

Abstract
Aims
To evaluate whether the potent CYP3A4 inhibitor ketoconazole has any influence on the pharmacokinetic and electrocardiographic parameters of the antimalarial co-artemether (artemether-lumefantrine) in healthy subjects.
Methods
Sixteen subjects were randomized in an open-label, two period crossover design study. Subjects received a single dose of co-artemether (day 1) either alone or in combination with multiple oral doses of ketoconazole (400 mg on day 1 followed by 200 mg o.d. for 4 additional days). Serial blood samples were taken and assayed for artemether and its main active metabolite dihydroartemisinin (DHA), and lumefantrine.
Results
The pharmacokinetics of artemether, its metabolite DHA, and lumefantrine were influenced by the presence of ketoconazole. AUC(0,∞) was increased from 320 to 740 ng ml−1 h (ratio 2.4, 90% CI 2.00, 2.86) for artemether, from 331 to 501 ng ml−1 h (ratio 1.7, 90% CI 1.40, 1.98) for DHA, and from 207 to 333 µg ml−1 h (ratio 1.7, 90% CI 1.23, 2.21) for lumefantrine in the presence of ketoconazole. Cmax also increased in similar proportions for the three compounds (ratio 2.2 (90% CI 1.78, 2.83), 1.4 (90% CI 1.12, 1.74), and 1.3 (90% CI 0.96, 1.64), respectively). The terminal elimination half-life was increased for artemether (2.5 vs 1.9 h, 90% CI 1.12, 1.72) and DHA (3.1 vs 2.1 h, 90% CI 0.02, 3.36), but remained unchanged for lumefantrine (88 vs 95 h, 90% CI 0.81, 1.04). These increases in exposure to the antimalarial combination were much smaller than observed with food intake (up to 16 fold), and were not associated with increased side-effects or changes in electrocardiographic parameters. The study medications were well tolerated.
Conclusions
The concurrent administration of ketoconazole with co-artemether led to modest increases in artemether, DHA, and lumefantrine exposure in healthy subjects. Dose adjustment of co-artemether is probably unnecessary in falciparum malaria patients when administered in association with ketoconazole or other potent CYP3A4 inhibitors.
Keywords: artemether, ECG, ketoconazole, lumefantrine, malaria, QTc
Introduction
Malaria is a leading cause of mortality and morbidity in developing areas of the world, and remains a major health problem in endemic regions. Resistance to available drugs is increasing, thus creating a clear need for new drugs that are well tolerated and simple to use.
Co-artemether is an oral fixed-dose combination tablet of artemether (a derivative of artemisinin) and lumefantrine, an antimalarial synthesized and developed by the Academy of Military Medical Sciences in Beijing, People's Republic of China. This combination was registered in China in 1992 for the treatment of Plasmodium falciparum malaria, and has been developed further by Novartis Pharmaceuticals (Coartem®/Riamet®). To date, Coartem®/Riamet® (Exafal® in Pakistan) has been ap-proved in more than 60 countries, mainly in Africa, Asia, South America, and Europe.
Artemether is characterized by a rapid onset of schizonticidal action, but has a short elimination half-life (2–3 h). However, recrudescence is frequent when artemether is employed as monotherapy [1], unless high dosages are given over several days [2–4]. By contrast, lumefantrine has a longer elimination half-life of several days [5, 6] and is associated with a low recrudescence rate [7, 8], but has a slower onset of action. The rationale for the drug combination is to combine the benefits of the rapid onset of action of artemether with the long duration of action and high cure rate of lumefantrine in a single oral formulation. The combination represents a short, simple to administer, and highly effective antimalarial treatment, and is also likely to improve compliance [8]. This combination approach of an artemisinin derivative with a long-acting other antimalarial is now recommended by the most recent guidelines (World Health Organization [WHO] Model List of Essential Drugs, 1999), since it protects more effectively against the development of resistance [9–14]. The therapeutic dosage regimen in adults consists of repeated administration over 2 days of four doses of co-artemether, comprising four consecutive doses of four tablets each (80 mg artemether +480 mg lumefantrine per dose). In areas of multiple drug-resistant malaria, such as Thailand, and for stand-by emergency treatment, an intensive 3 day course including two additional doses is recommended.
The pharmacokinetics of artemether, its active me-tabolite dihydroartemisinin (DHA), and lumefantrine have been characterized after oral doses of co-artemether in healthy subjects and in paediatric and adult patients with falciparum malaria [5, 6, 14–16]. In vitro and in vivo data indicated that artemether and lumefantrine are predominantly metabolized by cytochrome P450 3A4 (CYP3A4) [5, 17]. Therefore, concomitant intake of drugs that inhibit CYP3A4 activity could result in increased plasma concentrations of artemether and/or lumefantrine, potentially leading to unwanted pharmacodynamic effects.
Ketoconazole, an imidazole derivative and broad-spectrum antifungal agent, is among the most potent inhibitors of CYP3A4. It has been shown to impair the oxidative metabolism of a wide range of clinically used drugs such as antihistamines, immunosuppressives, benzodiazepines and HIV protease inhibitors [18–22]. Clinically significant interactions were reported between ketoconazole and terfenadine or astemizole or tolbutamide leading to QTc interval prolongation and torsade de pointes [23–27].
