

Abstract
Neurons contain abundant subsets of highly stable microtubules that resist depolymerizing conditions such as exposure to the cold. Stable microtubules are thought to be essential for neuronal development, maintenance, and function. Previous work has indicated an important role of the microtubule-associated protein STOP in the induction of microtubule cold stability. Here, we developed STOP null mice. These mice were devoid of cold-stable microtubules. In contrast to our expectations, STOP−/− mice had no detectable defects in brain anatomy but showed synaptic defects, with depleted synaptic vesicle pools and impaired synaptic plasticity, associated with severe behavioral disorders. A survey of the effects of psychotropic drugs on STOP−/− mice behavior showed a remarkable and specific effect of long-term administration of neuroleptics in alleviating these disorders. This study demonstrates that STOP is a major factor responsible for the intriguing stability properties of neuronal microtubules and is important for synaptic plasticity. Additionally, STOP−/− mice may yield a pertinent model for study of neuroleptics in illnesses such as schizophrenia, currently thought to result from synaptic defects.
Keywords: Microtubule, neuron, STOP, knock-out, synaptic plasticity, neuroleptic
Neurons contain abundant subpopulations of stable microtubules that resist depolymerizing conditions such as exposure to the cold and to depolymerizing drugs. It is thought that stable microtubules are important for the generation and maintenance of neuronal morphology and function (Baas and Heidemann 1986; Guillaud et al. 1998). Neuronal microtubules are stabilized by microtubule-associated proteins (MAPs). Among these MAPs, only a few exhibit the capacity to reconstitute the microtubule cold and drug stability observed in normal neurons (Houseweart and Cleveland 1999). Cold stability actually represents an extreme state of microtubule stabilization, and it has been argued that the functional importance of the various MAPs found in neurons could be related to their ability to induce microtubule cold stability (Houseweart and Cleveland 1999). Thus, eliminating MAPs such as tau, which cannot induce microtubule cold stability, has little effect in mice (Harada et al. 1994), whereas suppression of MAPs with cold-stabilizing activity such as BPAG1 (Yang et al. 1999) or doublecortin (Francis et al. 1999) has major detrimental effects on the maintenance or development of the nervous system. Although several MAPs can induce microtubule cold stability, previous works have indicated a major contribution of STOP proteins. STOP proteins are calmodulin-binding and calmodulin-regulated MAPs. STOPs are encoded by a single gene (Denarier et al. 1998a) but have several tissue- or developmental-specific variants. Neurons contain the major STOP variants, N-STOP and E-STOP (Bosc et al. 1996; Guillaud et al. 1998). Other cell types such as fibroblasts contain less abundant STOP variants such as F-STOP (Denarier et al. 1998b). Acute STOP inhibition by injection of specific blocking antibodies suppresses microtubule cold stability in both neuronal and nonneuronal cultured cells (Denarier et al. 1998b; Guillaud et al. 1998). Furthermore, in cultured neuronal cells, STOP inhibition impairs neuronal differentiation (Guillaud et al. 1998).
To evaluate the role of STOP directly, we used gene targeting and derived STOP null mice. The lack of STOP induced a dramatic loss of microtubule cold stability in both neuronal and nonneuronal microtubules. Although stable microtubules are thought to provide a key structural support for neuronal growth, adult STOP−/− mice display apparently normal brain histology. However, in the absence of STOP, synaptic abnormalities were observed with depleted synaptic vesicle pools and defects in both short- and long-term plasticity. These anomalies were associated with severe behavioral disorders that were specifically alleviated by long-term administration of neuroleptics. These results demonstrate that STOP is a major factor responsible for microtubule cold-stability and that it is important for synaptic function. Additionally, STOP−/− mice may provide novel opportunities for study of synaptic plasticity and response to neuroleptic drugs, in the context of recent models in which synaptic defects are a causal mechanism for neuroleptic-sensitive diseases such as schizophrenia (Mirnics et al. 2001).
Results
Generation of STOP-deficient mice
STOP-deficient mice (STOP−/− mice) were derived by gene targeting. Exon 1 was replaced with a cassette consisting of the neo gene that conferred positive selection and a LacZ reporter gene (Fig. 1A,a). Mating between heterozygous mice yielded the expected Mendelian ratio of mice bearing the STOP targeted gene. STOP−/− mice were viable, with no obvious anatomical defects. To verify that STOP proteins were absent in homozygous mutants, we compared STOP−/− mice to wild-type mice for STOP content. Immunoblot analysis of brain extracts from adult wild-type mice showed the presence of STOP proteins (E-STOP and N-STOP), whereas no signal was observed with corresponding extracts from STOP−/− mice (Fig. 1A,b). Immunological detection of STOP in brain sections from wild-type mice showed widespread labeling of neuronal tissues, whereas sections from STOP−/− mice remained unstained (Fig. 1A,c).
Figure 1.

STOP suppression in mice. (A) Generation of STOP−/− mice. (a) Genomic organization of STOP gene, showing disruption of STOP exon 1. EI, Eco RI; EV, Eco RV; TK, thymidine kinase. (b) Western blot analysis of STOP expression in wild-type and STOP−/− mice. Brain extracts on immunoblots were assayed for STOP content using polyclonal STOP antibody 23C (Guillaud et al. 1998). Equivalent loading is demonstrated using antibodies specific for β-tubulin. (c) STOP immunostaining in parasagittal brain sections from adult wild-type or STOP−/− mice. Bar, 5 mm. (B) Microtubule stability. Analysis of microtubule cold stability in primary cultures of neuronal cells (a) and of glial cells (b) from wild-type or STOP−/− embryos. Cells from both wild-type mice and STOP−/− mice were either maintained at 37°C or exposed to the cold (0°C for 45 min). Following free tubulin extraction by cell permeabilization (Guillaud et al. 1998), microtubules were stained with anti-β-tubulin (green) and nuclei with Hoescht 33258 (blue). Bars, 20 μm.
Microtubule stability in STOP−/− mice
To assess the consequence of STOP deficiency on microtubule stability, we compared microtubule cold stability in cells from wild-type and STOP−/− mouse embryos. In neuronal cells from wild-type mouse embryos, microtubules resisted exposure to the cold (45 min, 0°C). In contrast, in cells from STOP−/− embryos, all neuronal microtubules depolymerized during cold exposure (Fig. 1B,a). Glial cells from wild-type mouse embryos also contained abundant subpopulations of cold-stable cytoplasmic microtubules that were completely absent in the corresponding cells from STOP−/− mice (Fig. 1B,b). A similar loss of microtubule cold stability was observed in fibroblasts from STOP−/− mice (not shown). These results show a widespread multisystem loss of microtubule cold stability in STOP-deficient mice.
Brain anatomy of STOP−/− mice
Since cold-stable microtubules are believed to be important for neuronal growth (Baas and Heidemann 1986), we expected the brain anatomy of STOP−/− mice to be abnormal. However, systematic examination of the brain by light microscopy did not show obvious structural changes in STOP−/− mice nor evidenced neuronal degeneration (Fig. 1A,c). In wild-type mice, immunolabeling for STOP was most intense in the olfactory bulb glomeruli, hippocampus, and cerebellum (Fig. 2A). Examination of cell layers in these brain regions showed apparently normal organization in STOP−/− mice as illustrated in Figure 2B. Taking advantage of the reporter LacZ gene included in the targeting vector, we did X-gal staining on brain sections. Similar patterns of staining were observed in heterozygous and STOP−/− mice, showing that the cells with high STOP promoter activity were still present in STOP−/− mice (Fig. 2C). This argues against major developmental or degeneration problems associated with the absence of STOP.
Figure 2.

Brain anatomy in STOP−/−, STOP+/−, and wild-type adult mice. (A) STOP distribution in the olfactory bulb (OB), cerebellum (Cb), and hippocampus (Hip) from wild-type mice (wt). Parasagittal brain sections were stained with 23C STOP antibody. (B) Cell layer organization in wild-type (wt) and STOP−/− (−/−) mice, in the brain regions shown in A. Parasagittal brain sections were stained with cresyl-violet. (C) LacZ expression in parasagittal brain sections from heterozygous mice (+/−) and STOP−/− (−/−) mice, revealed by β-galactosidase activity. (D) Barrel field organization of the somatosensory cortex in wild-type (wt) and STOP−/− (−/−) mice. Tangential brain sections were stained to reveal the cytochrome oxidase activity pattern. (E) Mossy fiber organization in wild-type (wt) and STOP−/− (−/−) mice. Staining for zinc by the Timm sulfide-silver method showed similar mossy fiber pathway in wild-type and STOP−/− mice. (F) Dendritic organization of Purkinje cells in wild-type (wt) and STOP−/− (−/−) mice. Cerebellum sections stained for calbindin indicated normal dendritic arborization in STOP−/− mice. (G) Representative examples of CA1 pyramidal cells in wild-type (wt) and STOP−/− (−/−) mice. The pictures were obtained by confocal imaging of neurons intracellularly labeled with neurobiotin. The pyramidal cells shown in STOP−/− were both injected. A total number of 27 STOP−/− CA1 pyramidal cells were examined and exhibit normal somatodendritic organization. Bars, A–E, 0.5 mm; F, 20 μm; G, 40 μm.
