

Abstract
Aims
1) To develop an estimate of oral clearance (CLPx/F) for the antianginal agent perhexiline based on the ratio of cis-OH-perhexiline metabolite/parent perhexiline plasma concentrations at steady-state (COHPx,ss/CPx,ss). 2) To determine whether the ratio measured in the first fortnight of treatment 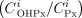 may be used to guide patient dosing with perhexiline, a drug with a narrow therapeutic index, long half-life and saturable metabolism via CYP2D6.
may be used to guide patient dosing with perhexiline, a drug with a narrow therapeutic index, long half-life and saturable metabolism via CYP2D6.
Methods
Two retrospective studies were conducted reviewing patient records and data obtained from routine monitoring of plasma perhexiline and cis-OH-perhexiline concentrations.
Results
Study 1 (n = 70). At steady-state, the frequency distributions of CLPx/F and COHPx,ss/CPx,ss were consistent with CYP2D6 metabolism. Putative poor metabolizers (approximately 8%) were identified by CLPx/F≤50 ml min−1 or COHPx,ss/CPx,ss ≤0.3. A group of patients with CLPx/F 950 ml min−1 may have been ultra-rapid metabolizers. In this group, the high CLPx/F values suggest extensive first-pass metabolism and poor bioavailability. In patients with therapeutic plasma perhexiline concentrations (0.15–0.60 mg l−1), the variability in dose appeared directly proportional to CLPx/F (r2=0.741, P < 0.0001). Study 2 (n=23). Using patients were tentatively identified as poor, extensive and ultra-rapid metabolizers, with CLPx/F of 23–72, 134–868 and 947–1462 ml min−1, respectively, requiring doses of 10–25, 100–250 and 300–500 mg day−1, respectively.
Conclusions
The cis-OH-perhexiline/perhexiline concentration ratio may be useful for optimizing individual patient treatment with the antianginal agent perhexiline.
Keywords: CYP2D6, metabolic ratio, perhexiline, therapeutic drug monitoring
Introduction
Perhexiline is a prophylactic antianginal agent currently recommended in Australia and New Zealand for the treatment of patients with intractable angina who are refractory or intolerant to other conventional therapy, or who are not suitable candidates for coronary bypass surgery. Its mechanism of action is not well understood, however, it is thought to act by inhibiting mitochondrial carnitine palmitoyltransferase-1 thereby shifting myocardial metabolism from fatty acid to glucose utilization which, for the same oxygen consumption, results in increased ATP production and consequently increased myocardial efficiency [1].
Although clinical trials have demonstrated that perhexiline produces significant objective improvement in angina symptoms in approximately 70% of patients [2, 3], its use remains relatively limited due to a potential to cause severe hepatic and neurological toxicity associated with CYP2D6 polymorphism [4, 5] and high plasma perhexiline concentrations [6–8]. It is now recognized that maintaining plasma perhexiline concentrations within the range of 0.15–0.60 mg l−1 minimizes the risk of clinically significant toxicity whilst still retaining clinical efficacy [7, 9], and therapeutic drug monitoring has become essential in guiding dosage, due to the high interpatient variability in perhexiline pharmacokinetics [10].
Perhexiline has only ever been available as an oral formulation, thus its bioavailability is unknown. However, it has been reported that in a small group of volunteers receiving 400 mg of perhexiline daily for 14 days, 24 h recoveries of unchanged perhexiline in faeces on days 12, 13 or 14 averaged 7.7% (range 0–32.5%), suggesting good absorption from the gastrointestinal tract [11]. The major determinant of perhexiline clearance appears to be hepatic metabolism, since in humans only approximately 0.1% of a dose is eliminated as unchanged drug in urine [12]. Perhexiline forms two primary monohydroxy (OH) metabolites, cis-OH-perhexiline and trans-OH-perhexiline, which can undergo further secondary metabolism to dihydroxy metabolites, as well as glucuronide conjugates [11]. Formation of the major primary metabolite, cis-OH-perhexiline is catalysed by CYP2D6 [13, 14], and this metabolic pathway is thought to give rise to both the saturability [15, 16] and genetic polymorphism in perhexiline clearance [13].
