

Abstract
Aims
To examine the stereoselective disposition of chlorpheniramine and to evaluate the role of CYP2D6 in chlorpheniramine pharmacokinetics in humans.
Methods
Eight healthy volunteers (six extensive metabolizers with respect to CYP2D6 and two poor metabolizers) received a single 8 mg oral dose of rac-chlorpheniramine either given alone or following administration of quinidine 50 mg every 6 h for 2 days prior to the study day and every 6 h thereafter until the end of the study. Plasma concentrations of (S)-(+)- and (R)-(−)-enantiomers of chlorpheniramine were determined using liquid chromatography/mass spectrometry.
Results
In extensive metabolizers, mean Cmax was greater (12.55±1.51 ng ml−1vs 5.38±0.44 ng ml−1) and CLoral was lower (0.49±0.08 l h−1 kg−1vs 1.07±0.15 l h−1 kg−1) for (S)-(+)- than for (R)-(−)-chlorpheniramine (P<0.005). For (S)-(+)-chlorpheniramine, administration of quinidine, an inhibitor of CYP2D6, resulted in an increase in Cmax to 13.94±1.51 (P<0.01), a reduction in CLoral to 0.22±0.03 l h−1 kg−1 (P<0.01), and a prolongation of elimination half-life from 18.0±2.0 h to 29.3±2.0 h (P<0.001). Administration of quinidine decreased CLoral for (R)-(−)-chlorpheniramine to 0.60±0.10 l h−1 kg−1 (P<0.005). In CYP2D6 poor metabolizers, systemic exposure was greater after chlorpheniramine alone than in extensive metabolizers, and administration of quinidine resulted in a slight increase in CLoral.
Conclusions
Stereoselective elimination of chlorpheniramine occurs in humans, with the most pharmacologically active (S)-(+)-enantiomer cleared more slowly than the (R)-(−)-enantiomer. CYP2D6 plays a role in the metabolism of chlorpheniramine in humans.
Keywords: chlorpheniramine, CYP2D6, pharmacokinetics, stereoselectivity
Introduction
Chlorpheniramine is a propylamine H1-receptor antagonist (antihistamine) that is available as an over-the-counter antiallergy drug. It is indicated for use in the common cold and for symptomatic treatment of allergies and is widely used. Although it has been available for more than 50 years and taken by millions of people, its pharmacokinetics have been incompletely characterized. Chlorpheniramine has a chiral carbon (see Figure 1) and although chlorpheniramine is usually given as a racemic mixture, it demonstrates stereoselectivity in the pharmacological response. In in vitro assays of histamine-induced guinea pig ileum contraction [1], in vivo studies of protection against histamine-induced lethality in guinea pigs [1], and in vitro receptor binding studies in a variety of tissues [2, 3], the (S)-(+)-enantiomer (the (+)-isomer) is approximately 100 times more potent than the R-(−)-enantiomer (the (−)-isomer). Most of the pharmacokinetic data obtained after administration in humans has been obtained using achiral methods of chemical detection [4–6]. Both the pharmacokinetics and pharmacodynamic response have shown a great deal of variability, with elimination half-lives ranging from 18 h to 43 h [5, 6]. The duration of the pharmacodynamic response shows similar variability [7]. Characterization of factors contributing to pharmacokinetic variability may be useful in identifying factors that could reduce variability in response.
Figure 1.

Structural formula of chlorpheniramine. The chiral carbon is identified with an asterisk.
We have previously characterized histamine H1-receptor antagonist activity in plasma of healthy volunteers following administration of rac-chlorpheniramine, using an in vitro radioreceptor assay [8]. The receptor occupancy assay is used to assess the composite ability of substances present in the plasma, following a dose of a drug, to bind to a specific receptor. Thus, it could detect the composite contribution of parent compound, active isomer, or active metabolite, if present. This is in contrast to a chemical detection method that has been designed to measure plasma concentrations of a known substance. We and others have used this bioassay to evaluate the relationship between pharmacokinetics and pharmacodynamics of drugs acting at a variety of receptors, including β-adrenergic receptors [9, 10], and adenosine A1 receptors [11]. In our previous study, a discrepancy between the time course of H1-receptor occupancy and the time course for rac-chlorpheniramine plasma concentrations, obtained concurrently using achiral determination, was observed [8]. Since the occupancy assay would detect the activity of the active (S)-(+)-enantiomer, but the pharmacokinetic data reflected plasma concentrations of both the (S)-(+)- and (R)-(−)-enantiomers, one factor that could account for this discrepancy would be stereoselectivity of the pharmacokinetics of chlorpheniramine. In addition, a difference in receptor occupancy was observed between individuals who were poor and extensive metabolizers with respect to cytochrome P450 2D6. Thus, the CYP2D6 polymorphism may be a factor in the variability of pharmacokinetics and pharmacodynamic response after administration of chlorpheniramine in humans. The role of CYP2D6 in chlorpheniramine elimination has not been previously characterized.