Olfactory bulb patterning was normal in STOP-deficient mice, both morphologically (Fig. 2B) and as assessed by quantification of the number of glomeruli per histological section (wild-type mice: 55 ± 3 and STOP−/− mice: 58 ± 4, mean ± s.e.m., n = 2 mice in each group) and by measurements of the glomerulus diameter (wild-type mice: 106 ± 4 μm and STOP−/− mice: 103 ± 2 μm, mean ± s.e.m., n = 2 mice in each group). Flattened sections of the somatosensory cortex showed normal organization of the barrel field in STOP−/− mice as revealed by staining for cytochrome oxidase (Fig. 2D). The detailed analysis of the posteromedial barrel subfield (corresponding to the whisker array) indicates a normal number of barrels (five rows and 32 barrels) in STOP−/− mice.
Lastly, we examined axonal and dendritic organization of neurons expressing a high level of STOP. Staining for zinc revealed a normal organization of the hippocampal mossy fibers in the stratum lucidum of CA3 region without sprouting in dentate gyrus (Fig. 2E). Somatodendritic configuration of CA1 pyramidal cells was also normal as revealed by intracellular labeling (Fig. 2G). Calbindin immunostaining indicated that the typical dendritic arborization of Purkinje cells was also preserved in STOP −/− mice (Fig. 2F). Together these results indicate that there is no disruption of cellular layering, sensory patterning, or axonal and dendritic organization in the brain of STOP−/− mice.
Electron microscopy analysis of synapses
To assess possible consequences of STOP deficiency at the ultrastructural level, we carried out an electron microscopy (EM) examination of the CA1 region of the hippocampus. The hippocampal ultrastructure (not shown) and organization of synapses (Fig. 3A,B) in the wild-type and the STOP−/− mice did not show any major difference. However, in the STOP−/− Schaffer collateral terminals, the number of synaptic vesicles seemed smaller than in the wild-type (Fig. 3A,B). This observation was confirmed by quantifying the surface density of the synaptic vesicles in the presynaptic nerve terminals, in the stratum radiatum region of CA1. A striking twofold decrease in the synaptic vesicle density was observed in STOP−/− mice compared to wild-type mice (Fig. 3E). In the wild-type hippocampus, cryoimmuno-EM studies of STOP showed a widespread, cytoplasmic distribution.STOP labeling was observed in both the dendritic spines in close proximity to postsynaptic densities (Fig. 3C) and in axons, where it seemed slightly more abundant (Fig. 3D).
Figure 3.
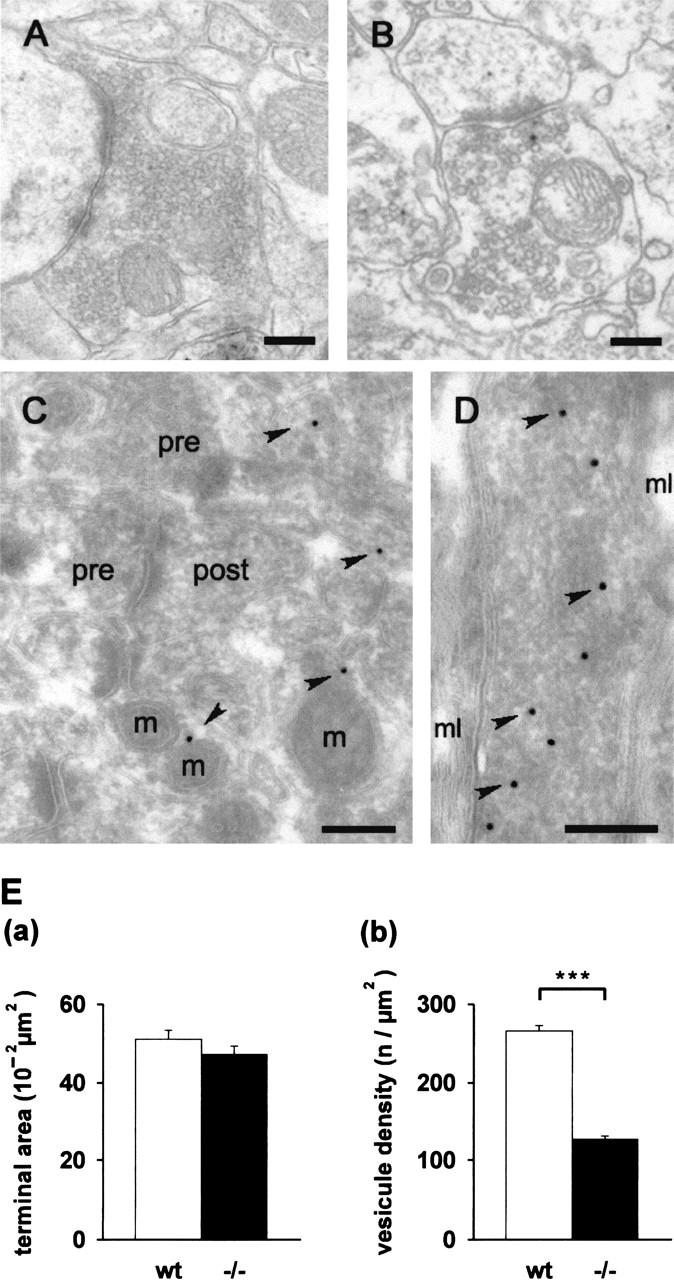
Ultrastructural analysis of synapses and localization of STOP in the hippocampus. Electron micrographs of the stratum radiatum area in CA1. (A,B) Epon-embedded sections. The axons terminals of wild-type (A) and of STOP−/− mice (B) showed similar general ultrastructural organization with the synaptic specialization clearly visible under both conditions. However, the number of synaptic vesicles per synapse area was markedly decreased in the STOP−/− mice. (C,D) Ultrathin cryosections. Immunogold localization of STOP in wild-type mice showed a widespread distribution in the neuronal extensions near the nerve terminals. (C) A dispersed labeling was found in dendritic spines. (D) A more concentrated labeling of STOP was found in the axoplasm, as shown in a longitudinal cross-section of a myelinated axon. The diameter of the gold particles, indicated by arrowheads, is 15 nm. pre, presynaptic terminal; post, postsynaptic terminal; m, mitochondria; mL, myelin sheath. Bars, 200 nm. (E) Quantitative analysis of synaptic vesicle density: (a) Surface of the presynaptic nerve terminals. (b) Synaptic vesicle density, calculated as the ratio of the number of vesicles/nerve terminal surface (after subtraction of the surface area occupied by mitochondria). Results (mean ± s.e.m.) are shown for pooled 150 measurements from three wild-type mice and similar measurements from three STOP−/− mice. The vesicle density was twofold lower in STOP−/− mice compared to wild-type. ***, P < 0.01, t-test.
Biochemical analysis of synaptosomes
We then investigated whether the depletion of the vesicle pool observed by EM in the active zone of synapses reflected a deficit in the import of synaptic proteins. In this case, we expected to see a perturbation of the protein composition of synaptosomes. However, an analysis of a battery of known synaptic proteins in synaptosomal fractions from wild-type and STOP−/− mice did not show detectable differences between the two groups (Fig. 4). Thus, with this method, there was no sign of defects in protein import in STOP−/− mice. In the same study, our assay of STOP showed the presence of the protein in synaptosomes, with a distinct enrichment in a fraction containing proteins associated with insoluble synaptic structures (Fig. 4B).
Figure 4.

Biochemical composition of synaptosomal fractions in STOP−/− mice. (A) Coomassie blue-stained SDS-PAGE of synaptosomal fractions from wild-type and STOP−/− mice. No obvious difference in protein content was found between the two fractions. Total synaptosomal proteins (Sy) and fractions S1, S2, and C3 from wild-type and STOP−/− mice (see Materials and Methods) were analyzed by quantitative immunoblotting with 125I-labeled secondary antibody and phosphorimager detection. (B) Representative immunoblots of synaptosomal proteins. Equal amounts of protein from fractions Sy and equal volumes of each fraction, S1, S2, and C3 from wild-type and STOP−/− mice were analyzed on the same immunoblot. With this procedure, signal intensities in fractions S1, S2, and C3 reflect the distribution of proteins among these fractions. In both phenotypes, protein profiles were similar (except for N-STOP). N-Cadherin, a pre- and postsynaptic protein, was found in all fractions; Rab3, a presynaptic protein, was found only in S1 fraction, and PSD-95, a postsynaptic protein tightly associated with the postsynaptic density, was found in the insoluble fraction C3. N-STOP protein was found in all fractions with a higher concentration in fraction C3. (C) Synaptic proteins profiles. The ratios of synaptic protein levels in STOP−/− vs. wild-type were plotted (n = 2). Results are shown for fraction Sy. The limits of the gray area corresponds to the 95% confidence interval, determined by ANOVA. All observed ratios were within this confidence interval. Similar analysis with fractions S1, S2, and C3 showed no significant difference (data not shown).
Synaptic transmission in STOP−/− mice
The vesicle depletion observed in STOP−/− mice prompted us to examine synaptic function. We first assayed synaptic transmission in the CA1 region of the hippocampus, where STOP expression is intense (Fig. 2A). To assess basic synaptic transmission, we examined the input-output relationship between the fiber excitability and the synaptic strength. The amplitude of fiber volley from Schaffer collaterals was compared to the slope of field excitatory postsynaptic potentials (EPSPs) in the CA1 region of the hippocampus. The experiment was run at different electrical stimulus intensities. Input-output curves were qualitatively similar in STOP−/− and wild-type mice (Fig. 5A,a,b). For quantitative analysis we compared the slopes of these input-output curves between six wild-type and six STOP−/− mice. No significant difference was observed (Fig. 5A,c).