In light of these findings, the present study was conducted to investigate the effect of ketoconazole on the pharmacokinetics of artemether, its active main metabolite DHA, and lumefantrine. Although co-artemether, in contrast to other antimalarials like quinine or halofantrine, has never been reported to cause any relevant cardiotoxicity when given alone [28–31] or in combination with mefloquine [32] or quinine [16], electrocardiographic parameters were closely monitored during this study.
Methods
Subjects
Sixteen healthy Caucasian nonsmokers, male and female subjects aged 19–49 years (mean 29.7) and weighing 61.4–87.4 kg (mean 78.6) participated in the trial. The study excluded subjects with a history of drug sensitivity or allergy, heart disease or significant ECG abnormalities (e.g. QTc intervals>440 ms), psychiatric disturbances, a recent history of alcohol abuse, and subjects who re-quired regular medication. Subjects were excluded if they had taken any prescription medication within 4 weeks, or over-the-counter medications within 2 weeks of the start of the study. Furthermore, only females surgically sterilized prior to study entry were admitted. The trial was conducted at MDS Pharma Services (MDSPS), Belfast, Northern Ireland, in accordance with the World Medical Association's Declaration of Helsinki, and Good Clinical Practice. Ethics Committee approval of the protocol, consent form, and volunteer information document was obtained from the Queen's University Independent Review Board. All subjects provided written informed consent before participating in the trial.
Dosage forms and doses
Co-artemether (Riamet®, Novartis Pharma AG, Basel, Switzerland): tablets, each containing 20 mg artemether and 120 mg lumefantrine. Co-artemether was administered on day 1 as a single dose corresponding to four tablets (total dose: 80 mg artemether/480 mg lumefantrine).
Ketoconazole (Nizoral®, Janssen-Cilag, Saunderton, High Wycombe, United Kingdom) was administered on day 1 (400 mg, 2 tablets), followed by 200 mg (1 tablet) once daily on days 2–5.
Study design and rationale
Either a single dose of artemether-lumefantrine (80/480 mg), or five subsequent once-daily doses of ketoconazole (400 mg on day 1 and 200 mg on days 2–5) in combination with co-artemether (80/480 mg) on day 1 were administered in a randomized, non-blind, two period crossover design, with a 7 week washout (t1/2 of lumefantrine is 3–6 days). Each subject participated in a screening period (day −21 to day −2), a baseline period (12 h prior to dosing), and the two treatment periods. Subjects were confined to the study site until after the 48 h PK and safety assessments. They returned to the study centre on days 3, 4 and 5 to receive their daily dose of ketoconazole and/or for blood sampling and safety assessments. Since food is known to increase the systemic exposure to both ketoconazole [33] and artemether-lumefantrine [5, 34], the study medications were taken with a high fat breakfast (2 eggs fried, 2 strips of bacon, 1 slice of toast with butter, 2 hash brown potatoes, 240 ml of full-fat milk). Fluids that did not contain caffeine or grapefruit juice were allowed ad libitum.
Pharmacokinetic analysis
Blood samples were taken for the analysis of artemether and its active metabolite DHA in plasma at predose (0 h), and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, and 48 h post dose of co-artemether. Blood samples were taken for the analysis of lumefantrine in plasma at predose (0 h), and at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 10, 12, 16, 24, 36, 48, 72, 96, 120, 168, 216, and 264 h post-dose of co-artemether. Blood was collected in lithium-heparinized tubes, and centrifuged without delay at 4 °C at 2200 g for 5 min. Plasma was then split into three aliquots that were kept in polypropylene screw-cap tubes at −70 °C until analysis. Artemether and DHA were measured by high-performance liquid chromatography (h.p.l.c.) – mass spectrometry [35]. Lumefantrine was measured by h.p.l.c. with ultraviolet detection [36]. The within-assay accuracy data of control samples (in percent of nominal concentration) were in the range 97–102% for artemether, 95% for DHA, and 102–107% for lumefantrine, with coefficients of variation (CV) of 10–12%, 9–16%, and 6–8%, respectively. Limits of quantifica-tion were 5 ng ml−1 for artemether and DHA, and 50 ng ml−1 for lumefantrine.
The following model-independent pharmacokinetic parameters (WinNonlin Professional, Version 3.1, Pharsight Corporation, Mountain View, CA, USA) were determined from plasma concentration data: Cmax, highest observed plasma concentration; tmax, time to reach Cmax; t1/2, apparent terminal elimination half-life (calculated as t1/2 = ln2/λz, where λz is the rate constant associated with the terminal phase of elimination, estimated by log-linear least-squares regression through the last concentration points different from zero); AUC(0,last), area under the plasma concentration-time curve calculated by the trapezoidal (linear-log) method over the time interval 0 h to the last time point with a concentration different from zero (Ct). AUC(0,∞), AUC extrapolated to time infinity calculated as AUC(0,last) + Ct/λz. Concentrations below the limit of quantification of the relevant analytical method were taken as zero in descriptive statistics and pharmacokinetic calculations.
