

Abstract
Branching morphogenesis in the mammalian lung and Drosophila trachea relies on the precise localization of secreted modulators of epithelial growth to select branch sites and direct branch elongation, but the intercellular signals that control blood vessel branching have not been previously identified. We found that VEGF120/120 mouse embryos, engineered to express solely an isoform of VEGF-A that lacks heparin-binding, and therefore extracellular matrix interaction domains, exhibited a specific decrease in capillary branch formation. This defect was not caused by isoform-specific differences in stimulating endothelial cell proliferation or by impaired isoform-specific signaling through the Nrp1 receptor. Rather, changes in the extracellular localization of VEGF-A in heparin-binding mutant embryos resulted in an altered distribution of endothelial cells within the growing vasculature. Instead of being recruited into additional branches, nascent endothelial cells were preferentially integrated within existing vessels to increase lumen caliber. The disruption of the normal VEGF-A concentration gradient also impaired the directed extension of endothelial cell filopodia, suggesting that heparin-binding VEGF-A isoforms normally provide spatially restricted stimulatory cues that polarize and thereby guide sprouting endothelial cells to initiate vascular branch formation. Consistent with this idea, we found opposing defects in embryos harboring only a heparin-binding isoform of VEGF-A, including excess endothelial filopodia and abnormally thin vessel branches in ectopic sites. We conclude that differential VEGF-A isoform localization in the extracellular space provides a control point for regulating vascular branching pattern.
Keywords: VEGF, blood vessel, angiogenesis, embryo, branching morphogenesis
The elaboration of complex pattern during embryonic development relies on the spatio-temporal organization of differentiation events through the localized expression of a large number of secreted signaling molecules (Lawrence and Struhl 1996). The importance of the precise intercellular localization of such molecules is exemplified during branching morphogenesis in several organs, including the mammalian kidney and lung and the Drosophila tracheal system. Here, the reiterative formation of branch points relies on the precisely coordinated expression of stimulatory and inhibitory extracellular cues to transform a simple epithelial tubule into an expanding tree-like structure (Hogan and Yingling 1998; Metzger and Krasnow 1999).
As we have learnt much about branching morphogenesis in the systems described above, it is surprising how little we understand about branching in the most pervasive network of vertebrates, the vasculature. Expanding blood vessel networks must branch frequently to ensure that all tissues of the developing embryo receive an adequate blood supply, but equally, they must obey strict patterning cues to match the precise architectural designs of the resident tissue. This patterning demand is perhaps best exemplified in the lung and kidney, in which juxtaposition of the bloodstream with alveoli or nephric tubules, respectively, is essential for organ function (Abrahamson et al. 1998; Gebb and Shannon 2000). Much progress has been made in the identification of intercellular signaling factors that control the assembly of blood vessels from primitive precursors (vasculogenesis) or can induce the extension of existing networks by sprouting growth (angiogenesis); yet, we still do not know which of these molecules control vessel branching (Risau 1997).
A conserved and well-studied feature of intercellular signaling in other branching epithelia is the precise spatial positioning of behavioral and gene activation cues, presumably to single out focal subsets of cells from the epithelia to select branch sites. In the Drosophila trachea, single cells situated around the periphery of a tracheal tubule secrete the fibroblast growth factor (FGF) branchless to induce the migration of tracheal cells toward the ligand source and thereby initiate new branch formation (Sutherland et al. 1996). In a similar manner, lung and kidney tubules branch in response to stimulatory signals from the juxtaposed mesenchyme, FGF10 and hepatocyte growth factor (HGF), respectively (Bellusci et al. 1997; Davies 2001).
In addition to their highly localized gene expression patterns, the secreted molecules described above share a common biochemical feature—a high affinity for the glycosaminoglycan heparin (Baeg and Perrimon 2000; Weaver et al. 2000). The ability to bind heparin is thought to signify the capacity of a secreted molecule to be deposited in the extracellular matrix or on the cell surface, most likely through binding to heparan sulfate proteoglycans (HSPGs; Esko and Lindahl 2001). Biochemical and cell culture assays suggest that HSPGs serve as extracellular depots for secreted signaling molecules, can shape the distribution of these signaling molecules, and act as coreceptors for ligand-specific receptor tyrosine kinases (RTKs; Vlodavsky et al. 1987; Yayon et al. 1991; Rapraeger 2000). The importance of HSPGs in modulating intercellular communication in vivo was demonstrated with the identification of mutations that impair heparan sulfate synthesis and phenocopy the defects observed when the signaling ligands or their receptors are lost (Perrimon and Bernfield 2000). Accordingly, the mutation of enzymes involved in HSPG synthesis impairs branching in the Drosophila tracheal system and the mouse kidney (Bullock et al. 1998; Lin et al. 1999).