The purpose of this study was to examine the stereoselective disposition of chlorpheniramine and to test the hypothesis that inhibition of CYP2D6 alters the pharmacokinetics and a biomarker for the pharmacodynamics of chlorpheniramine in healthy volunteers. Quinidine, a potent and selective inhibitor of CYP2D6 activity, was used to inhibit the activity of CYP2D6 in individuals proven to be extensive metabolizers.
Methods
Clinical study
Eight healthy volunteers participated in this study at the Clinical Research Center at Georgetown University Medical Center (Washington, D.C.) after written informed consent was obtained, a history taken and physical, and laboratory examinations were performed. Six volunteers had been previously characterized as extensive metabolizers of drugs metabolized by CYP2D6 and two volunteers were characterized as poor metabolizers using dextromethorphan and genotype methods previously described [12, 13], The poor metabolizers had a dextromethorphan metabolic ratio of greater than 0.3, and were homozygous for the CYP2D6*4 allele. This study was approved by the Georgetown University Institutional Review Board. No subject was taking medications known to interfere with cytochrome P450-mediated metabolism and no subject had taken H1-receptor antagonists within 4 weeks of the study.
This was a randomized crossover trial in which each study period was separated by a 3 week washout. Subjects were randomized to the order of the study drugs using a table of random numbers. Following drug administration, plasma samples were collected at fixed times over a period of 60 h. The study periods were as follows: Phase I: 8 mg (+/−)-chlorpheniramine maleate; Phase II: quinidine plus 8 mg (+/−)-chlorpheniramine maleate. Chlorpheniramine was given in each phase as a single oral dose given on the morning of the first study day of each phase. In Phase II, 50 mg quinidine sulphate was taken every 6 h for 2 days prior to the study day and every 6 h thereafter until the 60 h blood sample was collected.
Liquid chromatography/mass spectrometry determination of plasma concentrations
Enantioselective analysis of chlorpheniramine in plasma samples was performed using a validated liquid chromatography (LC)/mass spectrometry (MS) method using a Hewlett Packard series 1100 LC/MSD system [14]. Plasma samples (1 ml) were treated with 1 m sodium hydroxide and 3 ml of diethyl ether. An internal standard was not used. After mixing and centrifugation, the lower aqueous layer was frozen in an acetone/dry ice bath and the organic layer was transferred into a clean test tube containing 0.5% acetic acid. After mixing and centrifugation, the aqueous phase was frozen in an acetone/dry ice bath and the organic layer was discarded. After evaporation of the remaining ether, the thawed aqueous extract was transferred to autosampler vials and an aliquot of 75 µl was injected onto the chromatographic system.
The chiral separation was achieved using a 250×4.6 mm, 5 µ, β-cyclodextrin column (CYCLOBOND I, Advanced Separation Technologies, Whippany, NJ). The mobile phase was composed of 0.25% diethylamine (pH 4.4) : acetonitrile : methanol (50 : 50 v/v), 85 : 7.5 : 7.5 (v : v : v) at a flow rate of 0.5 ml min−1, with the column temperature set at 15° C. The MS was set for single ion monitoring at 275.1 m z−1 for chlorpheniramine with a drying gas flow rate of 13.0 l min−1, a nebulizer pressure of 30 psi, and a drying gas temperature of 350° C. The capillary voltage was set to 2800 V. Under these conditions, the (R)-(−)-enantiomer was eluted first, followed by the (S)-(+)-enantiomer, with retention times of 17.5±2.10 min and 18.9±2.20 min, respectively. The assay was sensitive to 0.05 ng ml−1 and the assay was linear to more than 50 ng ml−1 per enantiomer. Coefficients of variation (established at concentrations of 0.125, 0.25, 1.25, 3.75, 12.5, and 25 ng ml−1 per enantiomer) were less than 8% for intraday (n = 3) and 12% or less for interday (n = 14).