Safety assessments
Medical history, physical examination, vital signs, routine clinical laboratory tests, ECGs and urine screens for drugs and alcohol were performed at screening and at specific time points during the trial (ECGs were recorded frequently throughout the trial period), then at the follow up visit. Adverse events were recorded continuously throughout the trial, and the onset, duration, severity, and relationship to the trial drugs were noted.
Standard 12-lead ECGs were recorded at screening, baseline (both treatment periods), immediately prior to dosing (time 0 h), and at 2, 8, 10, 12, 24, 36, and 48 h postdose of co-artemether. QTc-interval was calculated by using the Bazett formula QTc = QT/√RR [37, 38] to correct for the influence of heart rate. For subjects’ safety, the investigator evaluated the PR-, QRS- and QT-intervals visually on an ongoing basis.
Statistical analysis
The power calculation for determination of sample size was based on detecting a two-fold difference in exposure (AUC). Based on a previous volunteer study [6], and assuming no drug–drug interaction, 13 subjects were required to demonstrate with 80% power that the 90% confidence interval (90% CI) for AUCcombination/AUCco-artemether lies within the interval 0.5–2.0. Because of the long duration of the study, 16 subjects were enrolled in case of withdrawals.
To investigate the pharmacokinetic interaction effects between treatments, AUC(0,last), Cmax, tmax, and t1/2 of artemether, DHA, and lumefantrine were compared by analysis of variance (anova). Values were transformed to the natural logarithm scale and then used as the dependent variable with treatment, sequence, subject within sequence, and period as factors. tmax was analysed by the Wilcoxon rank sum test. In accordance with recent FDA guidance for industry on In Vivo Drug Metabolism/Drug Interaction Studies (http://www.fda.gov/cder/guidance/2635fnl.pdf), comparison between treatment was based on 90% CIs. No P values were given according to this guidance.
The GLM procedure (SAS Release 8.1 for Open VMS, SAS Institute Inc.) was used in the above anova evaluations. Estimated differences and their standard errors between pairs of treatments (or other main effects) were obtained using the ESTIMATE statement. These were exponentiated, i.e. antilogarithmically transformed, to obtain estimated ratios. Similarly, 90% CIs for the ratios were also obtained. The intersubject coefficient of variation of the dependent variable was obtained by taking the square root of the residual variance after fitting the GLM model. The GLM procedure was used for Wilcoxon test of tmax. Only descriptive statistics were done for cardiographic parameters (QTc).
Results
Safety and tolerability
Co-artemether given as a single dose or coadministered with multiple doses of ketoconazole appeared to be safe and well tolerated in this healthy male and female population.
There were no serious adverse events (AEs). There were 9 AEs in 5 (31%) of the 16 subjects, mostly of mild intensity. These 9 AEs included sunburn, increased GGT, increased blood CK, and dizziness. The only AE considered to be possibly treatment-related was the episode of dizziness that occurred 24 h following co-artemether alone treatment and lasted 1 min. Three subjects withdrew from the study, one due to a bloodshot eye, abrasion, contusion, and hand fracture subsequent to a bike accident (unrelated to the study drugs), one due to increased GGT (unrelated to the study drugs), and one due to withdrawal of consent. No clinically significant deviations in laboratory parameters (haematology, blood chemistry and urinalysis) were observed in any of the treatment groups.
ECG parameters (PR-interval, QRS-complex, heart rate, QT- and QTc-interval) remained well within normal limits in both treatment groups. Except for one single borderline peak QTc value of 465 ms (causally unrelated to the study medication) observed in a male 2 h after co-artemether alone treatment, no clinically significant modifications in QTc intervals were observed in any subject and treatment, and no abnormal clinical signs or symptoms were reported. No clinically significant trends in ventricular rate, RR- or PR-interval were observed for any subject.
Pharmacokinetics
Artemether–Dihydroartemisinin
The mean plasma profiles of artemether and dihydroartemisinin are shown in Figure 1, and the pharmacokinetic parameters and statistical results are summarized in Table 1. Co-administration of co-artemether with ketoconazole was associated with an increase in overall artemether and DHA exposure compared to co-artemether alone. AUC(0,∞) increased by 2.4 fold (from 320 to 740 ng ml−1 h) for artemether, and 1.7 fold (from 331 to 501 ng ml−1 h) for DHA. Similarly, Cmax increased by 2.2 fold (from 104 to 225 ng ml−1) for artemether, and 1.4 fold (from 104 to 142 ng ml−1) for DHA. These changes were associated with a slight increase in t1/2 for both artemether (1.9 vs 2.5 h) and DHA (2.1 vs 3.1 h). There was no treatment difference in tmax for both artemether and DHA. Since the 90% CIs for ratios of Cmax and of AUC means both fell outside the no effect 0.80–1.25 range, the effect of ketoconazole was statistically significant.
Figure 1.