Figure 5.

Analysis of long- and short-term synaptic plasticity in STOP−/− mice. (A) Basic synaptic transmission. The input-output curves corresponding to Schaffer collateral synaptic responses were generated by computing fiber volley amplitude against field EPSP slope in a slice from one wild-type (a) or from one STOP−/− mouse (b). (c) Summary results of input-output curves obtained from six wild-type and six STOP−/− mice. The slopes of the curves were not significantly different, showing normal basic synaptic transmission in STOP−/− mice. (B) LTP experiments at the Schaffer collateral-CA1 pyramidal cell synapses. (a) A high-frequency stimulation (tetanus, four 100 Hz, duration 1 sec stimuli, given 10–20 sec apart) induced a long-term increase in the EPSP slope in a slice from wild-type mice. Sample traces (average of five successive sweeps) were taken 5 min before the tetanus (thin line) and at the end of the experiment (thick line). (b) A similar tetanus induced only a weak increase in the EPSP slope in a slice from STOP−/− mice. Sample traces were as in a. (c) Summary of LTP experiments (mean ± s.e.m.) in wild-type and STOP−/− mice. Initial EPSP slopes were normalized in each experiment using the averaged slope value during the control period (−10 to 0 min). Data were from 13 and nine slices obtained from seven wild-type and six STOP−/− mice, respectively. Results showed significant impairment of LTP in STOP−/− mice (P = 0.0007, unpaired t-test, computed at 30–40 min). (C) LTD experiments at the Schaffer collateral-CA1 pyramidal cell synapses. (a) A low-frequency stimulation (LFS, 1 Hz, 15 min) induced a long-term decrease in the EPSP slope in slices from wild-type mice. Samples traces (average of five successive sweeps) were taken 5 min before LFS (thin line) and at the end of the experiment (thick line). (b) LFS did not induce a long-term decrease in the EPSP slope in slices from STOP−/− mice. Samples traces were as in a. (c) Summary of LTD experiments (mean ± s.e.m.) in wild-type and STOP−/− mice. Data were from 15 and nine slices from nine wild-type and six STOP−/− mice, respectively. Results showed significant impairment of LTD in STOP−/− mice (P = 0.01, unpaired t-test, computed at 40–45 min). (D) Depolarization during tetanic stimulation of Schaffer collateral. Summary graph quantifying depolarization during tetanic stimulation. Depolarization was calculated at 300 msec from the beginning of the first 100 Hz stimulus. Experiments were run using 11 wild-type and eight STOP−/− slices obtained from seven wild-type and six STOP−/− mice, respectively. Results were not significantly different between wild-type and STOP−/− slices. (E) NMDA response at Schaffer collateral-CA1 pyramidal cell synapses. (a) Averaged traces of evoked EPSCs (average of 15 traces) obtained from a CA1 pyramidal cell recorded in a STOP−/− slice. (Left) Recordings first done in picrotoxin (PTX, Vhold = +30 mV), then in the presence of added 10 μM NBQX (PTX + NBQX) to isolate the NMDA receptor-mediated response. (Right) plot of the difference between PTX and PTX+NBQX traces (AMPA response). The PTX trace (NMDA + AMPA response) is also shown for comparison. (b) Summary graph of the NMDA/AMPA ratio obtained by computing NMDA EPSC amplitudes against AMPA EPSC amplitudes in seven and seven cells from four wild-type and four STOP−/− mice, respectively. No significant differences were observed between wild-type and STOP−/− cells. The NMDA/AMPA ratio was quantified for a stimulus intensity corresponding to two times the threshold for evoked EPSCs. (F) Quantal analysis of strontium-induced asynchronous EPSCs in CA1 pyramidal cells. Superimposed traces of asynchronous EPSCs obtained from neurons in wild-type and in STOP−/− mice (recordings done in 8 mM SrCl2 and in picrotoxin, Vhold = −70 mV). Summary graph of the asynchronous AMPA EPSCs obtained in nine and nine cells from five wild-type mice and five STOP−/− mice, respectively. No significant differences were observed between wild-type and STOP−/− cells. (G) Posttetanic potentiation (PTP) of Schaffer collateral synaptic transmission. A high-frequency stimulation in the presence of NMDA antagonist D-APV (50–100 μM) induced a transient increase in the EPSP slope. Data were from six and 10 slices obtained from four wild-type and five STOP−/− mice, respectively. Results showed impaired PTP in STOP−/− mice (P = 0.04, unpaired t-test from 0 to 30 sec after tetanus). (H) Paired pulse facilitation (PPF) of Schaffer collateral synaptic transmission. Data were from seven and 12 slices obtained from four wild-type and five STOP−/− mice, respectively. PPF was not significantly modified in STOP−/− mice. (I) Hippocampal mossy fiber frequency facilitation. Data were from 10 and 12 slices from six wild-type and seven STOP−/− mice, respectively. In wild-type mice, repetitive stimulation of mossy fiber synapses using stimulation frequencies from 0.033 to 1 Hz resulted in an over threefold increase in the amplitude of mossy fiber response. Facilitation was significantly impaired in STOP−/− mice (P = 0.03, unpaired t-test, data computed for 1-Hz stimulation frequency).
We then examined the quantal amplitude of the synaptic response in the mutant mice by using whole-cell patch clamp techniques. The quantal amplitude of AMPA-mediated synaptic response was estimated by measuring strontium-induced asynchronous excitatory postsynaptic currents (aEPSCs), in nine anatomically identified CA1 pyramidal cells from STOP−/− or wild-type mice. No difference in the amplitude of aEPSCs was observed (Fig. 5F). These results indicate normal basic synaptic transmission in STOP−/− mice.
We then examined synaptic plasticity. In synaptic plasticity experiments, the synaptic response to standard stimuli is measured by the slope of EPSP. Baseline values of the slopes are determined using repetitive stimulation at low frequencies (0.03–0.1 Hz). At time zero, a conditioning stimulation protocol is applied. Synaptic adaptation is evidenced by a stable deviation of EPSP slope values from the baseline values. A high-frequency conditioning protocol (100 Hz, duration 1 sec, tetanus) applied at the Schaffer collateral-CA1 pyramidal cell synapses produced a stable increase in EPSP slopes in slices from wild-type mice (Fig. 5B,a), demonstrating synaptic potentiation in these mice. This potentiation persisted over 30 min, and such a persistence defines long-term potentiation (LTP). The same protocol produced potentiation of synaptic transmission reduced by threefold in normalized experiments using slices from STOP−/− mice (Fig. 5B,b,c).
We then examined long-term depression (LTD) at the same Schaffer collateral-CA1 pyramidal cell synapses. We used the classical low-frequency conditioning protocol (LFS, 1 Hz, 900 pulses). Slices from STOP−/− mice showed a significant decrease in the amplitude of LTD compared to wild-type mice (Fig. 5C). Thus, both LTP and LTD were impaired in STOP−/− mice. LTP and LTD depend crucially on NMDA receptor (for review, see Bear and Malenka 1994), and a possible basis of this perturbation is an altered receptor activity in STOP−/− mice. However, the NMDA receptor activation during tetanus (Fig. 5D) and the basal NMDA receptor activity, measured as the ratio of NMDA/AMPA-evoked postsynaptic currents in whole-cell patch clamp recordings (Fig. 5E), were similar in wild-type and STOP−/− mice.
As shown in Figure 5B, synaptic potentiation in STOP−/− mice was altered during the first minutes following tetanus, as at later timepoints. This prompted us to test whether short-term plasticity was impaired at the Schaffer collateral-CA1 pyramidal cell synapses. We investigated posttetanic potentiation (PTP) and paired pulse facilitation (PPF). Like LTP, PTP is a form of synaptic potentiation following a tetanus (100 Hz, duration 1 sec, tetanus). However, PTP persists only for minutes following tetanus and, unlike LTP, is not blocked by NMDA receptor antagonists. PPF is another form of short-term synaptic plasticity observed when synapses are stimulated with paired stimuli. PPF is defined as an increase in the synaptic response following the second stimulus. PTP was reduced in STOP−/− mice (Fig. 5G). In contrast, PPF was unaffected over a large range of decreasing extracellular calcium concentrations in STOP−/− mice (Fig. 5H; data not shown).
We next examined synaptic plasticity at the mossy fiber-CA3 pyramidal cell synapses. No significant differences in either LTP or PPF between STOP−/− and wild-type mice were observed (data not shown). To further examine short-term plasticity, mossy fibers were stimulated using increasing frequencies from 0.033 to 1 Hz. This protocol normally induces a large, but transient, increase in the amplitude of mossy fiber EPSP, a phenomenon called frequency facilitation. The amplitude of frequency facilitation was significantly decreased in mutant mice compared to wild-type mice (Fig. 5I).
Taken together these results show that several distinct forms of long-term and short-term plasticity are affected in different regions of the hippocampus, in STOP−/− mice.