The secreted signaling protein vascular endothelial growth factor (VEGF-A) has several properties that make it a likely stimulatory patterning cue for vascular branching. VEGF-A is the most potent and specific vascular growth factor, and its expression heralds the growth of new blood vessels during both physiological and pathological angiogenesis (Ferrara and Alitalo 1999). VEGF-A levels are exquisitely regulated through transcriptional control and mRNA stability (Neufeld et al. 1999). Moreover, VEGF-A is made as several isoforms that are produced by alternative splicing from a single gene and whose relative abundance varies between different tissues (Fig. 1A; Tischer et al. 1991; Shima et al. 1996; Ng et al. 2001). These isoforms differ by the absence or presence of domains that confer the ability to bind heparin in vitro and heparan sulfate proteoglycans in the extracellular matrix of cultured cells, and they therefore are predicted to distribute differentially in the environment of a VEGF-secreting cell (Houck et al. 1992). In agreement with this idea, VEGF121 (termed VEGF120 in the mouse) is soluble, whereas VEGF189 (VEGF188 in the mouse) is found only on the cell surface or bound to the extracellular matrix, and VEGF 165 (VEGF164 in the mouse), which lacks one of the two heparin-binding domains present in VEGF189, has intermediate properties (Park et al. 1993). It is generally postulated that the differential localization of these isoforms might result in a distinct influence on the behavior of the responding endothelium, although this has not been demonstrated in vivo. In addition to their differential localization in the extracellular space, VEGF-A isoforms may elicit distinct responses in target cells through their binding to isoform-specific receptors. Most notably, the axon guidance molecule neuropilin-1 (Nrp-1) has been identified as a coreceptor for VEGF165 but not for VEGF121 (Soker et al. 1998). When coexpressed with the signal transducing VEGF-A receptor Flk1, Nrp-1 amplifies VEGF-A induced endothelial cell motility in culture models (Soker et al. 1998), and mice lacking Nrp-1 display severe cardiovascular defects (Kawasaki et al. 1999). These findings led to the suggestion that Nrp-1 might execute VEGF165-specific functions in responding endothelial cells (Miao and Klagsbrun 2000; Shima and Mailhos 2000).
Figure 1.

Loss of heparin-binding VEGF-A reduces vascular branching complexity during embryogenesis. (A) VEGF-A isoforms in the mouse; the isoforms differ by the absence or presence of heparin-binding domains encoded by exons 6 and 7 (highlighted in yellow); the VEGF120 isoform (asterisk) is the only isoform expressed when exons 6 and 7 are ablated. (B) RT-PCR of cDNA derived from 12.5-dpc hindbrains (wt/wt, wt/120, and 120/120) using oligonucleotides specific for VEGF-A or GAPDH; cloned cDNAs (pBS-120, pBS-164, and pBS-188) were used as controls. (C,D) Whole-mount immunohistochemistry of wt/wt (C) and 120/120 (D) littermate embryos at 11.25 dpc using an anti-PECAM mAb shows that loss of heparin-binding VEGF-A does not ablate vascularization during embryogenesis. (E–H) Vascular networks in the limb at 12.5 dpc (E,F) and 13.5 dpc (G,H). (I–L) Vascular networks in the hindbrain at 11.25 dpc (I,J) and in the posterior lateral hindbrain at 13.5 dpc (K,L). (M–P) Vascular networks in the stomach at 11.25 dpc (M,N) and 12.5 dpc (O,P). Blood vessels in stage matched wt/wt (C,E,G,I,K,M,O) and 120/120 (D,F,H,J,L,N,P) littermates were visualized by immunohistochemistry using an antibody to PECAM (C–F,I–P) or a Tie2LacZ reporter (G,H). Examples of vessel abnormalities are highlighted. Stretches devoid of branchpoints are labeled with black arrowheads; regions severely impaired in branching are indicated with Δ; vessel branches with coiling ends are labeled with black arrows; and a large vessel tuft is with a star. Bars: C,D, 1 mm; E–H, 500 μm; I,J, 500 μm; K,L, 100 μm; M,N, 100 μm; O,P, 200 μm.