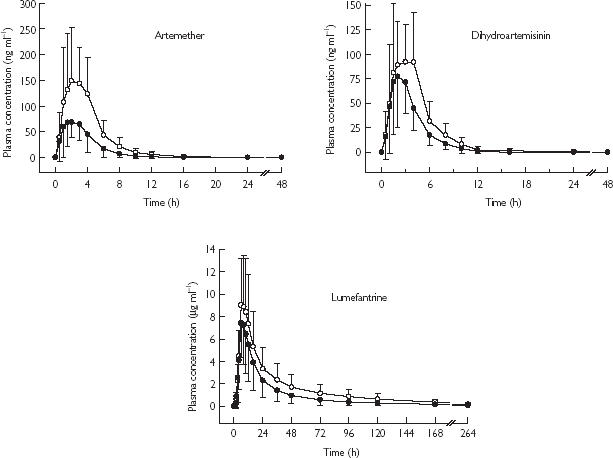
Plasma concentrations (mean ± s.d.) of artemether, dihydroartemisinin, and lumefantrine following administration of co-artemether alone (•, n = 14) or in combination with ketoconazole (○, n = 15).
Table 1.
Mean ± s.d. (n = 14–15) pharmacokinetic parameters for artemether, dihydroartemisinin, and lumefantrine following administration of co-artemether alone or with ketoconazole.
| Co-artemetheralone | Co-artemether+ketoconazole | Ratio1[90% CI] | |
|---|---|---|---|
| Artemether | |||
| Cmax (ng ml−1) | 104 ± 40 | 225 ± 77 | 2.24 [1.78, 2.83] |
| tmax2 (h) | 2.0 [1.0–4.0] | 2.0 [1.0–4.0] | |
| AUC(0,last) (ng ml−1 h) | 302 ± 135 | 718 ± 279 | 2.51 [2.07, 3.04] |
| AUC(0,∞) (ng ml−1 h) | 320 ± 138 | 740 ± 286 | 2.39 [2.00, 2.86] |
| t1/2 (h) | 1.9 ± 0.8 | 2.5 ± 1.1 | 1.42 [1.12, 1.72] |
| Dihydroartemisinin | |||
| Cmax (ng ml−1) | 104 ± 45 | 142 ± 55 | 1.40 [1.12, 1.74] |
| tmax2 (h) | 2.5 [1.0–4.0] | 2.0 [1.0–4.0] | |
| AUC(0,last) (ng ml−1 h) | 308 ± 112 | 475 ± 139 | 1.72 [1.42, 2.08] |
| AUC(0,∞) (ng ml−1 h) | 331 ± 111 | 501 ± 155 | 1.66 [1.40, 1.98] |
| t1/2 (h) | 2.1 ± 0.9 | 3.1 ± 3.8 | 1.69 [0.02, 3.36] |
| Lumefantrine | |||
| Cmax (µg ml−1) | 7.91 ± 3.49 | 10.1 ± 4.74 | 1.26 [0.96, 1.64] |
| tmax2 (h) | 6.0[4.0–10.0] | 6.0 [6.0–10.0] | |
| AUC(0,last) (µg ml−1 h) | 195 ± 119 | 312 ± 181 | 1.66 [1.23, 2.23] |
| AUC(0,∞) (µg ml−1 h) | 207 ± 123 | 333 ± 194 | 1.65 [1.23, 2.21] |
| t1/2 (h) | 95 ± 18 | 88 ± 15 | 0.93 [0.81, 1.04] |
median difference for tmax, CI = confidence interval
median [range] for tmax.
Lumefantrine
The mean plasma profiles of lumefantrine following the two treatments co-artemether alone or with ketoconazole are shown in Figure 1, and the pharmacokinetic parameters and statistical results are summarized in Table 1. Co-administration of co-artemether with ketoconazole was associated with an increase in lumefantrine exposure compared with co-artemether alone. AUC(0,∞) increased by 1.7 fold (from 207 to 333 µg ml−1 h) and Cmax by 1.3 fold (from 7.91 to 10.1 µg ml−1). No treatment difference was observed for either tmax (6.0 h following both treatments) or t1/2 (95 vs 88 h). Although the 90% CIs for the ratios of AUC(0,last) and of AUC(0,∞) did not encompass 1, and therefore indicated that the extent of exposure from the two treatments was statistically different, the lower boundaries of the 90% CIs for the ratios of AUC and of Cmax (for which the 90% CIs did encompass 1), were all below 1.25. Therefore, the drug–drug interaction between lumefantrine and ketoconazole was not considered statistically significant, although there was evidence of drug–drug interaction.
Discussion
The antimycotic agent ketoconazole is a potent inhibitor of hepatic and gastrointestinal cytochrome P450 isoenzyme CYP3A4, and of intestinal P-glycoprotein (P-gp), the multidrug transporter which pumps many drugs from cytoplasm back into the gastrointestinal tract [39, 40]. Co-administration of such potent inhibitors like ketoconazole with other drugs metabolized by CYP3A4 has been reported to lead to potentially highly clinically relevant interactions [22–27, 41]. Since artemether and lumefantrine are both predominantly metabolized by CYP3A4, this study in healthy volunteers aimed to in-vestigate whether coadministration of ketoconazole could affect the pharmacokinetics of artemether, dihydroartemisinin, and lumefantrine.