Histamine H1-receptor occupancy
Histamine H1-receptor occupancy by antagonist present in plasma was determined in samples from the extensive metabolizers using a method that has been previously described [8]. Briefly, H1-receptor-containing membranes were prepared from guinea pig lung by homogenization in 30 volumes of Na+-K+ phosphate buffer (pH 7.4) and filtered through two layers of cheesecloth. The filtrate was centrifuged at 30 000 g for 20 min, and the pellets were resuspended by homogenization at a concentration of 1 g wet weight/6.7 ml buffer.
Receptor binding studies were performed in a 0.5 ml total volume containing two-thirds volume plasma, 15 mg original wet weight of tissue and 3H-mepyramine (4.5 nmol l−1 final concentration). Nonspecific binding was determined in the presence of an excess (2 µm) of triprolidine. Samples were incubated for 40–50 min at 25° C. The reaction was stopped by the addition of 4 ml ice cold buffer. The samples were filtered by vacuum filtration through glass fibre filters (Schleicher & Schuell no. 30, Schleicher & Schuell Inc., Keene, N.H.) to retain membrane-bound radioligand. The filters were washed three times with 4 ml of ice cold buffer. Samples were analysed in triplicate.
The following equation was used to calculate the occupancy of H1-receptors by antagonist present in plasma samples:
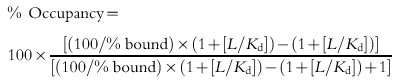 |
where % bound is the amount of radioligand specifically bound in the presence of drug (sample) relative to control (baseline) values. L is the concentration of radioligand used. Kd was previously determined to be 1.43 nmol l−1 in the presence of plasma [8].
Statistical analysis
Pharmacokinetic parameters were estimated using noncompartmental methods (WinNonlin, Pharsight Corporation, Mountain View, California). Area under the plasma concentration-time curve from 0 to infinity (AUC(0,∞)) was determined by linear trapezoidal rule, as was area under the occupancy-time curve from 0 to 60 h (AUC(0,60 h)) for H1-receptor occupancy. AUC(0,∞) was calculated as AUClast+(Clast/λz), where λz is the first order rate constant of the terminal portion of the curve and Clast is the concentration corresponding to the last time point. Apparent oral clearance (CLoral) was calculated as dose/AUC(0,∞). The paired t-test was used to compare the difference in results between Phase I and Phase II. P < 0.05 was considered to be statistically significant.
Results
Demographics
The six subjects that were extensive metabolizers of CYP2D6 ranged in age from 22 to 28 years old (25.3±0.9, mean±s.e. mean). The extensive metabolizers included three males and three females. The two poor metabolizers, one male and one female, were 24 and 21 years old, respectively.
Pharmacokinetics
The plasma concentration-time courses in extensive metabolizers for both Phase I and Phase II of the study are shown in Figure 2. The role of stereoselective elimination of the enantiomers of chlorpheniramine was evaluated by comparing, in extensive metabolizers with respect to CYP2D6, the plasma concentration-time course of (S)-(+)-chlorpheniramine with that of (R)-(−) chlorpheniramine following a single oral 8 mg dose of (±)-chlorpheniramine in Phase I of the study. Pharmacokinetic parameters from Phase I are shown in Table 1. Mean Cmax was greater [mean difference 7.18; 95% confidence intervals of the difference: (4.28, 10.07); P < 0.005], AUC(0,∞) was greater [mean difference −176; 95% confidence intervals of the difference: (−315, −38); P < 0.05], and CLoral was lower [mean difference 0.58; 95% confidence intervals of the difference: (0.39, 0.78); P < 0.005], for (S)-(+)-chlorpheniramine than for (R)-(−)-chlorpheniramine, with no difference in tmax or elimination half-life. The ratio of CLoral for the (R)-(−): (S)-(+) was 2.3±0.1 (mean±s.e. mean) in extensive metabolizers.
Figure 2.

Plasma concentration time course in CYP2D6 extensive metabolizers (n = 6) after administration of 8 mg racemic chlorpheniramine in the absence (closed symbols) or presence (open symbols) of quinidine. The R-(−)-enantiomer is represented by the circles (• or ○) and the S-(+)-enantiomer is represented by the triangles (▾ or ▿). Data are shown as mean±s.e. mean.
Table 1.