The analysis of null or conditional alleles for VEGF-A and its cognate RTKs, Flk1 and Flt1, has demonstrated that VEGF-A is required for the formation, remodeling, and survival of embryonic blood vessels (Ferrara et al. 1996; Carmeliet et al. 1999; Gerber et al. 1999). However, the strict dose-dependent requirement for VEGF-A during multiple stages of blood vessel development has prevented a detailed analysis of its precise functions during vascular network formation. We have focused our analysis of vascular development on mouse embryos genetically engineered to express solely the soluble VEGF-A isoform, VEGF120. In contrast to mice with reduced overall VEGF-A levels, VEGF120/120 mice develop to term but suffer from postnatal angiogenesis defects in several organs (Carmeliet et al. 1999; Stalmans and Ng 2002). We now show that these mutants undergo grossly normal embryonic vascular development, with the exception that they exhibit a subtle, widespread, and specific vessel patterning defect that results in the creation of capillary networks with fewer branch points and a larger luminal diameter. Importantly, the vascular defects in these mutants were distinct from those of embryos lacking solely VEGF164 or Nrp-1, suggesting that mechanisms other than VEGF164-dependent signaling via Nrp-1 must influence vascular branching morphogenesis. Accordingly, the detailed analysis of vascular branching patterns, endothelial cell behavior, and VEGF-A protein distribution in wild-type embryos and mutants producing solely VEGF120 or VEGF188 has revealed that the heparin-binding VEGF-A isoforms serve as spatially restricted stimulatory cues that elicit stereotypical branching behavior at the leading edge of the growing microvessel network, in analogy to the role of localized FGF sources in other branching organs.
Results
Heparin-binding VEGF-A is not essential for vasculogenesis or angiogenesis
To address if heparin-binding VEGF-A isoforms play a role in vascular pattern formation, we examined blood vessel development in mouse embryos that express VEGF120, but not VEGF164 or VEGF188, owing to the removal of exons 6 and 7 of the VEGF-A gene (Fig. 1A; Carmeliet et al. 1999). RT-PCR analysis of the relative ratios of the VEGF-A isoforms in whole embryos and dissected hindbrains at 12.5 days postcoitum (dpc; Fig. 1B; data not shown) demonstrated that VEGF120 and VEGF164 transcripts normally predominated over the barely detectable VEGF188 transcripts (see Ng et al. 2001). We also saw abundant expression of an exon 6-containing isoform that had not been described previously in the mouse and corresponded to human VEGF145 (termed VEGF144; Fig. 1B; Poltorak et al. 1997). As expected, VEGF120/120 embryos produced only VEGF120 mRNA (Fig. 1B). Interestingly, the targeting of just one VEGF-A allele (VEGFwt/120 animals) resulted in a reversal of the relative ratio of VEGF120 to VEGF164 mRNA, with VEGF120 now predominating. The relative increase in VEGF120 production in gene-targeted embryos compared to wild-type littermates was consistent with previous studies, which demonstrated that total VEGF-A mRNA levels (i.e., the sum of all isoforms) are similar in wild-type and VEGF120/120 newborns (Carmeliet et al. 1999).
In contrast to the severe cardiovascular defects of mouse embryos that produce 50% reduced or twofold elevated amounts of VEGF-A, the lack of heparin-binding VEGF-A isoforms did not cause gross malformations of the major vessels or the heart during early embryonic development (Carmeliet et al. 1996; Miquerol et al. 2000; data not shown). Moreover, all embryonic organs were abundantly vascularized in mutants, with no tissue damage or hemorrhage up to at least 15.5 dpc (Fig. 1C–P; data not shown). These results suggested that VEGF120 was sufficient to support both vasculogenesis and angiogenesis at levels that can sustain embryonic development. Importantly, neither homozygotes (VEGF120/120) nor heterozygotes (VEGFwt/120) were delayed in their development, as assessed by the normal progression of facial, somite, and motor neuron differentiation (Fig. 1, cf. C and D; data not shown).
Reduced vascular branching complexity and increased microvessel caliber in the absence of heparin-binding VEGF-A
The loss of heparin-binding VEGF-A caused a striking reduction in vascular branching complexity at the earliest stages of angiogenic invasion in several organs and resulted in the formation of microvessel networks with an unusually large mesh-size (Figs. 1, 2). For example, the developing limbs contained an extensive network of microvessels, which appeared to elongate as the digits extended, but formed fewer lateral branches (Fig. 1E–H). The timely development of the limb itself and the presence of microvessels throughout again indicated that the vascular branching defect was not caused by a general failure of organogenesis or delayed angiogenesis but reflected an abnormality of vascular patterning.
Figure 2.