Behavioral study of STOP-deficient mice
Although STOP−/− mice were fully viable, they consistently exhibited atypical behaviors. Phases of intense activity, apparently without goal orientation, accompanied by frequent random shifts between activities were common. Occasionally, the mice displayed crisis, lasting over 20 min, during which the animals continuously circled the cage or demonstrated burrowing motion. The same mice also went through periods of freezing behavior, during which they remained immobile and were unreactive to their environment. Such crises did not mimic epilepsy-like events and were never observed in wild-type mice. We used video recording for quantitative assessment of mouse behavior. The time spent by the mice feeding, sleeping, grooming, walking, or remaining still while awake was measured over a 3-h period during the night (Fig. 6A). Compared to wild-type mice, STOP−/− mice spent more time walking or remaining still while awake, with a decrease in the time spent feeding and sleeping (Fig. 6A,a). The mutant mice also displayed increased shifting between behavioral states, mainly due to a significantly larger number of periods of walking and stillness (Fig. 6A,b). State shifts in STOP−/− mice often broke characteristic sequences of activity. For instance, in wild-type mice, 71% of the sleeping phases were preceded by a phase of grooming. The corresponding frequency was 47% in STOP−/− mice (Fig. 6A,c), when the expected value for random activities was 35%. These quantitative results substantiated the global impression of purposeless and disorganized activity observed in STOP−/− mice.
Figure 6.

Behavioral defects in STOP−/− mice. (A) Mouse activities. Mouse activities (sleeping, feeding, grooming, walking, and remaining still while awake) were videotaped during a 3-h period. n = 11 for both wild-type and STOP−/− mice. (a) Time spent in each activity. Each block corresponds to an activity as indicated (left panel). STOP−/− mice spent more time walking and remaining still than wild-type mice, with a decrease in the time spent sleeping and feeding. (b) Number of occurrences of each activity. Compared to wild-type mice, STOP−/− mice showed a higher number of activity shifts, with increased occurrence of phases of walk and stillness. (c) Percent of grooming phases followed by a sleeping phase (GS) over total number of sleeping phases (S; mean ± s.e.m). Percents were calculated for each mouse and averaged. The GS sequence typical of wild-type mice was often broken in STOP−/− mice. (B) Anxiety-related behavior in STOP−/− mice. Anxiety-related behavior was assessed in eight wild-type mice and six STOP−/− mice using the light/dark test. (a) Time spent in the lit compartment over a 5-min period (mean ± s.e.m.). (b) Number of transitions between the two compartments (mean ± s.e.m.). Compared to wild-type mice, STOP−/− mice spent less time in the lit compartment and showed a smaller number of transitions between compartments. (C) Social behavior in STOP−/− mice. Social behavior was assessed using the resident-intruder test. (a) Social investigation. Time spent by the resident male actively pursuing social investigation of the intruder male (mean ± s.e.m.; n = 11 for wild-type mice and n = 13 for STOP−/− mice). (b,c) Intermale aggression. Aggression tests were conducted for two consecutive days and were recorded on day 2. (b) Number of aggressive encounters. (c) Time spent in fighting (mean ± s.e.m.; n = 11 for wild-type and n = 10 for STOP−/−). −/−, STOP−/− mice; wt, wild-type mice; *, P<0.05; **, P<0.02; ***, P<0.01, Mann-Whitney nonparametric U-test. (D) Nurturing defects in STOP−/− mice. (a) Photographs of wild-type and STOP−/− primiparous females showing typical postures shortly after giving birth. Wild-type mice: the pups are in the nest. STOP−/− mice: the pups are scattered within the bedding. (b) Pup retrieving test in wild-type and STOP−/− mice. Pup retrieving tests were run using virgin females or males as indicated. Trained mice were exposed to three pups placed in the three corners of the cage distant from the nest, and the number of pups retrieved by each mouse was scored. Scores were averaged for each genotype and are plotted on this figure (mean ± s.e.m.). n = 9 for both wild-type and STOP−/− females. n = 10 for wild-type males and n = 9 for STOP−/− males. −/−, STOP−/− mice; wt, wild-type mice.
To evaluate the major classes of behavioral tasks (Crawley 1999), we applied a series of standard behavioral tests. Anxiety-like behavior was evaluated using the light–dark crossing task (Misslin et al. 1990) in which the animals are allowed to choose between a dark and an adjacent bright compartment. Although mice prefer the dark compartment, they normally exhibit brief incursions into the bright compartment. Anxiety-like behavior is manifested by a decrease in the time spent in the bright compartment and in the number of transitions between the two boxes. Compared to wild-type mice, STOP−/− mice showed dramatic signs of anxiety-like behavior, spending virtually all of the time in the dark compartment with very few transitions between the light and dark compartments (Fig. 6B). To assess short-term memory and learning in wild-type and STOP−/− mice, we used an object recognition task (Meziane et al. 1998). In this test, the animals were challenged to discriminate between a familiar object and a novel object. Wild-type mice (n = 6) performed the test successfully, whereas STOP−/− mice (n = 8) were unable to perform the test (data not shown). These mice showed no apparent interest in the objects, suggesting perturbed interactions with their physical environment. To assess social behavior, we monitored both social investigation and intermale aggression, using the resident–intruder test (Nelson et al. 1995; Mohn et al. 1999). Both active social investigation and aggressive encounters were dramatically reduced in STOP−/− mice compared to controls (Fig. 6C), indicating severe social withdrawal in mutant mice.
Postnatal death in the progeny from STOP−/− female mice
Progeny from heterozygous males and females are viable and develop normally. However, pups born from heterozygous males crossed with STOP−/− females died in the perinatal period within 24–48 h after birth, regardless of their genotype. Among 161 pups from 20 STOP−/− females, none survived the perinatal period. The dead pups were never cannibalized. When cross-fostered to wild-type mothers, pups from STOP−/− mothers were raised to weaning. Thus, pup death was directly related to the mother's genotype. To determine the cause of death, we first examined obvious explanations such as suckling defects due to a lack of olfactory cues in the nipples of STOP−/− females. When pups from STOP−/− mice were left with their mother but repetitively placed in nursing position by human intervention, all of the pups showed nipple searching and attachment behavior. The normal expression of this guided behavior suggests that the STOP−/− females emit the olfactory cues necessary for suckling. In addition, in these conditions, milk could be observed in the pups' stomachs. Thus, pup death was not due to lactation defects in STOP−/− females.
The most likely possibility then was that pup death was due to an abnormal behavior of STOP−/− mothers. Indeed, STOP−/− females, whether from BALBc/129 SvPas or pure 129 SvPas backgrounds, showed severe defects in their nurturing behavior. Normal nurturing behavior in mice includes building a nest and retrieving pups. Such behavior was uniformly observed in both wild-type (Fig. 6D,a) and heterozygous mice (data not shown). Although STOP−/− mothers were never aggressive toward the pups, they showed a consistent defect in maternal behavior (Fig. 6D,a). The nurturing behavior of STOP−/− mothers did not improve with multiple pregnancies. Retrieving defects could be due to a failure of the STOP−/− mothers to recognize their progeny. The major sensory cue in this behavior is thought to be olfaction (Gandelman et al. 1971); however, STOP−/− females showed normal behavior in the hidden food olfactory test (data not shown) and, when placed close to their progeny, obviously sniffed the pups. To test whether the nurturing deficiency observed in STOP−/− mice was related to the hormonal status, nurturing behavior was examined in virgin females and in males, using standard tests of pup retrieval (Brown et al. 1996). Pup retrieval was significantly impaired in such female and male STOP−/− mice (Fig. 6D,b). Thus, the nurturing defect observed in STOP−/− mice was independent of obvious organic defects and of the hormonal status.
Effects of anxiolytics and neuroleptics on STOP−/− mice behavior
The synaptic defects and the multiple behavioral disorders observed in STOP-deficient mice were reminiscent of current models of schizophrenia (Mohn et al. 1999; Mirnics et al. 2001). This prompted us to test the action of psychotropic drugs on STOP−/− mice behavior. Because maternal behavior is highly conserved among mammals (Fleming 1989), it provided a stringent criterion to evaluate drug influence in a complex deficit. In pilot experiments (data not shown), STOP−/− mice were treated with different dosages of either anxiolytic (diazepam) or neuroleptics (chlorpromazine, haloperidol, or clozapine) using a variety of schedules. Both types of drugs administered at the time of or shortly following delivery were apparently ineffective in improving nurturing behavior in STOP−/− mothers. When mice were treated with various drugs beginning at least 6 d prepartum there was a positive effect, although incomplete, of neuroleptics on nest building, maternal care, and pup retrieving in STOP−/− mice, with best results yielded by a combination of chlorpromazine and haloperidol. For quantitative assessment of treatment effects, wild-type and STOP−/− mice were subjected to the test of pup retrieval, following various drug treatments. The severe impairment in pup retrieval observed in untreated STOP−/− mothers was only slightly and not significantly improved by diazepam administration (Fig. 7A,a). In contrast, both STOP−/− mothers treated with neuroleptics and wild-type mothers succeeded in the pup retrieval test (Fig. 7A,a,b). However, no pup survival was observed in either treated or untreated groups of STOP−/− females. These results indicate a limited but specific beneficial effect of short-term administration of neuroleptics on behavior in STOP−/− mice.
Figure 7.