Since ketoconazole is a very potent CYP3A4 inhibitor, only a single dose of co-artemether was given in order to avoid any potential adverse effects of the latter. Hepatic CYP3A4 activity is nearly abolished after a single dose of ketoconazole [42], and since lumefantrine undergoes slow absorption with a 2 h lag time, pretreatment by ketoconazole prior to administration of co-artemether was not considered necessary. Treatment by ketoconazole for several days (after the dose of co-artemether) was considered to better distinguish between metabolism occurring in the liver compared to the intestine.
In the present study, coadministration of ketoconazole and co-artemether resulted in an increase in the exposure to the three active principles artemether, DHA, and lumefantrine. However, we did not consider the magnitude of this effect to be clinically important, because of the wide safety margin of co-artemether. The extent of this change was within the between-subject variability in artemether, DHA, and lumefantrine pharmacokinetics observed in previous clinical trials, and, in addition, no relationship has so far been observed in the clinical setting between AUC (or Cmax) and adverse effects [43]. Moreover, the influence of ketoconazole on artemether-lumefantrine exposure (1.3–2.5 fold increase) was much smaller than the 16-fold increase in plasma lumefantrine concentrations observed when co-artemether is given with food, as recommended [5, 34].
There are three possible mechanisms by which ketoconazole can increase exposure to artemether, DHA, and lumefantrine, namely inhibition of hepatic CYP3A4 resulting in reduction in systemic metabolism, inhibition of gastrointestinal CYP3A4 resulting in increased absorption, and/or inhibition of P-gp resulting in increased systemic bioavailability [44]. That the half-life of lumefantrine was not increased when co-artemether was given with ketoconazole suggests that the main effect of ketoconazole was to increase lumefantrine bioavalability through inhibition of presystemic CYP3A4 metabolism in the gut, rather than that in the liver. The results for artemether (i.e. increased AUC and t1/2) suggest strongly that ketoconazole inhibits artemether metabolism mainly through its effect on hepatic CYP3A4. Similar data for dihydroartemisinin, although of smaller magnitude than these observed for its parent, artemether, provide evidence that dihydroartemisinin is also a substrate for CYP3A4, as reported for artemisinin [45].
These data demonstrate that potent inhibition of CYP3A4 activity has only a modest effect in vivo on the pharmacokinetics of artemether and lumefantrine, even though ketoconazole treatment was continued for several consecutive days. However, the present findings were not totally unexpected, since in addition to the predominant role of CYP3A4, in vitro data showed that CYP1A1, CYP1A2, CYP2B6, CYP2C9, and CYP2C19 may also contribute to the biotransformation of co-artemether [5, 6]. Our data are in agreement with a previous report showing that grapefruit juice, another inhibitor of CYP3A4 present in the mucosa cells of the gut wall, increased by nearly two-fold the systemic exposure to artemether after a dose of 100 mg [17].
The two treatments were safe and well tolerated in healthy subjects. Of particular importance is that no prolongations of the QTc interval were observed with co-artemether alone or in combination with ketoconazole, indicating that the observed increase in systemic exposure to artemether, DHA and lumefantrine had no detectable influence on the safety/tolerability profile of co-artemether.
All reports on co-artemether (Coartem®/Riamet®) show that the drug is safe and effective [16, 46, 47]. In contrast to several other antimalarials of the aryl amino alcohol class, co-artemether causes no cardiotoxicity, even when given in combination with quinine [16], or mefloquine [6, 32] an antimalarial known to enhance the QTc prolonging effect of halofantrine [48, 49]. QTc changes are common in malaria, and are variously ascribed to drug or disease effects [1]. A prospective population pharmacokinetic and pharmacodynamic analysis of lumefantrine in 266 Thai malaria patients showed that the drug had no effect on the QTc interval, and that there were no other adverse effects that could be ascribed unequivocally to the drug [43]. Similarly, lumefantrine was observed to have little or no effect on ventricular repolarization, suggesting that there is no a priori reason to withhold this drug from patients with a long QTc or any other cardiac abnormality [28].
In conclusion, overlapping therapy with the antimalarial artemether-lumefantrine and the potent CYP3A4 inhibitor ketoconazole is unlikely to increase the risk of adverse effects in the treatment of Plasmodium falciparum malaria. The pharmacokinetics of artemether and its active metabolite DHA, and lumefantrine were increased following the combined treatment with ketoconazole, but the difference was not considered to be of clinical importance. Artemether-lumefantrine alone had no effect on the ECG parameters, nor did the combination with ketoconazole. If these data in healthy subjects can be extrapolated to the clinical setting, no dose adjustment of co-artemether is recommended in malaria patients also receiving ketoconazole or other potent CYP3A4 inhibitors.