Due to its variable clearance and long and variable half-life, which ranges from 1 to 2 days in extensive metabolizers up to 30–40 days in poor metabolizers [10, 12, 14, 15], attainment of steady-state plasma perhexiline concentrations within the recommended therapeutic range can be a protracted process, potentially exposing patients to periods of transient toxicity, subtherapeutic concentrations and the inconvenience of multiple venepunctures for monitoring plasma perhexiline concentrations. The aim of this study was to investigate whether the ratio of plasma cis-OH-perhexiline/perhexiline concentrations might provide an estimate of perhexiline clearance in individual patients and therefore their dosage requirements.
Methods
Steady-state pharmacokinetics of perhexiline and cis-OH-perhexiline
Over a 3 month period, blood specimens received for routine monitoring of plasma perhexiline concentrations were used to identify patients who had attained steady-state with respect to perhexiline and cis-OH-perhexiline concentrations. During this period, each assay result was returned to the requesting medical officer together with a short questionnaire requesting information on the patient's current daily perhexiline dose and the length of time the patient had been on this dose. In addition, records of any previous requests for monitoring perhexiline concentrations were also reviewed.
Patients at steady-state were identified as having been on the same perhexiline dose for at least 3 months and/or having two consecutive plasma perhexiline and cis-OH-perhexiline concentrations varying by less than 20%. For each patient, the quotient of perhexiline plasma clearance (CLPx) and bioavailability (F) was calculated as:
| (1) |
where DR is the perhexiline dose rate (mg day−1), and CPx,ss is the average steady-state plasma concentration of perhexiline (mg l−1). Due to its long half-life, only a small difference between peak and trough perhexiline concentrations was anticipated at steady-state, and samples drawn at any time after the dose were accepted as measures of CPx,ss. At steady-state the rate of formation of cis-OH-perhexiline should equal its rate of elimination, so that
| (2) |
where 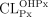 is the partial clearance of perhexiline via formation of cis-OH-perhexiline, COHPx,ss is the average steady-state plasma concentration of cis-OH-perhexiline, and CLOHPx is the plasma clearance of cis-OH-perhexiline. Thus, the metabolic ratio of COHPx,ss/CPx,ss was calculated for each patient since, as indicated by equation 2, it may serve as an estimate of , assuming there is relatively little interindividual variability in CLOHPx,Px. Like perhexiline, cis-OH-perhexiline also has a long half-life in humans, ranging from 31 to 53 h in extensive metabolizers, and much longer in poor metabolizers [14, 17]. Therefore, samples drawn at any time after the dose were accepted as measures of COHPx,ss.
is the partial clearance of perhexiline via formation of cis-OH-perhexiline, COHPx,ss is the average steady-state plasma concentration of cis-OH-perhexiline, and CLOHPx is the plasma clearance of cis-OH-perhexiline. Thus, the metabolic ratio of COHPx,ss/CPx,ss was calculated for each patient since, as indicated by equation 2, it may serve as an estimate of , assuming there is relatively little interindividual variability in CLOHPx,Px. Like perhexiline, cis-OH-perhexiline also has a long half-life in humans, ranging from 31 to 53 h in extensive metabolizers, and much longer in poor metabolizers [14, 17]. Therefore, samples drawn at any time after the dose were accepted as measures of COHPx,ss.
Estimating individual perhexiline dose requirements from early plasma perhexiline and cis-OH-perhexiline concentrations
A second retrospective study was carried out to determine whether the ratio of plasma cis-OH-perhexiline to perhexiline concentrations, measured during the first fortnight of commencing perhexiline treatment (), could be used as a guide to the dose of perhexiline required to attain steady-state plasma concentrations within the recommended therapeutic range.
A review of patient records of perhexiline and cis-OH-perhexiline concentrations was carried out to identify those patients at steady-state, as described above. A subgroup of patients was then identified whose plasma perhexiline and cis-OH-perhexiline concentrations were also determined within the first 2 weeks of commencing perhexiline therapy. Other relevant clinical data such as concomitant medications at the time of commencing perhexiline treatment, as well as at steady-state, were also recorded.
Quantification of plasma perhexiline and cis-OH-perhexiline concentrations
Perhexiline maleate and dansyl chloride were purchased from Sigma Chemical Company (St Louis, MO, USA). Hexadiline and cis-OH-perhexiline were supplied by Merrell Dow Pharmaceuticals Australasia Pty. Ltd. All other reagents were of analytical grade.