Comparison of pharmacokinetics of (S)-(+) and (R)-(−)-chlorpheniramine in extensive metabolizers in absence (Phase I) or presence (Phase II) of quinidine.
| (R)-(−)-chlorpheniramine | (S)-(+)-chlorpheniramine | |||||||
|---|---|---|---|---|---|---|---|---|
| Phase I | Phase II | aMean difference (95% CI) | aP value | Phase I | Phase II | aMean difference (95% CI) | aP value | |
| tmax (h) | 3.5 (2–4) | 3.0 (2–4) | 0.5 (−0.07, 1.1) | NS | 3.0 (2–4) | 3.0 (2–6) | −0.3 (−1.9,1.2) | NS |
| Cmax (ng ml−1) | 5.38±0.44 | 5.78±0.69 | −4.0 (−1.27, 0.47) | NS | 12.55±1.51 | 13.94±1.51 | −1.38 (−2.21, −0.56) | <0.01 |
| AUC(0,∞) (ng ml−1 h) | 124±24 | 224±41 | −100 (−158, −43) | <0.01 | 300±76 | 625±146 | −325 (−527, −124) | <0.01 |
| CLoral (l h−1 kg−1) | 1.07±0.15 | 0.60±0.10 | 0.47 (0.25, 0.68) | <0.005 | 0.49±0.08 | 0.22±0.03 | 0.27 (0.11, 0.43) | <0.01 |
| t½ (h) | 19.5±1.4 | 25.2±2.8 | −5.7 (−14.1, 2.7) | NS | 18.0±2.0 | 29.3±2.0 | −11.3 (−15.2, −7.4) | <0.001 |
NS, Not statistically significant. Data are mean values ±s.e. mean with the exception of tmax which is reported as median (range).
These values represent comparisons of Phase I and Phase II data.
The role of CYP2D6 in the pharmacokinetics of chlorpheniramine was evaluated by comparing pharmacokinetics in Phase I to pharmacokinetics in Phase II. Results for the extensive metabolizers are shown in Table 1. Administration of quinidine to study subjects resulted in a statistically significant increase in (R)-(−)-chlorpheniramine AUC(0,∞) and a statistically significant decrease in CLoral. For the (S)-(+)-enantiomer, statistically significant increases were observed in Cmax, AUC(0,∞) and elimination half-life, with a reduction in CLoral. In Phase II, the ratio of CLoral for (R)-(−): (S)-(+) was 2.6±0.2 [mean±s.e. mean; mean difference −0.3; 95% confidence intervals of the difference: (−0.5, −0.01); P < 0.05] compared with the (R)-(−): (S)-(+) ratio in Phase (I).
The pharmacokinetics of chlorpheniramine were evaluated in two poor metabolizers with respect to CYP2D6 (Table 2, Figure 3). In contrast to results with the extensive metabolizers, exposure to quinidine did not result in a substantial increase in AUC(0,∞) or a reduction in CLoral. The systemic exposure to both the (S)-(+) and (R)-(−) enantiomers appeared to be greater in the poor metabolizers than in the extensive metabolizers in the absence of quinidine. In both subjects a small decrease in AUC(0,∞) with a slight increase in CLoral of both enantiomers after administration of quinidine was observed. Plasma samples were not collected for a long enough period of time after dosing to permit for accurate estimation of the terminal half-life in the poor metabolizers. However, it was roughly estimated to be 29 h or longer for the (S)-(+)-enantiomer.
Table 2.
Pharmacokinetics of (S)-(+) and (R)-(−)-chlorpheniramine in poor metabolizers.
| tmax (h) | Cmax (ng ml−1) | AUC(0,∞) (ng ml−1 h) | CLoral (l h−1 kg−1) | |
|---|---|---|---|---|
| PM1 (R)-(−)-enantiomer | ||||
| Phase I | 8 | 5.61 | 412 | 0.24 |
| Phase II | 6 | 6.08 | 325 | 0.31 |
| (S)-(+)-enantiomer | ||||
| Phase I | 3 | 14.85 | 1098 | 0.09 |
| Phase II | 6 | 13.01 | 600 | 0.17 |
| PM2 (R)-(−)-enantiomer | ||||
| Phase I | 1 | 10.4 | 192 | 0.57 |
| Phase II | 3 | 5.59 | 164 | 0.67 |
| (S)-(+)-enantiomer | ||||
| Phase I | 1 | 29.94 | 836 | 0.13 |
| Phase II | 2 | 14.23 | 630 | 0.17 |
Figure 3.