Acknowledgments
We thank the staff of MDS Pharma Services, for conducting the study and their support in collecting and handling of plasma samples. We thank N. Gauducheau, N. Sandrenan, J.J. Courte, and P. Mangoni (Novartis Pharma SA, Rueil-Malmaison, France) for their expert technical assistance in analysing artemether, DHA, and lumefantrine in plasma samples. This study was supported by a grant from Novartis Pharma AG.
References
- 1.von Seidlein L, Jaffar S, Pinder M, et al. Treatment of African children with uncomplicated falciparum malaria with a new antimalarial drug, CGP 56697. J Infect Dis. 1997;176:1113–1116. doi: 10.1086/516524. [DOI] [PubMed] [Google Scholar]
- 2.Bunnag D, Viravan C, Looareesuwan S, Karbwang J, Harinasuta T. Clinical trial of artesunate and artemether on multidrug resistant falciparum malaria in Thailand. A preliminary report. Southeast Asian J Trop Med Public Health. 1991;22:380–385. [PubMed] [Google Scholar]
- 3.Karbwang J, Bangchang K, Thanavibul A, Back D, Bunnag D, Harinasuta T. Pharmacokinetics of mefloquine alone or in combination with artesunate. Bull World Health Organ. 1994;72:83–87. [PMC free article] [PubMed] [Google Scholar]
- 4.White NJ. The treatment of malaria. N Engl J Med. 1996;335:800–806. doi: 10.1056/NEJM199609123351107. [DOI] [PubMed] [Google Scholar]
- 5.Lefèvre G, Thomsen MS. Clinical pharmacokinetics of artemether and lumefantrine (Riamet®) Clin Drug Invest. 1999;18:467–480. [Google Scholar]
- 6.Lefèvre G, Bindschedler M, Ezzet F, Schaeffer N, Meyer I, Thomsen M. Pharmacokinetic interaction trial between co-artemether and mefloquine. Eur J Pharm Sci. 2000;10:141–151. doi: 10.1016/s0928-0987(00)00060-9. [DOI] [PubMed] [Google Scholar]
- 7.Ezzet F, Mull R, Karbwang J. Population pharmacokinetics and therapeutic response of CGP 56697 (artemether + benflumetol) in malaria patients. Br J Clin Pharmacol. 1998;46:553–561. doi: 10.1046/j.1365-2125.1998.00830.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Skelton-Stroud P, Mull R. The Novartis Co-artemether International Development Team. Positioning, labelling, and medical information control of co-artemether tablets (CGP 56697): a fixed novel combination of artemether and benflumetol. Med Trop. 1998;58:77S–81S. [PubMed] [Google Scholar]
- 9.White NJ. Assessments of the pharmacodynamic properties of antimalarial drugs in vivo. Antimicrob Agents Chemother. 1997;41:638–644. doi: 10.1128/aac.41.7.1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.White NJ. Preventing antimalarial drug resistance through combinations. Drug Resistance Updates. 1998;1:3–9. doi: 10.1016/s1368-7646(98)80208-2. [DOI] [PubMed] [Google Scholar]
- 11.Ambroise-Thomas P. Current data on major novel antimalaria drugs: artemisinin (qinghaosu) derivatives. Bull Acad Natl Med. 1999;183:797–800. [PubMed] [Google Scholar]
- 12.Price RN. Artemisinin drugs: novel antimalarial agents. Expert Opin Invest Drugs. 2000;9:1815–1827. doi: 10.1517/13543784.9.8.1815. [DOI] [PubMed] [Google Scholar]
- 13.Peters W, Robinson BL. The chemotherapy of rodent malaria. LVIII. Drug combinations to impede the selection of drug resistance, Part. 2: The new generation-artemisinin or artesunate with long-acting, blood schizontocides. Ann Trop Med Parasitol. 2000;94:23–35. doi: 10.1080/00034980057581. [DOI] [PubMed] [Google Scholar]
- 14.White NJ, van Vugt M, Ezzet F. Clinical pharmacokinetics and pharmacodynamics of artemether-lumefantrine. Clin Pharmacokinet. 1999;37:105–125. doi: 10.2165/00003088-199937020-00002. [DOI] [PubMed] [Google Scholar]
- 15.van Agtmael M, Cheng-Qi S, Qing JX, Mull R, van Boxtel CJ. Multiple dose pharmacokinetics of artemether in Chinese patients with uncomplicated falciparum malaria. Int J Antimicrob Agents. 1999;12:151–158. doi: 10.1016/s0924-8579(99)00063-1. [DOI] [PubMed] [Google Scholar]
- 16.Lefèvre G, Looareesuwan S, Treeprasertsuk S, et al. A Clinical and pharmacokinetic trial of six doses of artemether–lumefantrine for multidrug-resistant Plasmodium falciparum malaria in Thailand. Am J Trop Med Hyg. 2001;64:247–256. doi: 10.4269/ajtmh.2001.64.247. [DOI] [PubMed] [Google Scholar]