Plasma concentrations of perhexiline and cis-OH-perhexiline were measured using a modification of a previously published method [10]. Briefly, 500 µl of plasma sample (calibrators, controls or patient specimens) and 200 µl of internal standard solution (hexadiline, 1.4 mg l−1) were made basic by the addition of 50 µl of 2 m NaOH and extracted into 4 ml of a solution of 10% ethylacetate and 90% n-hexane. The organic layer was separated, dried and the residue incubated for 20 min at 37° C with 200 µl of 0.1 m NaHCO3 and 200 µl of 5 mm dansyl chloride in acetone. Following incubation, samples were extracted with 3 ml of n-hexane, the organic layer was separated, dried and the residue reconstituted in 100 µl of mobile phase, 50 µl of which were injected onto the h.p.l.c. column. Chromatographic separation was achieved using a Merck Purospher RP-18E column (5 µm, 125 × 4 mm) maintained at 45° C, with a mobile phase consisting of 90% methanol and 10% glass distilled water, pumped at 1.0 ml min−1, and fluorescence detection with excitation and emission wavelengths of 360 and 470 nm, respectively.
The retention times for cis-OH-perhexiline and perhexiline were 2.9 and 9.9 min, respectively (Figure 2). The internal standard eluted as two isomers with retention times of 8.1 and 8.6 min, and the first peak was routinely used for quantification. There was no chromatographic interference with any of the peaks of interest from either endogenous compounds in plasma (Figure 1) or other potentially coadministered drugs including amiodarone (and desethylamiodarone), amitriptyline, atenolol, captopril, diltiazem (and its major metabolites), digoxin, enalaprilat, fluoxetine, glibenclamide, metformin, prednisolone, prednisone, sertraline, theophylline, verapamil and warfarin (data not shown). In plasma specimens from patients administered perhexiline a small peak eluting at approximately 2.7 min was also evident (Figure 1c). This peak was likely to represent trans-OH-perhexiline or di-hydroxy metabolite(s). However, without pure reference compounds, we were unable to positively identify or quantify it. Inter- and intra-assay accuracy and precision for the quantification of perhexiline and cis-OH-perhexiline are shown in Table 1.
Figure 2.

Frequency distributions of (a) log CLPx/F (ml min−1) and (b) log COHPx,ss/CPx,ss in 70 steady-state patients administered perhexiline.
Figure 1.

H.p.l.c. chromatograms of (a) drug-free human plasma, (b) drug free human plasma spiked with cis-OH-perhexiline, hexadiline and perhexiline, and (c) plasma from a patient administered perhexiline. Retention times were 2.9 min for cis-OH-perhexiline, 8.1 and 8.6 min for the two peaks formed by hexadiline, and 9.9 min for perhexiline.
Table 1.
Accuracy and precision for the quantification of perhexiline and cis-OH-perhexiline in human plasma.
| Perhexiline | cis-OH-Perhexiline | |||||
|---|---|---|---|---|---|---|
| Target (mg l−1) | Mean (mg l−1) | C.V. (%) | Bias (%) | Mean (mg l−1) | C.V. (%) | Bias (%) |
| Intra-assay (n=6) | ||||||
| 3.00 | 2.911 | 1.6 | −3.1 | 2.869 | 4.4 | −4.6 |
| 0.05 | 0.043 | 6.1 | −17.1 | 0.051 | 7.3 | 2.0 |
| Inter-assay (n=10) | ||||||
| 3.00 | 2.971 | 0.6 | −1.0 | 3.008 | 3.4 | 0.3 |
| 0.50 | 0.492 | 8.9 | −1.7 | 0.509 | 8.6 | 1.8 |
| 0.05 | 0.039 | 10.7 | −22.8 | 0.051 | 12.3 | 1.0 |
Results
Steady-state pharmacokinetics of perhexiline and cis-OH-perhexiline
A total of 70 patients were identified as having attained steady-state perhexiline and cis-OH-perhexiline concentrations. Steady-state plasma perhexiline concentrations ranged from 0.06 to 3.91 mg l−1 and Figure 2 shows the frequency distributions for both CLPx/F and COHPx,ss/CPx,ss in the patient group. There was an extremely high interindividual variability in both CLPx/F and COHPx,ss/CPx,ss, with values for CLPx/F ranging from less than 100 ml min−1 to 1500 ml min−1, i.e. approaching human hepatic blood flow rate. In addition, both CLPx/F and COHPx,ss/CPx,ss did not display a normal distribution (Figure 2). The distribution of CLPx/F appeared bimodal with an antimode at 50 ml min−1 and 5/70 patients falling into a discrete subpopulation with CLPx/F ≤50 ml min−1. The remainder of the data appeared skewed towards the right, suggesting the presence of a third subpopulation. Similarly, in the frequency distribution of COHPx,ss/CPx,ss, a discrete subpopulation with ratios <0.30 was evident, comprising 6/70 patients. Regression analysis of CLPx/F vs the ratio COHPx,ss/CPx,ss showed a statistically significant linear correlation between the two parameters (Figure 3a).