Plasma concentration time course in CYP2D6 poor metabolizers after administration of 8 mg racemic chlorpheniramine in the absence (closed symbols) or presence (open symbols) of quinidine. Figure 3 (upper panel) represents PM1 and Figure 3 (lower panel) represents PM2. The R-(−)-enantiomer is represented by the circles (• or ○) and the S-(+)-enantiomer is represented by the triangles (▾ or ▿).
Pharmacodynamics
H1-histamine receptor occupancy by antagonist present in the plasma of extensive metabolizers was evaluated. Following 3 day administration of quinidine, receptor occupancy was greater in Phase II than in Phase I, notably at the time points more than 12 h after the chlorpheniramine dose (Figure 4). The AUC(0,60 h) for occupancy was 2210±600 (mean±s.e. mean) for Phase I and 3782±314 (mean±s.e. mean) for Phase II [mean difference −1572; 95% confidence intervals of the difference: (−3274, 130); P = 0.06].
Figure 4.

Time course of H1-receptor occupancy by antagonist present in plasma in CYP2D6 extensive metabolizers (n = 6) after administration of 8 mg racemic chlorpheniramine in the absence (•) or presence (○) of quinidine. Data shown are mean±s.e. mean.
Discussion
In the present study, chlorpheniramine plasma concentrations were evaluated using an enantioselective assay for chlorpheniramine plasma concentrations in humans that directly determined the concentrations of both (S)-(+)- and (R)-(−)-chlorpheniramine after administration of racemic chlorpheniramine. Stereoselective pharmacokinetics were observed, with higher plasma concentrations and lower CLoral of the (S)-(+)-enantiomer. The results are in agreement with those of Koch et al. [15] who reported an approximate two-fold higher AUC and lower renal clearance of (S)-(+)-chlorpheniramine than for (R)-(−)-chlorpheniramine following administration of 4 mg chlorpheniramine in healthy volunteers, based on estimated concentrations of (S)-(+)- and (R)-(−)-chlorpheniramine determined using an enantiomeric ratio. Miyzaki & Abuki [16] also evaluated the stereoselective pharmacokinetics of chlorpheniramine in humans by giving the deuterium labelled (+)-isomer and nonlabled (−)-chlorpheniramine to three healthy volunteers. These authors reported a (+)/(−) concentration ratio of 1.9–3.1 over a 48 h period after administration of 3 mg of each isomer. Thus the results of these studies are in agreement with the present results showing that chlorpheniramine is stereoselectively eliminated in humans, resulting in slower clearance of the (S)-(+)-enantiomer.
In the present study we have found that coadministration of low-dose quinidine with rac-chlopheniramine in CYP2D6 extensive metabolizers resulted in a marked increase in plasma (R)-(−)- and (S)-(+)-chlorpheniramine systemic exposure as compared with that observed in the same subjects receiving chlorpheniramine alone. This is most likely attributable to a reduction in chlorpheniramine metabolic clearance as opposed to chlorpheniramine absorption since an increase in the mean plasma AUC and the terminal elimination half-life values was observed, whereas the increase in Cmax was modest. Quinidine is a potent competitive inhibitor of O-demethylation of dextromethorphan [17] that is catalysed by CYP2D6. In the present study and in other studies of drug metabolism [18–20], low dosages of quinidine effectively convert the extensive metabolizers to the poor metabolizer phenotype. Thus the results of the present study are consistent with a role for CYP2D6 in the metabolism of chlorpheniramine. In the presence of quinidine, H1-receptor occupancy of more than 50% was observed for approximately 36 h after a single 8 mg dose of chlorpheniramine. This is in agreement with our previous study in which we observed approximately 60% occupancy from 12 to 30 h after a dose in five poor metabolizers with respect to CYP2D6 [8]. In the present study, the AUC(0,∞) and CLoral in the two poor metabolizers resemble those results from the extensive metabolizers in the presence of quinidine. The changes in chlorpheniramine pharmacokinetics as well as increased H1-receptor antagonist activity observed with inhibition of CYP2D6 activity suggest the potential for prolonged pharmacodynamic effects following administration of chlorpheniramine in poor metabolizers or when given in combination with other drugs that can inhibit CYP2D6 activity.