Effects of neuroleptics on behavioral and synaptic defects in STOP−/− mice. (A) Effect of short-term drug treatment on pup retrieving in wild-type and STOP−/− postpartum females. Pup retrieving was tested within the first day postpartum, in mice treated with neuroleptics (haloperidol-chlorpromazine) or anxiolytic (diazepam), or in untreated mice. Drug administration began 6–8 d before delivery, and continued through delivery. Females were exposed to three pups from their own litter, and the number of pups retrieved by each female over a 30-min period was scored. (a) Retrieving scores. Scores were averaged for each genotype and treatment (mean ± s.e.m.; n = 6 for both wild-type and STOP−/− mice in each group). −/−, STOP−/− mice; wt, wild-type mice; *, P<0.05; **, P<0.02; ***, P<0.01, Mann-Whitney nonparametric U-test. (b) Photographs showing the end result of pup retrieving tests in wild-type and STOP−/− females. In the absence of treatment, STOP−/− females failed to retrieve pups, whereas neuroleptic-treated STOP−/− mice and wild-type females retrieved the three pups, which are underneath the mothers. (B) Effect of long-term neuroleptic treatment on pup survival in wild-type and STOP−/− mice. Pup survival was never observed in untreated STOP−/− mice (n = 20) or in short-term neuroleptic-treated STOP−/− mice (n = 8). In contrast, pup survival was observed in four of seven STOP−/− mice subjected to long-term (4 mo) neuroleptic treatment. −/−, STOP−/− mice; wt, wild-type mice; *, P<0.05; **, P<0.02; ***, P<0.01, Fisher exact test. (C) Effect of long-term neuroleptic treatment on synaptic plasticity in wild-type and STOP−/− mice. Mice were treated with neuroleptics for 4 mo starting at weaning. No neuroleptics were added during the slice recording. (a) LTP experiments at the Schaffer collateral-CA1 pyramidal cell synapses in wild-type and STOP−/− mice. Initial EPSP slopes were normalized in each experiment using the averaged slope value during the control period (−10–0 min). Data (mean ± s.e.m.) were from eight and seven slices obtained from five wild-type and five STOP−/− mice, respectively. LTP was still impaired in STOP−/− mice (P = 0.018, t-test computed at 30–40 min). (b) Posttetanic potentiation (PTP) of Schaffer collateral synaptic transmission. Data were from nine and 14 slices obtained from six wild-type and seven STOP−/− mice, respectively. Following long-term neuroleptic treatment, PTP was not significantly different in wild-type and in STOP−/− mice (P = 0.79, unpaired t-test from 0 to 30 sec after tetanus).
We then tested whether a longer neuroleptic treatment could induce pup survival in STOP−/− mothers. Seven STOP−/− and seven wild-type females were subjected to daily administration of a combination of chlorpromazine and haloperidol, for 4 mo, starting at weaning. This was maintained through subsequent pregnancy, delivery, and the postpartum period. The seven wild-type mice showed normal maternal behavior and all of their pups survived (Fig. 7B). Remarkably, in four of the seven STOP−/− females subjected to this long-term regime, nurturing improvement was sufficient to permit pup survival with ratios of surviving pups to newborns of 3/11, 4/8, 2/4, and 1/5 for the four mice. The occurrence of pup survival in long-term neuroleptic-treated STOP−/− mice was striking because, in the absence of such treatment, pup survival never occurs among the progeny from STOP−/− mothers (Fig. 7B). These results indicate a remarkable capacity of long-term neuroleptic administration in reestablishing maternal abilities compatible with pup survival in STOP−/− mice.
We next tested whether neuroleptics' effects on behavior were accompanied by modifications of synaptic response. On the input-output curves of field EPSPs, we found no change in basic synaptic transmission in the CA1 region of the hippocampus following long-term neuroleptic treatment whether in wild-type or STOP−/− mice (data not shown). In LTP experiments (Fig. 7C), there was still a significant difference between wild-type and STOP−/− mice at 30–40 min following long-term neuroleptic administration, but potentiation was apparently improved at early times following tetanus in STOP−/− mice. Based on these results, we tested the effect of long-term neuroleptic treatments on PTP. Indeed, PTP experiments showed an apparent increase in the synaptic response in STOP−/− mice following neuroleptic administration (Fig. 7C).
Discussion
Microtubules assembled from pure tubulin in vitro are dynamic structures that exhibit both treadmilling behavior and spontaneous length fluctuations (Margolis and Wilson 1978; Mitchison and Kirshner 1984). Such dynamic microtubules are strongly sensitive to modifications in their physicochemical environment. For instance, microtubule exposure to the cold or to depolymerizing drugs induces rapid polymer disassembly. The intrinsic instability of microtubules seems to be intimately related to many of their basic functions in cells (Desai and Mitchison 1997). However, stable polymers resistant to exposure to the cold and to depolymerizing drugs are also encountered among living cells (Baas et al. 1994; Lieuvin et al. 1994). For instance, in mature neurons virtually all microtubules are cold-stable. The origin of such microtubule stability has been a long-standing enigma. The marked reduction of cold-stable microtubules observed in STOP−/− mice establishes a major role for STOP proteins in microtubule cold stability.
It has long been thought that cold-stable, nondynamic microtubules in neurons were required to sustain neurite extensions using a microtubule cytoskeleton whose maintenance did not require continuous energy consumption (Baas and Heidemann 1986; Guillaud et al. 1998). Remarkably, in the present study, we demonstrate that mice completely devoid of cold-stable microtubules fail to display any obvious disturbance of neuronal cell morphology, differentiation, or survival. The absence of cold-stable polymers seemed to principally affect synaptic transmission.
Because synaptic plasticity involves morphogenetic events at synapses and spines, and vesicle trafficking, it is believed that microtubules and microtubule effectors are important for this aspect of synaptic transmission (for review, see van Rossum and Hanisch 1999). In the present study we found a twofold reduction of the synaptic vesicle pool in CA1 hippocampal nerve terminals. Our biochemical data argue against a major perturbation of synaptic proteins import from the soma through microtubule-dependent vesicle transport (for review, see Goldstein and Yang 2000). In the absence of major defects in protein import, the size of the synaptic vesicle pool may depend on the kinetics of vesicle recycling from the apical membrane and of vesicle budding from the endoplasmic reticulum (for review, see Calakos and Scheller 1996). STOP deficiency may perturb synaptic vesicle turnover by affecting microtubule dynamics or microtubule interaction with motors.
The observed reduction in the size of vesicle pools may account for the perturbations of hippocampal CA1 PTP and mossy fiber frequency facilitation in STOP-deficient mice, because both forms of synaptic plasticity involve presynaptic vesicle trafficking (Salin et al. 1996; Thomson 2000). In contrast, PPF, which is unaffected in STOP−/− mice, only involves ready-to-go vesicles (Thomson 2000). Both microscopy and biochemical data indicate the presence of STOP in synapses. The deleterious effect of STOP deficiency on LTP and LTD in the CA1 synapses of the hippocampus indicates that STOP has functions in the postsynaptic compartment. Both CA1 LTP and CA1 LTD involve Ca++- and calmodulin-dependent cascades in the postsynaptic compartment (for review, see Soderling and Derkach 2000). STOP proteins are unique Ca++-calmodulin-binding proteins (Bosc et al. 2001) and could be involved in these cascades. In contrast, mossy fiber LTP, which is normal in STOP−/− mice, is independent of postsynaptic Ca++-dependent cascades (Mellor and Nicoll 2001).
The synaptic defects observed in STOP−/− mice offer an obvious basis for the behavioral disorders exhibited by these mice. STOP deficiency affects a wide variety of basic behavioral tasks, including disorganized activity, nurturing defects, anxiety-related behavior, inability to perform object recognition tests, and social withdrawal, and consequently cannot be attributed either to limited or defined brain structures or to a single neurotransmitter pathway. For example, the different aspects of maternal behavior that are perturbed in STOP−/− mice depend on adaptive neuronal responses in different brain regions such as olfactory bulb and preoptic area of hypothalamus (Brown et al. 1996; Numan and Sheehan 1997). The apparently nonfocal origin of the behavioral deficiencies observed in STOP−/− mice suggests that the synaptic defects that we observed in the hippocampus are widespread in the brain.
The pattern of synaptic defects in STOP-deficient mice is such that the mice can live but exhibit multiple behavioral disorders that can be modulated by long-term treatment with neuroleptics. These drugs are major antipsychotic agents principally used in schizophrenia. There are still important pending questions regarding the mechanism of action and use of neuroleptics. For instance, there is an intriguing dissociation between the rapid blocking effect of neuroleptics on dopaminergic receptors and the apparent delayed effects of the drugs on symptoms (Kandel et al. 2000). The behavioral disorders observed in STOP-deficient mice are to our knowledge unique in showing a delayed sensitivity to neuroleptic treatment mimicking that observed in schizophrenia. Neuroleptics are principally known as dopamine receptor blockers (Seeman et al. 1976) but apparently act on disorders affecting other neurotransmitter pathways (Mohn et al. 1999; Leveque et al. 2000). Our data also indicate that some synaptic defects can be alleviated by neuroleptic treatment. This result is of particular interest in view of recent models in which schizophrenia arises from synaptic disorders (Harrison 1997; Mirnics et al. 2001). STOP−/− mice thus appear to be an interesting model to investigate neuroleptic effects in animals showing synaptic defects associated with severe behavioral disorders. Interestingly, the human STOP gene, located at position 11q14 (GenBank accession no. AP000588), lies within a region which has been linked to major mental diseases including schizoid disorders (Holland and Gosden 1990; St. Clair et al. 1990; Brzustowicz et al. 2000).