Figure 3.

Linear regression analysis in 70 steady-state patients administered perhexiline of (a) CLPx/F vs COHPx,ss/CPx,ss (y = 0.150x+0.953, r2 = 0.687, P < 0.0001); and in a subgroup of 54 patients with perhexiline concentrations within the therapeutic range (0.15–0.60 mg l−1) linear regression analysis of steady-state dose vs (b) steady-state plasma perhexiline concentration (r2 = 0.0007, P = 0.852), or (c) CLPx/F (y=3.19x−89.05, r2 = 0.741, P < 0.0001).
From the 70 patients at steady-state, a subgroup of 54 were identified as having plasma perhexiline concentrations within the recommended therapeutic range of 0.15–0.60 mg l−1. In these patients, there was no relationship between perhexiline dose and plasma perhexiline concentrations (Figure 3b), however, the dose required to attain the narrow range of ‘therapeutic’ perhexiline concentrations appeared directly proportional to CLPx/F (Figure 3c).
Estimating individual perhexiline dose requirements from early plasma perhexiline and cis-OH-perhexiline concentrations
A total of 23 patients who commenced perhexiline treatment at The Queen Elizabeth Hospital were included in this study. Starting doses of perhexiline ranged from 100 mg once a day to 200 mg twice daily (400 mg day−1). The first perhexiline and cis-OH-perhexiline concentrations were measured within a mean±s.d. of 6±4 days after starting treatment, at which time 10 patients had perhexiline concentrations <0.15 mg l−1 and 6 patients had concentrations ≥ 0.60 mg l−1. At the time the steady-state samples were drawn, patients had received perhexiline for between 2 and 16 months (mean 8.7 months), doses ranged from 100 mg once a week (14.3 mg day−1) up to 500 mg day−1, and all plasma perhexiline concentrations were within 0.15–0.60 mg l−1. All patients were on polytherapy, taking an average of 10 other medications in addition to perhexiline. Five patients were taking other medications metabolized by CYP2D6, one taking fluoxetine at the start of treatment but not at steady-state, three taking either amitriptyline, carvedilol, or haloperidol plus citalopram at steady-state but not at the start of treatment, and one taking metoprolol throughout the study period.
Patients could be clearly separated into three distinct groups based on either , CLPx/F or the daily dose required to attain steady-state perhexiline concentrations within the therapeutic range (Table 2). In the group as a whole, was significantly higher than COHPx,ss/CPx,ss (Figure 4, Table 2). However, the difference between the two ratios was only significant in patients with ratios >0.30, and no difference was apparent in patients with ratios below this value (Figure 4, Table 2).
Table 2.
Classification of patients as poor, extensive and ultra-rapid metabolizers based on observed perhexiline pharmacokinetic parameters, presented as mean±s.d. (range).
| Poor (n = 5) | Extensive (n = 15) | Ultra-rapid (n = 3) | |
|---|---|---|---|
| CLPX/F | 50.2±20.7 | 428.3±213.2 | 1202.1±257.5 |
| (ml min−1) | (23–72) | (134–868) | (947–1462) |
| Steady-state dosage | 17.7±6.9 | 173.3±56.3 | 400.0±100.0 |
| (mg/day) | (10–25) | (100–250) | (300–500) |
| COHPx,ss/CPx,ss | 0.01±0.02 | 11.2±7.6 | 34.4±1.4 |
| (0–0.05) | (1.0–22.0) | (33.3–36.0) | |
| 0.06±0.13 | 7.4±4.0* | 14.8±3.4** | |
| (0–0.3) | (3.1–16.3) | (12.2–18.6) |
Denotes P < 0.05 compared with using Wilcoxon matched pairs test.
