] [Google Scholar]
- 32.Zuidema J, Hilbers-Modderman ES, Merkus FW. Clinical pharmacokinetics of dapsone. Clin Pharmacokinet. 1986;11:299–315. doi: 10.2165/00003088-198611040-00003. [DOI] [PubMed] [Google Scholar]
- 33.Zhang L, Aoki K, Yoshida T, Kuroiwa Y. Deacetylation of cinobufagin by rat liver. Drug Metab Dispos. 1992;20:52–55. [PubMed] [Google Scholar]
- 34.LeBoeuf E, Grech-Belanger O. Deacetylation of diltiazem by rat liver. Drug Metab Dispos. 1987;15:122–126. [PubMed] [Google Scholar]
- 35.Zemaitis MA, Greene FE. Impairment of hepatic microsomal and plasma esterases of the rat by disulfiram and diethyldithiocarbamate. Biochem Pharmacol. 1976;25:453–459. doi: 10.1016/0006-2952(76)90349-x. [DOI] [PubMed] [Google Scholar]
- 36.Preuss CV, Svensson CK. Arylacetamide deacetylase activity towards monoacetyldapsone. Species comparison, factors that influence activity, and comparison with 2-acetylaminofluorene and p-nitrophenyl acetate hydrolysis. Biochem Pharmacol. 1996;51:1661–1668. doi: 10.1016/0006-2952(96)00134-7. [DOI] [PubMed] [Google Scholar]
- 37.Mitra AK, Thummel KE, Kalhorn TF, Kharasch ED, Unadkat JD, Slattery JT. Metabolism of dapsone to its hydroxylamine by CYP2E1 in vitro and in vivo. Clin Pharmacol Ther. 1995;58:556–566. doi: 10.1016/0009-9236(95)90176-0. [DOI] [PubMed] [Google Scholar]
- 38.Gill HJ, Tingle MD, Park BK. N-hydroxylation of dapsone by multiple enzymes of cytochrome P450: Implications for inhibition of haemotoxicity. Br J Clin Pharmacol. 1995;40:531–538. doi: 10.1111/j.1365-2125.1995.tb05797.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winter HR, Wang Y, Unadkat JD. CYP2c8/9 mediate dapsone n-hydroxylation at clinical concentrations of dapsone. Drug Metab Dispos. 2000;28:865–868. [PubMed] [Google Scholar]
- 40.Svendsen TL, Kristensen MB, Hansen JM, Skovsted L. The influence of disulfiram on the half life and metabolic clearance rate of diphenylhydantoin and tolbtamide in man. Eur J Clin Pharmacol. 1976;09:439–441. doi: 10.1007/BF00606562. [DOI] [PubMed] [Google Scholar]
- 41.Diquet B, Gujadhur L, Lamiable D, Warot D, Hayoun H, Choisy H. Lack of interaction between disulfiram and alprazolam in alcoholic patients. Eur J Clin Pharmacol. 1990;38:157–160. doi: 10.1007/BF00265976. [DOI] [PubMed] [Google Scholar]