- 17.van Agtmael M, Gupta V, van der Wösten TH, Rutten J-PB, van Boxtel CJ. Grapefruit juice increases the bioavailability of artemether. Eur J Clin Pharmacol. 1999;55:405–410. doi: 10.1007/s002280050648. [DOI] [PubMed] [Google Scholar]
- 18.Thummel KE, Wilkinson GR. In vitro and in vivo drug interactions involving human CYP3A. Ann Rev Pharmacol Toxicol. 1998;38:389–430. doi: 10.1146/annurev.pharmtox.38.1.389. [DOI] [PubMed] [Google Scholar]
- 19.Bennett JE. Antimicrobial agents: antifungal agents. In: Hardman JJG, Limbird LE, Molinoff PB, Ruddon RW, Goodman Gilman A, editors. The Pharmalogical Basis of Therapeutics. 8. New York: McGraw-Hill; 1996. pp. 1175–1190. [Google Scholar]
- 20.Ament PW, Paterson A. Drug interactions with the nonsedating antihistamines. Am Fam Physician. 1997;56:223–231. [PubMed] [Google Scholar]
- 21.Sadowski DC. Drug interactions with antacids. Mechanisms and clinical significance. Drug Safety. 1994;11:395–407. doi: 10.2165/00002018-199411060-00002. [DOI] [PubMed] [Google Scholar]
- 22.Heinig R, Adelmann HG, Ahr G. The effect of ketoconazole on the pharmacokinetics, pharmacodynamics and safety of nisoldipine. Eur J Clin Pharmacol. 1999;55:57–60. doi: 10.1007/s002280050593. [DOI] [PubMed] [Google Scholar]
- 23.Monahan BP, Ferguson CL, Killeavy ES, Lloyd BK, Troy J, Cantilena LR. Torsades de pointes occurring in association with terfenadine use. JAMA. 1990;264:2788–2790. [PubMed] [Google Scholar]
- 24.Honig PK, Wortham DC, Hull R, Zamani K, Smith JE, Cantilena LR. Itraconazole affects single-dose terfenadine pharmacokinetics and cardiac repolarization pharmacodynamics. J Clin Pharmacol. 1993;33:1201–1206. doi: 10.1002/j.1552-4604.1993.tb03920.x. [DOI] [PubMed] [Google Scholar]
- 25.Honig PK, Wortham DC, Hull R, et al. The effect of fluconazole on the steady-state pharmacokinetics and electrocardiographic pharmacodynamics of terfenadine in humans. Clin Pharmacol Ther. 1993;53:630–636. doi: 10.1038/clpt.1993.83. [DOI] [PubMed] [Google Scholar]
- 26.Honig PK, Wortham DC, Hull R, et al. Terfenadine–ketoconazole interaction. Pharmacokinetic and electrocardiographic consequences. JAMA. 1993;269:1513–1518. [Published erratum appears in JAMA 1993; 269: 2088; see comments in JAMA 1993; 269: 1550–1152] [PubMed] [Google Scholar]
- 27.Simons FE, Kesselman MS, Giddins NG, Pelech AN, Simons KJ. Astemizole-induced torsades de pointes. Lancet. 1988;ii:624. doi: 10.1016/s0140-6736(88)90656-3. [DOI] [PubMed] [Google Scholar]
- 28.van Vugt M, Ezzet F, Nosten F, et al. No evidence of cardiotoxicity during antimalarial treatment with artemether-lumefantrine. Am J Trop Med Hyg. 1999;61:964–967. doi: 10.4269/ajtmh.1999.61.964. [DOI] [PubMed] [Google Scholar]
- 29.van Agtmael M, Bouchaud O, Malvy D, et al. The comparative efficacy and tolerability of CGP 56697 (artemether + lumefantrine) versus halofantrine in the treatment of uncomplicated falciparum malaria in travelers returning from the Tropics to the Netherlands and France. Int J Antimicrob Agents. 1999;12:159–169. doi: 10.1016/s0924-8579(99)00070-9. [DOI] [PubMed] [Google Scholar]
- 30.Kshirsagar NA, Gogtay NJ, Moorthy NS, et al. A randomized, double-blind, parallel group, comparative safety and efficacy trial of oral co-artemether versus oral chloroquine in the treatment of acute, uncompliacted P. falciparum malaria in adults in India. Am J Trop Med Hyg. 2000;62:402–408. doi: 10.4269/ajtmh.2000.62.402. [DOI] [PubMed] [Google Scholar]
- 31.Bindschedler M, Lefèvre G, Degen P, Sioufi A. Comparison of the cardiac effects of the antimalarials co-artemether and halofantrine in healthy participants. Am J Trop Med Hyg. 2002;66:295–300. doi: 10.4269/ajtmh.2002.66.293. [DOI] [PubMed] [Google Scholar]
- 32.Bindschedler M, Lefèvre G, Ezzet F, Schaeffer N, Meyer I, Thomsen M. Cardiac effects of co-artemether (artemether/lumefantrine) and mefloquine given alone or in combination to healthy volunteers. Eur J Clin Pharmacol. 2000;56:375–381. doi: 10.1007/s002280000155. [DOI] [PubMed] [Google Scholar]