Denotes P < 0.01 compared with using Wilcoxon matched pairs test.
Figure 4.

The plasma cis-OH-perhexiline/perhexiline concentration ratio in 23 patients measured during the first 1–2 weeks of perhexiline treatment vs steady-state (P=0.005 initial vs steady-state value, Wilcoxon matched pairs test), showing putative poor (•), extensive (^) and ultra-rapid metabolizers (▴).
Discussion
Although clearly effective as an antianginal agent, clinical use of perhexiline has been limited by its narrow therapeutic index and large inter- and intraindividual variability in pharmacokinetics. A number of early studies of perhexiline metabolism have suggested that this variability is due primarily to saturability and genetic polymorphism in CYP2D6-catalysed formation of cis-OH-perhexiline [13, 15, 16, 18]. Indeed, the significant linear correlation observed between COHPx,ss/CPx,ss and CLPx/F in the first retrospective study (Figure 3a) supports the formation of cis-OH-metabolite as the primary determinant of perhexiline clearance, and additionally suggests that there was relatively little interindividual variability in the clearance of cis-OH-perhexiline. The very large interindividual variability in CLPx/F observed in both of the retrospective studies highlights the effects of saturability and genetic polymorphism on the pharmacokinetics of perhexiline, and is consistent with previous studies [10].
The Km of perhexiline for CYP2D6 metabolism in humans has not been investigated directly, however, its Ki for competitive inhibition of CYP2D6 metabolism by human liver microsomes has been reported as 0.4 µm, or approximately 0.1 mg l−1 [19]. Thus, the therapeutic plasma concentration range of perhexiline (0.15–0.60 mg l−1) easily exceeds its Km for CYP2D6 hydroxylation, giving rise to dose-dependent, or saturable, clearance.
Polymorphic metabolism by CYP2D6 in humans has been described for a large number of drugs and is characterized by the distribution of a population into poor, extensive and ultra-rapid metabolizers [20–22]. Consistent with the major contribution of CYP2D6 metabolism to perhexiline clearance, the frequency histograms of CLPx/F and COHPx,ss/CPx,ss in this study did not display a normal distribution (Figure 2). A subpopulation of patients with very low perhexiline clearances was easily identifiable using either CLPx/F or COHPx,ss/CPx,ss, accounting for 7.1% or 8.6% of patients, respectively. The frequency of CYP2D6 poor metabolizers in Caucasian populations has previously been estimated at 7–10% [20], thus, the subgroup of patients with CLPx/F values≤50 ml min−1 or COHPx,ss/CPx,ss values ≤ 0.30 were likely to be poor metabolizers. However, due to the retrospective nature of this study, we were unable to genotype patients and therefore could not rule out possible effects of coadministered inhibitors of CYP2D6 metabolism.
Patients with very high perhexiline clearances were evident in both Figures 2 and 3a. It is likely that the patients with CLPx/F values ≥950 ml min−1 represent a group of ultra-rapid metabolizers. However, unlike poor metabolizers, there was no clear separation of ultra-rapid and extensive metabolizers in either of the frequency distributions (Figure 2). Published studies of debrisoquine metabolism also similarly report that ultra-rapid metabolizers, identified by genotyping, are not clearly distinguishable from extensive metabolizers as a separate subpopulation on the basis of phenotyping alone [23, 24].
In the patients that were most likely ultra-rapid metabolizers, CLPx/F approached 1500 ml min−1, or hepatic blood flow rate. Such a high value suggests that, at least in this subgroup of patients who also all had perhexiline concentrations ≤0.22 mg l−1, the bioavailability of perhexiline may be quite low. Although its absolute bioavailability is unknown, perhexiline is thought to be well absorbed from the gastrointestinal tract in most patients [11]. Thus, since CLPx is primarily hepatic clearance, the high CLPx/F values suggest a high hepatic extraction ratio in this group. Amoah et al. have also previously suggested extensive presystemic metabolism of perhexiline, based on the early appearance of cis-OH-perhexiline in plasma at concentrations approximately an order of magnitude higher than those of perhexiline [12]. Therefore, the large intersubject variability in perhexiline pharmacokinetics may also result from variability in bioavailability, encompassing ultra-rapid metabolizers at one extreme with high clearances and low bioavailability, and poor metabolizers at the other extreme with low clearances and high bioavailability, a concept further supported by recent population modelling of perhexiline pharmacokinetics [25]. Importantly, like clearance, bioavailability is also concentration-dependent. Thus, even in ultra-rapid metabolizers, as perhexiline doses and plasma concentrations are increased, bioavailability will also increase. Concentration-dependent clearance and bioavailability may, in part, explain the difficulty in separating extensive and ultra-rapid metabolizers of perhexiline, using phenotyping alone.