Materials and methods
STOP targeting construct and generation of STOP−/− mice
We isolated genomic clone DNA containing the STOP locus from a mouse strain 129 SvPas genomic DNA library (Denarier et al. 1998a). As all characterized STOP cDNAs contain exon 1, we targeted exon 1 to suppress STOP proteins. In a 7.2-kb genomic clone (Fig. 1A), the exon 1 was replaced by LacZ gene under the control of the endogenous STOP promoter followed by a phosphoglycerate kinase-driven neomycin resistance (pGK-neo) cassette [plasmid pWH9, a generous gift from E. Hirsch (Torino, Italy)]. We used either R1 ES cells kindly provided by Andras Nagy (Nagy et al. 1993) or AT1 ES cells raised in the transgenic facility of our department. The AT1 ES line was established from (129 SvPas × 129 SvPas) F1, 3.5-d blastocysts. The targeting vector was electroporated into R1 or AT1 ES cells. Transfectants were plated on mitomycin-treated neomycin-resistant primary embryonic fibroblasts as feeder layer and cultured in DMEM high glucose (Invitrogen) supplemented with 15% fetal calf serum and 1000 U/mL leukemia inhibitory factor (Esgro, Chemicon). Forty-eight hours after transfection, 250 μg/mL G-418 (Invitrogen) was added to the medium. Selection with 2 μg/mL gancyclovir (Syntex) was performed from day 4 to day 8 postelectroporation. Resistant colonies were picked at day 10 and expanded for freezing and DNA analysis. Recombinant ES clones were identified by Eco RV digestion of genomic DNA and hybridization with Eco RV-Eco RI probe located immediately in the 5′ position of the homologous region of the targeting construct (Fig. 1A). Two recombinant ES clones (R1–7 and AT1–76) were aggregated with OF1 morula to generate chimaeric mice (Nagy and Rossant 1993). Germline transmitting mice from both R1–7 and AT1–76 ES clones were mated to either BALBc or 129 SvPas mice to produce heterozygous mutant mice on either mixed BALBc/129 SvPas or pure 129 SvPas backgrounds. All wild-type mice and mice homozygous for the mutant STOP gene (STOP−/−) were obtained from the intercross of heterozygous animals. BALBc and 129 SvPas animals were purchased from Charles River Laboratories.
Analysis of brain anatomy
For histological analysis, we perfused mice aged 8–12 wk with 4% paraformaldehyde (PFA). Brains were postfixed 2 h in 4% PFA at 4°C. Brain slices (50 μm) were stained with cresyl violet or assayed for cytochrome oxidase (Liu et al. 1993). For analysis of olfactory bulb glomeruli and barrel field, littermate mice were used. The examination was done blind to genotype. For the olfactory bulb, sections throughout the full length were examined. The number and the size of glomeruli were assessed on 20 coronal sections (50 μm) located after the point where the lateral ventricle first appeared. Measurements were not corrected for shrinkage. For barrel field, the examination was done on sections (80 μm) tangential to the barrels. For STOP staining, brains were prepared as above and then cryoprotected in 20% sucrose in PBS. Brain slices (20 μm) were successively incubated with 1% H2O2 (15 min), 3% BSA (30 min), affinity-purified STOP antibody 23C (100 μg/mL overnight; Guillaud et al. 1998) and with anti-rabbit peroxydase-conjugated antibody, and then revealed with ethylcarbazole (DAKO AEC). For mossy fiber staining, mice were perfused first with 50 mM sodium sulfide followed by 4% PFA. Hippocampi were then dissected, sliced (50 μm), and revealed according to Danscher (1981). For β-galactosidase activity staining, animals were perfused with 0.2% glutaraldehyde and 2% formaldehyde in PBS. Brain slices (50 μm) were incubated in PBS containing 5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM magnesium chloride and 1 mg/mL X-Gal as substrate for 3–5 h at 30°C.
Floating sections of cerebellum (50 μm) were successively incubated with calbindin antibody (generous gift of J. Baudier, Grenoble, France), 10 μg/mL overnight) and with anti-rabbit Cy3 antibody.
Electron microscopy analysis
The hippocampus was dissected out from transcardially fixed mice. Perfusion was performed with 2% PFA and 0.2% glutaraldehyde in 0.1 M phosphate-buffer at pH 6.9. For morphometric analyses, hippocampal slices were postfixed in 2.5% glutaraldehyde in 0.1% cacodylate buffer for 2 h, fixed with 1 % OsO4 for 1 h at 4°C, and embedded in Epon. Matching areas from the hippocampus were identified in semithin sections (500 nm), and for the comparison of the wild-type and the mutant mice ultrathin sections (70 nm) were cut, prepared, and viewed under a transmission electron microscope (JEOL 1200EX). To determine the surface density of synaptic vesicles, cross-sections of 50 synapses per animal made of CA1 region beneath the pyramidal cell layer were photographed randomly, and the numbers of synaptic vesicles in each nerve preterminal were counted on the electron micrographs. For this analysis, three wild-type and three STOP−/− hippocampi were used.
For localization of STOP, ultrathin frozen sections were prepared from hippocampus slices fixed in 2% PFA and 0.2% glutaraldehyde in 0.1 M phosphate-buffer at pH 6.9. Briefly, blocks of hippocampus were embedded in gelatin, and infiltrated in sucrose before freezing in liquid nitrogen. Ultrathin sections were cut at −120°C, and the retrieval of sections from the knife was achieved with a 1:1 mixture of 2% methylcellulose and 2.3 M sucrose according to the method of Liou et al. (1996). Labeling against STOP was performed with affinity-purified STOP antibody 23C, and localization was detected by 15-nm gold particle conjugated to protein A, using the STOP−/− knockout hippocampus as a negative control.
Synaptic proteins quantification
Crude synaptosomes were prepared from three adult brains from either wild-type or STOP−/− mice (Philips et al. 2001). Synaptic proteins were then sequentially extracted, resulting in three fractions (Philips et al. 2001). Synaptosomes were extracted sequentially with 1% Triton X-100 (TX-100) at pH 6.0 followed by pH 8.0. Synaptosomes (6 mL) were diluted 1:10 with ice-cold 0.1 mm CaCl2. The suspension was brought to a final concentration of 20 mM Tris at pH 6.0 and 1% TX-100. The extraction was performed for 30 min at 4°C, and the insoluble material was pelleted (40,000g, 30 min). The supernatant was decanted and proteins precipitated with 10 volumes acetone at −20°C and recovered by centrifugation at 15,000g for 30 min. The insoluble pellet was resuspended (10 mL of 20 mM Tris at pH 8.0, 1% TX-100), extracted, and centrifuged, and the supernatant was reprecipitated as above. The insoluble pellet as well as the proteins from the two extractions were dissolved in an equal volume of 2% SDS. Proteins resulting from pH 6.0 extraction at pH 8.0 extraction, or insoluble are referred to as S1, S2, and C3, respectively. All protein purifications were done in parallel for wild-type and STOP−/− mice. Equal volumes, from wild-type and STOP−/−, of crude synaptosomal (Sy), S1, S2, and C3 fractions were analyzed by SDS-PAGE and quantitative immunoblotting with 125I-labeled secondary antibodies (Amersham Pharmacia) and phosphorimager detection. Primary antibodies are from BD Biosciences, except for Glu R1 (Chemicon), synaptophysin (rabbit polyclonal H-93, Santa Cruz Biotechnology), N-cadherin (Zymed), and STOP 23C (Guillaud et al. 1998).
Electrophysiological analysis
For preparation of hippocampal slices, mice (1–5-mo-old) were deeply anesthetized with Nembutal. Brain slices (300–400 μm) were cut in a cold artificial cerebrospinal fluid (4°–8°C). The artificial cerebrospinal fluid (ACSF) contained 124 mM NaCl, 26 mM NaHCO3, 2.5 mM KCl, 1.25 mM NaH2PO4, 2.5 mM CaCl2, 1.3 mM MgCl2, and 10 mM glucose. Slices were maintained at room temperature for at least 1 h in a submerged chamber containing ACSF equilibrated with 95% O2 and 5% CO2 and then transferred to a superfusing chamber.
Field EPSPs were recorded using microelectrodes (1–3 MΩ) filled with ACSF. Recordings were done at 22°–25°C. Bipolar stainless steel electrodes were used for electrical stimulation of Schaffer collaterals and mossy fibers (0.1 msec, 10–100 μA pulses, intertrial intervals of 10–30 sec). Whole-cell patch clamp recordings from hippocampal pyramidal cells were performed with the Axopatch 200A amplifier (Axon Instruments) under visual control with an upright microscope (BX50WI, Olympus). Patch pipettes were filled with the following solution: 120 mM Cs-gluconate, 10 mM CsCl, 2 mM MgCl2, 8 mM NaCl, and 10 mM HEPES (300 mOsm at pH 7.2). Microelectrodes had a resistance of ∼7 MΩ. Neurobiotin (0.5%) was routinely added to the electrode solution to allow morphological identification of the recorded cells. The neurobiotin labeling was developed by using avidin conjugated to Texas red D. Labeled neurons were examined by using a laser confocal microscope (TCS-SP2, Leica).
For all the experiments in the CA1 region, stimulating and extracellular recording electrodes were placed in the stratum radiatum, and GABA-A receptor antagonist picrotoxin (100 μM) was added to the ACSF. In this series of experiments, the CA1 region was separated from the CA3 region by a knife section of the brain slice before recording.