The two poor metabolizers with respect to CYP2D6 were included as controls in the present study, as quinidine would not be expected to produce any further inhibition of CYP2D6 in those subjects. However, a slight decrease in AUC(0,∞) and a slight increase in CLoral was observed for both the (R)-(−)- and the (S)-(+)-enantiomers following administration of quinidine, although it is not possible to draw a firm conclusion from such a small number of subjects. Quinidine has been shown in vitro to increase the CYP3A-mediated 5′methylhydroxylation of meloxicam [21], as well as the CYP3A-mediated N-dealkylation of fentanyl [22]. It is possible that such a CYP3A-mediated mechanism could account for the increase in CLoral observed in the presence of quinidine in the two CYP2D6 poor metabolizers in the present study. This could be a minor pathway that becomes more apparent in the absence of CYP2D6. However, the role of CYP3A in chlorpheniramine metabolism and a potential interaction with quinidine via this mechanism requires further evaluation.
The long elimination half-life of chlorpheniramine in the present study is in agreement with that reported by others using both achiral and chiral assays [4–6, 15, 16], The prolonged H1-receptor occupancy observed, particularly following blockade of CYP2D6 activity, suggests that there may be a prolonged H1-receptor effect as well. This could have important implications in coadministration of chlorpheniramine with other medications that can inhibit CYP2D6. In addition to the contributions of (S)-(+)-chlorpheniramine to H1-receptor occupancy, there may also be an undetected active metabolite in the plasma that is formed by a pathway not catalysed by CYP2D6. Five metabolites of chlorpheniramine have been detected in urine or plasma between 0 and 48 h after dosing in humans [4]. These include N-desmethyl- and didesmethyl-chlorpheniramine, as well as unidentified polar metabolites. Further studies are required to characterize the metabolic pathways involved in the disposition of chlorpheniramine in humans, and to define the activity of the metabolites and their contribution to clinical effect.
In summary, the present study has demonstrated stereoselective elimination and a role for CYP2D6 in the metabolism of chlorpheniramine in humans. The data presented here begin to explain the basis for variability that has been observed in chlorpheniramine pharmacokinetics and pharmacodynamics. Full characterization of the clinical pharmacology of chlorpheniramine will ultimately require evaluation of the relationships between clinical response and the factors identified in this study, namely chiral elimination, drug interactions, and polymorphic metabolism.
Acknowledgments
This work was supported in part by Clinical Research Facility Contract 223-93-3011 from the Food and Drug Administration, 1 M01-RR13297 from the General Clinical Research Center Program of the National Center for Research Resources, National Institutes of Health, and 1 U18 HS 10385–01 from Agency for Healthcare Research and Quality.
References
- 1.Roth FE, Govier WM. Comparative pharmacology of chlorpheniramine (Chlor-Trimeton) and its optical isomers. J Pharmacol Exp Ther. 1958;124:347–349. [PubMed] [Google Scholar]
- 2.Tran VT, Chang RSL, Snyder SH. Histamine H1 receptors identified in mammalian brain membranes with [3H]-mepyramine. Proc Natl Acad Sci USA. 1978;75:6290–6294. doi: 10.1073/pnas.75.12.6290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Chang RSL, Tran VT, Snyder SH. Characteristics of histamine H1-receptors in peripheral tissues labeled with [3H]mepyramine. J Pharmacol Exp Ther. 1979;209:437–442. [PubMed] [Google Scholar]
- 4.Peets EA, Jackson M, Symchowicz S. Metabolism of chlorpheniramine maleate in man. J Pharmacol Exp Ther. 1972;180:464–474. [PubMed] [Google Scholar]
- 5.Huang SM, Athanikar NK, Sridhar K, Huang YC, Chiou WL. Pharmacokinetics of chlorpheniramine after intravenous and oral administration in normal adults. Eur J Clin Pharmacol. 1982;22:359–365. doi: 10.1007/BF00548406. [DOI] [PubMed] [Google Scholar]