Despite the complex pharmacokinetics of perhexiline, the first retrospective steady-state study demonstrates the simple linear relationship expected between CLPx/F and dose when steady-state perhexiline concentrations are kept constant (equation 1), or as in the subgroup of 54 patients in this study, were within the narrow therapeutic target of 0.15–0.60 mg l−1 (Figure 3c). By maintaining relatively constant perhexiline concentrations and studying each patient at only one steady-state perhexiline concentration, the effects of saturable metabolism were minimized in this subgroup and pharmacokinetic variability was primarily due to genetic polymorphism. Importantly, since COHPx,ss/CPx,ss appeared to be a good estimate of CLPx/F, the data suggest that it may be possible to use the plasma cis-OH-perhexiline/perhexiline concentration ratio as a guide to the dosage required for any individual to attain the narrow range of therapeutic plasma perhexiline concentrations at steady-state. Thus, the second retrospective study was carried out, over a narrow range of initial perhexiline doses, to determine whether measured within the first 1–2 weeks of commencing treatment might be also be useful in identifying poor, extensive and ultra-rapid metabolizers and titrating doses for these metabolizer populations.
Ultra-rapid metabolizers were tentatively identified as those with steady-state CLPx/F values ≥950 ml min−1, corresponding to three patients with initial perhexiline concentrations <0.15 mg l−1 and a ratio >30 (Figure 4, Table 2). Whilst approximately 10–30% of ultra-rapid metabolizers inherit alleles with more than one copy of functional CYP2D6 gene, the underlying defects for the remainder are as yet unknown and may include a role for the CYP2D6*35 allele, as well as the possibility of other mutations, including ones in the promoter region or transcription factor binding sites [21, 22]. Therfore, in some ultra-rapid metabolizers, high perhexiline clearance could theoretically be due to increased Vmax asssociated with high concentrations of CYP2D6 protein, rather than significantly altered Km, and saturation of metabolism would be expected at similar perhexiline concentrations as extensive metabolizers. Indeed, saturability in perhexiline metabolism was clearly evident in this group of patients with the plasma metabolite/perhexiline ratio decreasing from its initial measurement to steady-state (Figure 4, Table 2), as a result of increased plasma perhexiline concentrations at steady-state (P = 0.04, Wilcoxon matched pairs test, data not shown).
Poor metabolizers were tentatively identified as those with a COHPx,ss/CPx,ss ratio ≤ 0.30, corresponding to five patients with initial perhexiline concentrations ≥0.6 mg l−1 and a ratio ≤0.30 (Figure 4, Table 2). In these patients the metabolite/parent perhexiline concentration ratio remained ≤ 0.3, even when perhexiline concentrations decreased to within the target therapeutic range (Figure 4), supporting a low ratio due to lack of CYP2D6 metabolizing capacity rather than saturated metabolism.
Even though all patients were on polytherapy, of the different drugs taken only fluoxetine has been reported to cause clinically significant inhibition of CYP2D6 [26, 27], and of perhexiline metabolism [28], and only one patient was coadministered fluoxetine, at the start of treatment when was calculated, but not at steady-state. Interestingly, the ratio for this patient was the second lowest of the extensive metabolizer group (ratio = 2.4), consistent with CYP2D6 inhibition by fluoxetine. However, this patient could not have been mistaken for a poor metabolizer using the ratio.
The phenotyping based on as described in this paper was preliminary and will need to be confirmed in a prospective clinical study, preferably with concurrent genotyping. Nevertheless, the data suggest that poor metabolizers should not require doses greater than 25 mg day−1, typically administered as 50 mg perhexiline on alternate days, or 100 mg perhexiline once a week. In contrast, ultra-rapid metabolizers may require doses in the range of 300–500 mg day−1, and the metabolite/parent perhexiline concentration ratio may aid in differentiating these patients from noncompliers. Between these two extremes, the majority of patients appear to require doses of 100–250 mg day−1. Thus, the ratio of plasma cis-OH-perhexiline/perhexiline concentrations may be a useful clinical tool for early optimization of individual patient dosing regimens and may become a part of routine monitoring of patients treated with perhexiline.