For the quantitative analysis shown in the input-output curves, the fiber volleys were characterized after blockade of the AMPA receptor activation by using the selective antagonist NBQX (10 μM). For the assessment of the NMDA/AMPA ratio, evoked EPSCs were recorded at a Vhold of +30 mV in the control ACSF (including picrotoxin). The amplitude of NMDA receptor-mediated responses was quantified after the bath application of NBQX (10 μM). The amplitude of AMPA receptor-mediated responses was obtained by subtracting the average NMDA receptor-mediated response to the average evoked EPSC. For the amplitude analysis of strontium-induced asynchronous EPSCs, we used a protocol described by Bekkers and Clements (1999). Briefly, the following ACSF was used: 110 mM NaCl, 26 mM NaHCO3, 3 mM KCl, 2.5 mM NaH2PO4, 8 mM SrCl2, 3 mM MgCl2, and 10 mM glucose (solution adjusted to 300 mOsm). CA1 pyramidal cells were voltage clamped at −70 mV. For the two last experiments, the stimulating electrode constituted a patch pipette filled with ACSF, which was placed very close to the recorded neuron in the stratum radiatum.
For the PTP experiments, a high dose of D-APV (50–100 μM) was added to the bath solution at least 10 min before tetanus.
For the paired pulse experiments, Schaffer collaterals were stimulated repetitively by two stimuli of the same intensity separated by different short time intervals. Paired pulse ratio was calculated by averaging 15–20 responses at each interval and by computing the ratio of the amplitude of the response for the second stimulus against the amplitude of the response for the first stimulus.
Mossy fiber responses were identified in wild-type and STOP−/− mice by using the group 2-metabotropic glutamate receptor selective agonist DCG IV. Inhibitory effects of DCG IV (10 μM) on mossy fiber inputs were similar. NBQX (5–10 μM) was applied at the end of each mossy fiber experiment to assess the amplitude of the fiber volleys.
Data acquisition and analysis for LTP and LTD experiments were done blind to genotype. On- and off-line data analyses were carried out with Acquis1 (G. Sadoc, CNRS-ANVAR, France). All summary data are expressed as mean ± s.e.m. Analyses of asynchronous EPSCs were done 20–2000 msec after the stimulus artifact. For the pharmacological experiments, data were computed at least 10 min after the beginning of the drug application. The following drugs were used: NBQX, D-APV, DCGIV (Tocris), picrotoxin (Sigma), neurobiotin, and avidin Texas red D (Vector Laboratories, AbCys, France).
Behavioral studies
Animal experiments were conducted in agreement with the French (Ministère de l'Agriculture et de la Forêt) and European Community guidelines for care of laboratory animals. In all tests performed, STOP−/− mice and wild-type control littermate mice arose from the same colony (BALBc/129 Sv background). All experiments were done blind to genotype.
Light–dark box
The apparatus consisted of two polyvinylcarbonate boxes (20 × 20 × 14 cm) covered with Plexiglas. One of these boxes was darkened. A light from a 100W bulb lighted the other box (about 4400 Lux). An opaque plastic tunnel (5 × 7 × 10 cm) separated the dark box from the illuminated one. The animals were placed individually in the lit box, with head directed towards the tunnel. This test was done by Neurofit Co. (Strasbourg, France).
Resident-intruder assay
Social investigation
Male wild-type or STOP−/− mice (4-wk-old) were housed individually or in groups of 10 for 10 d prior testing. A group-housed male (intruder) was placed in the home cage of the individually housed male (resident), and their behavior was recorded for 6 min. We scored the time spent in active social investigation of the intruder (sniffing and sexual behaviors) by the resident.
Intermale aggression
The assay was as above except that the resident males were housed individually for one month. In this test we scored the number of attacks initiated by the resident male and the duration of each fighting bout. The aggression test was conducted for two consecutive days for a 5-min period. The number of attacks and the time spent in fighting performed by the resident were scored.
Pup retrieving tests
For virgin females, mice aged 28–49 d were housed individually for at least one day prior to the experiment and provided with cotton to build a nest. On day one, each female was exposed to three 1–3-d-old pups. Pups were placed in three corners of the cage distant from the nest. After 30 min, the pups were returned to their natural mother. On day two, each female was again exposed to three pups. The number of pups retrieved during 30 min by each female was scored.
For males, mice aged 30–45 d were used. Experiments were performed as for virgin females except that the exposure time was for two consecutive days before testing on day three.
For postpartum females (second pregnancy), females were housed individually once pregnant. On the day of delivery, the newborn pups were removed and kept warm for 1 h in another cage on a heating pad. The mother was then removed from her home cage; three pups from her own litter were placed in each corner of the cage distant from the nest, and the mother was returned to her nest. The number of pups retrieved over a 30-min period was monitored.
Drug administration
After determination of the daily drinking intake of the mice, haloperidol (Haldol, Janssen-Cilag), and diazepam (Valium, Roche) were delivered to the mice in the drinking water at 0.5 mg/d/kg. Chlorpromazine (Largactil, Rhone-Poulenc) was delivered at 5 mg/d/kg. The presence of the drug in the drinking water had no detectable influence on the mouse water intake.
Acknowledgments
We thank Dr. A. Nagy, N. Nagy, and W. Abramow-Newerly for the gift of R1 ES cells, Dr. J. Molgo and Dr. E. Benoit for examining peripheral nervous functions, Dr. J.-L. Puel for examining audition in our mice, Dr. B. Griffon for preliminary EM investigation, F. Leboeuf for analyzing anatomical data, S. Graal for help in anatomical processing, I. Parada for advice on neuronal labeling, Dr. P. Huber and Y. Saoudi for help in imaging, R. Scriwanek and M. van Peski for expert preparation of the electron micrographs, Drs. T. Galli and C. Dumontet for help and advice, and Drs. R.L. Margolis, A. Peterson, M. Scanziani, I. Jakob, and B. Schaal for critical reading of our manuscript. This work was supported in part by grants from ARC (no. 9041) to M.V., and from La Ligue Nationale contre le Cancer to D.J. PK was supported by grant ALW8PJ/00-31 from the Research Council for Earth and Lifesciences that was assigned to J.K.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL djob@cea.fr; FAX 33-43-878-5057.
Article and Publication are at httP://www.genesdev.org/cgi/doi/10.1101/gad.223302.
References
- Baas PW, Heidemann SR. Microtubule reassembly from nucleating fragments during the regrowth of amputated neurites. J Cell Biol. 1986;103:917–927. doi: 10.1083/jcb.103.3.917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baas PW, Pienkowski TP, Cimbalnik KA, Toyama K, Bakalis S, Ahmad FJ, Kosik KS. Tau confers drug stability but not cold stability to microtubules in living cells. J Cell Sci. 1994;107:135–143. doi: 10.1242/jcs.107.1.135. [DOI] [PubMed] [Google Scholar]
- Bear MF, Malenka RC. Synaptic plasticity: LTP and LTD. Curr Opin Neurobiol. 1994;4:389–399. doi: 10.1016/0959-4388(94)90101-5. [DOI] [PubMed] [Google Scholar]
- Bekkers JM, Clements JD. Quantal amplitude and quantal variance of strontium-induced asynchronous EPSCs in rat dentate granule neurons. J Physiol. 1999;516:227–248. doi: 10.1111/j.1469-7793.1999.227aa.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosc C, Cronk JD, Pirollet F, Watterson DM, Haiech J, Job D, Margolis RL. Cloning, expression, and properties of the microtubule-stabilizing protein STOP. Proc Natl Acad Sci. 1996;93:2125–2130. doi: 10.1073/pnas.93.5.2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosc C, Frank R, Denarier E, Ronjat M, Schweitzer A, Wehland J, Job D. Identification of novel bifunctional calmodulin-binding and microtubule-stabilizing motifs in STOP proteins. J Biol Chem. 2001;276:30904–30913. doi: 10.1074/jbc.M011614200. [DOI] [PubMed] [Google Scholar]
- Brown JR, Ye H, Bronson RT, Dikkes P, Greenberg ME. A defect in nurturing in mice lacking the immediate early gene fosB. Cell. 1996;86:297–309. doi: 10.1016/s0092-8674(00)80101-4. [DOI] [PubMed] [Google Scholar]
- Brzustowicz LM, Hodgkinson KA, Chow EW, Honer WG, Bassett AS. Location of a major susceptibility locus for familial schizophrenia on chromosome 1q21-q22. Science. 2000;288:678–682. doi: 10.1126/science.288.5466.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calakos N, Scheller RH. Synaptic vesicle biogenesis, docking, and fusion: A molecular description. Physiol Rev. 1996;76:1–29. doi: 10.1152/physrev.1996.76.1.1. [DOI] [PubMed] [Google Scholar]