- 33.Daneshmend TK, Warnock DW, Ene MD, et al. Influence of food on the pharmacokinetics of ketoconazole. Antimicrob Agents Chemother. 1984;25:1–3. doi: 10.1128/aac.25.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bindschedler M, Degen P, Lu ZL, Jiao XQ, Liu GY, Fan F. Proceedings of the Xivth International Congress for Tropical Medicine and Malaria. Nagasaki, Japan: 1996. Comparative bioavailability of benflumetol after administration of single oral doses of co-artemether under fed and fasted conditions to healthy subjects (abstract P-01–96) pp. 17–22. November. [Google Scholar]
- 35.Souppart C, Gauducheau N, Sandrenan N, Richard F. Development and validation of a high-performance liquid chromatography – mass spectrometry assay for the determination of artemether and its dihydroartemisinin metabolite in human plasma. J Chromatogr B. 2002 doi: 10.1016/s1570-0232(02)00207-6. in press. [DOI] [PubMed] [Google Scholar]
- 36.Mansor SM, Navaratnam V, Yahaya N, Nair NK, Wernsdorfer WH, Degen PH. Determination of a new antimalarial drug, benflumetol, in blood plasma by reverse-phase high-performance liquid chromatography with ultraviolet detection. J Chromatogr. 1996;682:321–325. doi: 10.1016/0378-4347(96)00086-2. [DOI] [PubMed] [Google Scholar]
- 37.Bazett HC. An analysis of the time relations of electrocardiograms. Heart. 1920;VII:354–369. [Google Scholar]
- 38.Ahnve S. Correction of QT interval for heart rate: review of different formulas and the use of Bazett's formula in myocardial infarction. Am Heart J. 1985;109:568–574. doi: 10.1016/0002-8703(85)90564-2. [DOI] [PubMed] [Google Scholar]
- 39.Siegsmund MJ, Cardarelli C, Aksentijevivh M, Sugimoto Y, Pastan I, Gottesman MM. Ketoconazole effectively reverses multidrug resistance in highly resistant KB cell. J Urol. 1994;151:485–491. doi: 10.1016/s0022-5347(17)34999-6. [DOI] [PubMed] [Google Scholar]
- 40.Takano M, Hasegawa R, Fukuda T, Yumoto R, Nagai J, Murakami T. Interaction with P-glycoprotein and transport of erythromycin, midazolam and ketoconazole in Caco-2. Eur J Pharmacol. 1998;358:289–294. doi: 10.1016/s0014-2999(98)00607-4. [DOI] [PubMed] [Google Scholar]
- 41.Kremens B, Brendel E, Bald M, Czyborra P, Michel MC. Loss of blood pressure control on withdrawal of fluoconazole during nifedipine therapy. Br J Clin Pharmacol. 1999;47:707–708. doi: 10.1046/j.1365-2125.1999.00965.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Polk RE, Crouch MA, Israel DS, et al. Pharmacokinetic interaction between ketoconazole and amprenavir after single doses in healthy men. Pharmacother. 1999;19:1379–1384. doi: 10.1592/phco.19.18.1378.30905. [DOI] [PubMed] [Google Scholar]
- 43.Ezzet F, van Vugt M, Nosten F, Looareesuwan S, White NJ. Pharmacokinetics and pharmacodynamics of lumefantrine (benflumetol) in acute falciparum malaria. Antimicrob Agents Chemother. 2000;44:697–704. doi: 10.1128/aac.44.3.697-704.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Hall SD, Thummel KE, Watkins PB, et al. Molecular and physical mechanisms of first-pass extraction. Drug Metab Dispos. 1999;27:161–166. [PubMed] [Google Scholar]
- 45.Svensson USH, Ashton M. Identification of the human cytochrome P450 enzymes involved in the in vitro metabolism of artemisinin. Br J Clin Pharmacol. 1999;48:528–535. doi: 10.1046/j.1365-2125.1999.00044.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.van Vugt M, Wilairatana P, Gemperli B, et al. Efficacy of six doses of artemether-lumefantrine (benflumetol) in multi-drug resistant falciparum malaria. Am J Trop Med Hyg. 1999;60:936–942. doi: 10.4269/ajtmh.1999.60.936. [DOI] [PubMed] [Google Scholar]
- 47.Bakshi R, Hermeling-Fritz I, Gathmann I, Alteri E. An integrated assessment of the clinical safety of artemether-lumefantrine: a new oral fixed-dose combination antimalarial drug. Trans R Soc Trop Med Hyg. 2000;94:419–424. doi: 10.1016/s0035-9203(00)90126-3. [DOI] [PubMed] [Google Scholar]
- 48.Nosten F, ter Kuile F, Luxemburger C, et al. Cardiac effects of antimalarial treatment with halofantrine. Lancet. 1993;341:1054–1056. doi: 10.1016/0140-6736(93)92412-m. [DOI] [PubMed] [Google Scholar]
- 49.Coyne PE, Ajayi F, Harris J, Wiley T, Worthham D, Cantilena LR. Pharmacodynamics and pharmacokinetics of halofantrine and mefloquine (abstract PII-7). American Society for Clinical Pharmacology and Therapeutics. Clin Pharmacol Ther. 1996;59:160. [Google Scholar]