References
- 1.Kennedy JA, Unger SA, Horowitz JD. Inhibition of carnitine palmitoyltransferase-1 in rat heart and liver by perhexiline and amiodarone. Biochem Pharmacol. 1996;52:273–280. doi: 10.1016/0006-2952(96)00204-3. [DOI] [PubMed] [Google Scholar]
- 2.Afzal Mir M, Kafetzakis EM. Assessment of perhexiline maleate in angiographically proven intractable angina: a double-blind trial. Am Heart J. 1978;96:350–354. [Google Scholar]
- 3.White H, Lowe J. Antianginal efficacy of perhexiline maleate in patients refractory to beta-adrenoreceptor blockade. Int J Cardiol. 1983;3:145–455. doi: 10.1016/0167-5273(83)90030-x. [DOI] [PubMed] [Google Scholar]
- 4.Shah RR, Oates NS, Idle JR, Smith RL, Lockhart JDF. Impaired oxidation of debrisoquine in patients with perhexiline neuropathy. Br Med J. 1982;284:295–299. doi: 10.1136/bmj.284.6312.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Morgan MY, Reshef R, Shah RR, Oates NS, Smith RL, Sherlock S. Impaired oxidation of debrisoquine in patients with perhexiline liver injury. Gut. 1984;25:1057–1064. doi: 10.1136/gut.25.10.1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Singlas E, Goujet MA, Simon P. Pharmacokinetics of perhexiline maleate in anginal patients with and without peripheral neuropathy. Eur J Clin Pharmacol. 1978;14:195–201. doi: 10.1007/BF02089960. [DOI] [PubMed] [Google Scholar]
- 7.Horowitz JD, Sia STB, Macdonald PS, Goble AJ, Louis WJ. Perhexiline maleate treatment for severe angina pectoris – correlations with pharmacokinetics. Int J Cardiol. 1986;13:219–229. doi: 10.1016/0167-5273(86)90146-4. [DOI] [PubMed] [Google Scholar]
- 8.Pilcher J, Cooper JDH, Turnell DC, Matenga J, Paul R, Lockhart JDF. Investigations of long-term treatment with perhexiline maleate using therapeutic monitoring and electromyography. Ther Drug Monit. 1985;7:54–60. doi: 10.1097/00007691-198503000-00009. [DOI] [PubMed] [Google Scholar]
- 9.Cole PL, Beamer AD, McGowan N, et al. Efficacy and safety of perhexiline maleate in refractory angina. A double-blind placebo-controlled clinical trial of a novel antianginal agent. Circulation. 1990;81:1260–1270. doi: 10.1161/01.cir.81.4.1260. [DOI] [PubMed] [Google Scholar]
- 10.Morris RG, Sallustio BC, Saccoia NC, Mangas S, Fergusson LK, Kassapidis C. Application of an improved HPLC perhexiline assay to human plasma specimens. J Liquid Chromatogr. 1992;15:3219–3232. [Google Scholar]
- 11.Wright GJ, Zeiger AV, Leeson GA, Lang JF. The absorption, excretion and metabolism of perhexiline maleate by the human. Postgrad Med J. 1973;49:8–15. [PubMed] [Google Scholar]
- 12.Amoah AGB, Gould BJ, Parke DV, Lockhart JDF. Further studies on the pharmacokinetics of perhexiline maleate in humans. Xenobiotica. 1986;16:63–68. doi: 10.3109/00498258609043506. [DOI] [PubMed] [Google Scholar]
- 13.Cooper RG, Evans DAP, Whibley EJ. Polymorphic hydroxylation of perhexiline maleate in man. J Med Genet. 1984;21:27–33. doi: 10.1136/jmg.21.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cooper RG, Evans DAP, Price AH. Studies on the metabolism of perhexiline in man. Eur J Clin Pharmacol. 1987;32:569–576. doi: 10.1007/BF02455990. [DOI] [PubMed] [Google Scholar]
- 15.Horowitz JD, Morris PM, Drummer OH, Goble AJ, Louis WJ. High-performance liquid chromatographic assay of perhexiline maleate in plasma. J Pharm Sci. 1981;70:320–322. doi: 10.1002/jps.2600700325. [DOI] [PubMed] [Google Scholar]