- 6.Vallner JJ, Needham TE, Chan W, Viswanathan CT. Intravenous administration of chlorpheniramine to seven subjects. Curr Ther Res. 1979;26:449–453. [Google Scholar]
- 7.Arbesman CE. Report of committee on therapy. J Allergy. 1950;21:255–258. [Google Scholar]
- 8.Yasuda SU, Wellstein A, Likhari P, Barbey JT, Woosley RL. Chlorpheniramine plasma concentration and histamine H1-receptor occupancy. Clin Pharmacol Ther. 1995;58:210–220. doi: 10.1016/0009-9236(95)90199-X. [DOI] [PubMed] [Google Scholar]
- 9.Yasuda SU, Barbey JT, Funck-Brentano C, Wellstein A, Woosley RL. d-Sotalol reduces heart rate in vivo through a β-adrenergic receptor-independent mechanism. Clin Pharmacol Ther. 1993;53:436–442. doi: 10.1038/clpt.1993.48. [DOI] [PubMed] [Google Scholar]
- 10.Wellstein A, Palm D, Pitschner HF, Belz GG. Receptor binding of propranolol is the missing link between plasma concentration kinetics and the effect-time course in man. Eur J Clin Pharmacol. 1985;29:131–147. doi: 10.1007/BF00547412. [DOI] [PubMed] [Google Scholar]
- 11.Yasuda SU, Nagashima S, Douyon E, Benton RE, Woosley RL, Barbey JT. Adenosine A1-receptor occupancy predicts A1-receptor antagonist effects of N-0861. Clin Pharmacol Ther. 1998;64:536–541. doi: 10.1016/S0009-9236(98)90136-9. [DOI] [PubMed] [Google Scholar]
- 12.Hou ZY, Pickle LW, Meyer PS, Woosley RL. Salivary analysis for determination of dextromethorphan metabolic phenotype. Clin Pharmacol Ther. 1991;49:410–419. doi: 10.1038/clpt.1991.48. [DOI] [PubMed] [Google Scholar]
- 13.Buchert ET, Woosley RL, Swain SM, et al. Relationship of CYP2D6 (debrisoquine hydroxylase) genotype to breast cancer susceptibility. Pharmacogenetics. 1993;3:322–327. doi: 10.1097/00008571-199312000-00006. [DOI] [PubMed] [Google Scholar]
- 14.Fried KM, Young AE, Yasuda SU, Wainer IW. The enantioselective determination of chlorpheniramine and its major metabolites in human plasma using chiral chromatography on a β-cyclodextrin chiral stationery phase and mass spectrometric detection. JPharmBiomedAnal. 2002;27:479–488. doi: 10.1016/s0731-7085(01)00570-2. [DOI] [PubMed] [Google Scholar]
- 15.Koch KM, O'Connor RL, Davis IM, Yin Y. Stereoselective pharmacokinetics of chlorpheniramine and the effect of ranitidine. J Pharm Sci. 1998;87:1097–1100. doi: 10.1021/js980045m. [DOI] [PubMed] [Google Scholar]
- 16.Miyazaki H, Abuki H. Mass fragmentographic determination of d- and l-chlorpheniramine with aid of the stable isotope technique. Chem Pharm Bull. 1976;24:2572–2574. doi: 10.1248/cpb.24.2572. [DOI] [PubMed] [Google Scholar]
- 17.Broly F, Libersa C, Lhermitte M, Bechtel P, Dupuis B. Effect of quinidine on the dextromethorphan O-demethylase activity of microsomal fractions from human liver. Br J Clin Pharmacol. 1989;28:29–36. doi: 10.1111/j.1365-2125.1989.tb03502.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Funck-Brentano C, Turgeon J, Woosley RL, Roden DM. Effect of low dose quinidine on encainide pharmacokinetics and pharmacodynamics. Influence of genetic polymorphism. J Pharmacol Exp Ther. 1989;249:134–142. [PubMed] [Google Scholar]
- 19.Funck-Brentano C, Kroemer HK, Pavlou H, Woosley RL, Roden DM. Genetically-determined interaction between propafenone and low dose quinidine: role of active metabolites in modulating net drug effect. Br J Clin Pharmacol. 1989;27:435–444. doi: 10.1111/j.1365-2125.1989.tb05391.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Turgeon J, Pavlou HN, Wong W, Funck-Brentano C, Roden DM. Genetically determined steady-state interaction between encainide and quinidine in patients with arrhythmias. J Pharmacol Exp Ther. 1990;255:642–649. [PubMed] [Google Scholar]
- 21.Ludwig E, Schmid J, Beschke K, Ebner T. Activation of human cytochrome P-450 3A4-catalyzed meloxicam 5′-methylhydroxylation by quinidine and hydroquinidine in vitro. J Pharmacol Exp Ther. 1999;290:1–8. [PubMed] [Google Scholar]
- 22.Feierman DE, Lasker JM. Metabolism of fentanyl, a synthetic opioid analgesic, by human liver microsomes. Role of CYP3A4. Drug Metab Disp. 1996;24:932–939. [PubMed] [Google Scholar]