- Crawley JN. Behavioral phenotyping of transgenic and knockout mice: Experimental design and evaluation of general health, sensory functions, motor abilities, and specific behavioral tests. Brain Res. 1999;835:18–26. doi: 10.1016/s0006-8993(98)01258-x. [DOI] [PubMed] [Google Scholar]
- Danscher G. Histochemical demonstration of heavy metals. A revised version of the sulphide silver method suitable for both light and electronmicroscopy. Histochemistry. 1981;71:1–16. doi: 10.1007/BF00592566. [DOI] [PubMed] [Google Scholar]
- Denarier E, Aguezzoul M, Jolly C, Vourc'h C, Roure A, Andrieux A, Bosc C, Job D. Genomic structure and chromosomal mapping of the mouse STOP gene (Mtap6) Biochem Biophys Res Commun. 1998a;243:791–796. doi: 10.1006/bbrc.1998.8179. [DOI] [PubMed] [Google Scholar]
- Denarier E, Fourest-Lieuvin A, Bosc C, Pirollet F, Chapel A, Margolis RL, Job D. Nonneuronal isoforms of STOP protein are responsible for microtubule cold stability in mammalian fibroblasts. Proc Natl Acad Sci. 1998b;95:6055–6060. doi: 10.1073/pnas.95.11.6055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai A, Mitchison TJ. Microtubule polymerization dynamics. Annu Rev Cell Dev Biol. 1997;13:83–117. doi: 10.1146/annurev.cellbio.13.1.83. [DOI] [PubMed] [Google Scholar]
- Fleming AS. Maternal responsiveness in human and animal mothers. New Dir Child Dev. 1989;43:31–47. doi: 10.1002/cd.23219894305. [DOI] [PubMed] [Google Scholar]
- Francis F, Koulakoff A, Boucher D, Chafey P, Schaar B, Vinet MC, Friocourt G, McDonnell N, Reiner O, Kahn A, et al. Doublecortin is a developmentally regulated, microtubule-associated protein expressed in migrating and differentiating neurons. Neuron. 1999;23:247–256. doi: 10.1016/s0896-6273(00)80777-1. [DOI] [PubMed] [Google Scholar]
- Gandelman R, Zarrow MX, Denenberg VH, Myers M. Olfactory bulb removal eliminates maternal behavior in the mouse. Science. 1971;171:10–211. doi: 10.1126/science.171.3967.210. [DOI] [PubMed] [Google Scholar]
- Goldstein LS, Yang Z. Microtubule-based transport systems in neurons: The roles of kinesins and dyneins. Annu Rev Neurosci. 2000;23:39–71. doi: 10.1146/annurev.neuro.23.1.39. [DOI] [PubMed] [Google Scholar]
- Guillaud L, Bosc C, Fourest-Lieuvin A, Denarier E, Pirollet F, Lafanechère L, Job D. STOP proteins are responsible for the high degree of microtubule stabilization observed in neuronal cells. J Cell Biol. 1998;142:167–179. doi: 10.1083/jcb.142.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada A, Oguchi K, Okabe S, Kuno J, Terada S, Ohshima T, Sato-Yoshitake R, Takei Y, Noda T, Hirokawa N. Altered microtubule organization in small-calibre axons of mice lacking tau protein. Nature. 1994;369:488–491. doi: 10.1038/369488a0. [DOI] [PubMed] [Google Scholar]
- Harrison PJ. Schizophrenia: A disorder of neurodevelopment? Curr Opin Neurobiol. 1997;7:285–289. doi: 10.1016/s0959-4388(97)80018-9. [DOI] [PubMed] [Google Scholar]
- Holland T, Gosden C. A balanced chromosomal translocation partially co-segregating with psychotic illness in a family. Psychiatry Res. 1990;32:1–8. doi: 10.1016/0165-1781(90)90129-s. [DOI] [PubMed] [Google Scholar]
- Houseweart MK, Cleveland DW. Cytoskeletal linkers: New MAPs for old destinations. Curr Biol. 1999;9:R864–R866. doi: 10.1016/s0960-9822(00)80048-2. [DOI] [PubMed] [Google Scholar]
- Kandel ER, Schwartz JH, Jessel TM. Principles of neural science. 4th ed. East Norwalk, CT: Appleton & Lange; 2000. pp. 1204–1206. [Google Scholar]
- Leveque JC, Macías W, Rajadhyaksha A, Carlson RR, Barczak A, Kang S, Li XM, Coyle JT, Huganir RL, Heckers S, et al. Intracellular modulation of NMDA receptor function by antipsychotic drugs. J Neurosci. 2000;20:4011–4020. doi: 10.1523/JNEUROSCI.20-11-04011.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieuvin A, Labbé J-C, Dorée M, Job D. Intrinsic microtubule stability in interphase cells. J Cell Biol. 1994;124:985–996. doi: 10.1083/jcb.124.6.985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou W, Geuze HJ, Slot J W. Improving structural integrity of cryosections. Histochem Cell Biol. 1996;106:41–58. doi: 10.1007/BF02473201. [DOI] [PubMed] [Google Scholar]
- Liu Y, Gu Q, Cynader MS. An improved staining technique for cytochrome C oxidase. J Neurosci Methods. 1993;49:181–184. doi: 10.1016/0165-0270(93)90122-8. [DOI] [PubMed] [Google Scholar]
- Margolis RL, Wilson L. Opposite end assembly and disassembly of microtubule at steady state in vitro. Cell. 1978;13:1–8. doi: 10.1016/0092-8674(78)90132-0. [DOI] [PubMed] [Google Scholar]
- Mellor J, Nicoll RA. Hippocampal mossy fiber LTP is independent of postsynaptic calcium. Nat Neurosci. 2001;4:125–126. doi: 10.1038/83941. [DOI] [PubMed] [Google Scholar]
- Meziane H, Dodart JC, Mathis C, Little S, Clemens J, Paul SM, Ungerer A. Memory-enhancing effects of secreted forms of the beta-amyloid precursor protein in normal and amnestic mice. Proc Natl Acad Sci. 1998;95:12683–12688. doi: 10.1073/pnas.95.21.12683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirnics K, Middleton FA, Lewis DA, Levitt P. Analysis of complex brain disorders with gene expression microarrays: Schizophrenia as a disease of the synapse. Trends Neurosci. 2001;24:479–486. doi: 10.1016/s0166-2236(00)01862-2. [DOI] [PubMed] [Google Scholar]
- Misslin R, Griebel G, Saffroy-Spittler M, Vogel E. Anxiolytic and sedative effects of 5-HT1A ligands, 8-OH-DPAT and MDL 73005EF, in mice. Neuroreport. 1990;1:267–270. doi: 10.1097/00001756-199011000-00025. [DOI] [PubMed] [Google Scholar]
- Mitchison T, Kirschner M. Dynamic instability of microtubule growth. Nature. 1984;312:237–242. doi: 10.1038/312237a0. [DOI] [PubMed] [Google Scholar]
- Mohn AR, Gainetdinov RR, Caron MG, Koller BH. Mice with reduced NMDA receptor expression display behaviors related to schizophrenia. Cell. 1999;98:427–436. doi: 10.1016/s0092-8674(00)81972-8. [DOI] [PubMed] [Google Scholar]
- Nagy A, Rossant J. Production of completely ES cell-derived fetuses. In: Joyner AL, editor. Gene targeting: A practical approach. New York, NY: Oxford University; 1993. pp. 147–179. [Google Scholar]
- Nagy A, Rossant J, Nagy R, Abramow-Newerly W, Roder JC. Derivation of completely cell culture-derived mice from early-passage embryonic stem cells. Proc Natl Acad Sci. 1993;90:8424–8428. doi: 10.1073/pnas.90.18.8424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson RJ, Demas GE, Huang PL, Fishman MC, Dawson VL, Dawson TM, Snyder SH. Behavioral abnormalities in male mice lacking neuronal nitric oxide synthase. Nature. 1995;378:383–386. doi: 10.1038/378383a0. [DOI] [PubMed] [Google Scholar]
- Numan M, Sheehan TP. Neuroanatomical circuitry for mammalian maternal behavior. Ann N Y Acad Sci. 1997;807:101–125. doi: 10.1111/j.1749-6632.1997.tb51915.x. [DOI] [PubMed] [Google Scholar]
- Phillips GR, Huang JK, Wang Y, Tanaka H, Shapiro L, Zhang W, Shan WS, Arndt K, Frank M, Gordon, et al. The presynaptic particle web: Ultrastructure, composition, dissolution, and reconstitution. Neuron. 2001;32:63–77. doi: 10.1016/s0896-6273(01)00450-0. [DOI] [PubMed] [Google Scholar]
- Salin PA, Scanziani M, Malenka RC, Nicoll RA. Distinct short-term plasticity at two excitatory synapses in the hippocampus. Proc Natl Acad Sci. 1996;93:13304–13309. doi: 10.1073/pnas.93.23.13304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Lee T, Chau-Wong M, Wong K. Antipsychotic drug doses and neuroleptic/dopamine receptors. Nature. 1976;261:717–719. doi: 10.1038/261717a0. [DOI] [PubMed] [Google Scholar]
- Soderling TR, Derkach VA. Postsynaptic protein phosphorylation and LTP. Trends Neurosci. 2000;23:75–80. doi: 10.1016/s0166-2236(99)01490-3. [DOI] [PubMed] [Google Scholar]
- St Clair D, Blackwood D, Muir W, Carothers A, Walker M, Spowart G, Gosden C, Evans HJ. Association within a family of a balanced autosomal translocation with major mental illness. Lancet. 1990;336:13–16. doi: 10.1016/0140-6736(90)91520-k. [DOI] [PubMed] [Google Scholar]
- Thomson AM. Molecular frequency filters at central synapses. Prog Neurobiol. 2000;62:159–196. doi: 10.1016/s0301-0082(00)00008-3. [DOI] [PubMed] [Google Scholar]
- Van Rossum D, Hanisch UK. Cytoskeletal dynamics in dendritic spines: Direct modulation by glutamate receptors? Trends Neurosci. 1999;22:290–295. doi: 10.1016/s0166-2236(99)01404-6. [DOI] [PubMed] [Google Scholar]
- Yang Y, Bauer C, Strasser G, Wollman R, Julien JP, Fuchs E. Integrators of the cytoskeleton that stabilize microtubules. Cell. 1999;98:229–238. doi: 10.1016/s0092-8674(00)81017-x. [DOI] [PubMed] [Google Scholar]