- 16.Cooper JDH, Turnell DC, Pilcher J, Lockhart D. Therapeutic monitoring of the anti-anginal drug perhexiline maleate. Ann Clin Biochem. 1985;22:614–617. doi: 10.1177/000456328502200611. [DOI] [PubMed] [Google Scholar]
- 17.Cooper RG, Harper G, Price AH, Evans DAP, Lockhart D. Simultaneous determination of perhexiline and its monohydroxy metabolites in biological fluids by gas chromatography – electron capture detection. J Chromatogr. 1986;381:305–314. doi: 10.1016/s0378-4347(00)83596-3. [DOI] [PubMed] [Google Scholar]
- 18.Amoah AGB, Gould BJ, Parke DV. Single-dose pharmacokinetics of perhexiline administered orally to humans. J Chromatogr. 1984;305:401–409. doi: 10.1016/s0378-4347(00)83354-x. [DOI] [PubMed] [Google Scholar]
- 19.Kerry NL, Somogyi AA, Bochner F, Mikus G. The role of CYP2D6 in primary and secondary oxidative metabolism of dextromethorphan: in vitro studies using human liver microsomes. Br J Clin Pharmacol. 1994;38:243–248. doi: 10.1111/j.1365-2125.1994.tb04348.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guttendorf RJ, Wedlund PJ. Genetic aspects of drug disposition and therapeutics. J Clin Pharmacol. 1992;32:107–117. doi: 10.1002/j.1552-4604.1992.tb03814.x. [DOI] [PubMed] [Google Scholar]
- 21.Ingelman-Sundberg M. Duplication, multiplication, and amplification of genes encoding drug-metabolizing enzymes: evolutionary, toxicological, and clinical pharmacological aspects. Drug Metab Rev. 1999;31:449–459. doi: 10.1081/dmr-100101930. [DOI] [PubMed] [Google Scholar]
- 22.Lovile R, Daly AK, Matre GE, Molven A, Steen VM. Polymorphisms in CYP2D6 duplication-negative individuals with the ultrarapid metabolizer phenotype. A role for the CYP2D6*35 allele in ultrarapid metabolism. Pharmacogenetics. 2001;11:45–55. doi: 10.1097/00008571-200102000-00006. [DOI] [PubMed] [Google Scholar]
- 23.Dahl M-L, Johansson I, Bertilsson L, Ingelman-Sundberg M, Sjoqvist F. Ultrarapid hydroxylation of debrisoquine in a Swedish population. Analysis of the molecular genetic basis. J Pharmacol Exp Ther. 1995;274:516–520. [PubMed] [Google Scholar]
- 24.Aklillu E, Persson I, Bertilsson L, Johansson I, Rodrigues F, Ingelman-Sundberg M. Frequent distribution of ultrarapid metabolizers of debrisoquine in an Ethiopian population carrying duplicated and multiduplicated functional CYP2D6 alleles. J Pharmacol Exp Ther. 1996;278:441–446. [PubMed] [Google Scholar]
- 25.Hussein R, Charles BG, Morris RG, Rasiah RL. Population pharmacokinetics of perhexiline from routine monitoring data. Ther Drug Monit. 2001;23:636–643. doi: 10.1097/00007691-200112000-00007. [DOI] [PubMed] [Google Scholar]
- 26.Ereshefsky L, Riesenman C, Lam YWF. Antidepressant drug interactions and the cytochrome P450 system. The role of cytochrome P450 2D6. Clin Pharmacokinet. 1995;29(Suppl 1):10–19. doi: 10.2165/00003088-199500291-00004. [DOI] [PubMed] [Google Scholar]
- 27.Alfaro CL, Lam YWF, Simpson J, Ereshefsky L. CYP2D6 inhibition by fluoxetine, paroxetine, sertraline, and venlafaxine in a crossover study: intraindividual variability and plasma concentration correlations. J Clin Pharmacol. 2000;40:58–66. doi: 10.1177/00912700022008702. [DOI] [PubMed] [Google Scholar]
- 28.Alderman CP, Hundertmark JD, Soetrama TW. Interaction of serotonin re-uptake inhibitors with perhexiline. Aust NZ J Psych. 1997;31:601–603. doi: 10.3109/00048679709065084. [DOI] [PubMed] [Google Scholar]