The balanced expression of heparin-binding VEGF-A versus VEGF120 controls microvessel branching and vessel caliber. (A) Schematic representation of hindbrain vascularization between 10.0 (1) and 10.5 (4) dpc; between 9.5 and 10.0 dpc, the perineural vascular plexus in the pial membrane begins to extend sprouts into the neural tube (1), which grow perpendicularly toward the ventricular zone (2), where they branch out to form the subventricular vascular plexus (3,4). (B,C) Microvessel appearance on the pial and ventricular sides of a flat-mounted 12.5-dpc hindbrain; the midline region is indicated with an asterisk; the pial side of the hindbrain with P, the ventricular side with V. (D–F) Visualization of vascular networks in representative 500-μm2 areas of the 13.5-dpc midbrain of wt/wt (D), wt/120 (E), and 120/120 (F) littermates; blood vessels were visualized with a Tie2LacZ reporter. (G) Number of microvessel branch points per 500-μm2 area in the subventricular zone of the embryonic central nervous system in wt/wt (black), wt/120 (red), and 120/120 (green) littermates; n = 3 for each specimen; 10.75-dpc hindbrain (1); 11.25-dpc hindbrain (2); 13.5-dpc hindbrain (3); 11.25 spinal cord (4); and 13.5-dpc midbrain (5). (H,I) Ultrastructure of microvessels in wt/wt (H) and 120/120 (I) hindbrains of littermates at 10.75 dpc, visualized by transmission electron microscopy (TEM); endothelial nuclei are indicated with red stars. (J) Number of endothelial cell nuclei per capillary cross section in the hindbrain in wt/wt (n = 96) and 120/120 (n = 106) littermates at 10.75 dpc (P ≤ 0.0001, Mann-Whitney test). (K,L) Endothelial cell proliferation in hindbrain vessel cross sections of wt/wt (K) and 120/120 (L) littermates at 11.25 dpc; PECAM-positive vessels are shown in green, BrdU-positive endothelial nuclei (white stars) in red. (M) HUVEC proliferation assay; VEGF-A (VEGF121 or VEGF165) concentration: 0.5 ng/mL (1) , 2 ng/mL (2) , 10 ng/mL (3) , 20 ng/mL (4); and basic fibroblast growth factor concentration 3 ng/mL (5). Bars: B,C, 500 μm; D–F, 100 μm; K,L, 5 μm. Magnification: H,I, 5500×.
Vessel branch points were more widely spaced throughout all other mutant tissues examined, including brain, gut, and somites (Figs. 1I–P, 7A,B, see below). Moreover, in the posterior hindbrain and spinal cord, we often observed large tuft-like structures or coiling ends, suggesting that vessel endothelium grew efficiently but had lost directionality during its extension (Fig. 1K,L). Similar results were obtained when blood vessels were visualized by use of an anti-PECAM (platelet-endothelial cell adhesion molecule) antibody, isolectin B4, or a Tie2LacZ reporter, and also in a number of different genetic backgrounds with 100% penetrance (Figs. 1, 2; data not shown).
The embryonic central nervous system is vascularized by angiogenic sprouting in a highly stereotypical fashion (Bär 1983) and produces a vascular plexus that can be readily examined for vascular patterning defects (Fig. 2A–C). Between 10.5 and 13.5 dpc, a vascular network forms in the subventricular zone that can be visualized as a two-dimensional array when the roofplate is opened and the neural tube is flat-mounted (Fig. 2C). To directly compare the branching complexity of the developing subventricular plexus in the presence and absence of heparin-binding VEGF-A, we counted the number of microvessel branch points in random 500-μm2 areas of midbrains, hindbrains, or spinal cords. We found that branching density was decreased in the absence of heparin-binding VEGF-A in all regions of the central nervous system, with a reduction of 15% to 40% in heterozygotes and 60% to 70% in homozygotes (Fig. 2G). As development proceeded, new branches were added into the existing subventricular network, but the branching deficit failed to recover in gene-targeted embryos (Fig. 2G, cf. experiments 1–3). The intermediate decrease in branching complexity of the subventricular plexus in heterozygotes, compared with wild-type and mutant littermates (Fig. 2, cf. D–F), suggested that vascular patterning was exquisitely controlled by the ratio of soluble to heparin-binding VEGF-A (see above, Fig. 1B).
The reduction in vascular branching complexity was accompanied by a uniform increase in microvessel caliber (Fig. 1, cf. H and L), which was already noticeable in heterozygotes (Fig. 2, cf. D and E,F). Quantitative analysis of transmission electron micrographs confirmed that the larger vessel size was caused by an increase in the number of endothelial cells per microvessel cross-section rather than vessel dilation (Fig. 2J) and illustrated the structure of the abnormal vessel tufts observed in the mutants (Fig. 2, cf. H and I; see above, Fig. 1H).
The presence of excess endothelial cells in mutant vessels suggested that the decrease in vascular branching was unlikely caused by differences in the ability of the various VEGF-A isoforms to support endothelial proliferation. Consistent with this idea, VEGF121 and VEGF165 promoted endothelial cell proliferation in vitro in a dose-dependent manner with comparable efficiency (Fig. 2M). Furthermore, when we compared the number of BrdU-labeled endothelial cell nuclei in branching networks from normal hindbrains and those that contain VEGF120 only, we found that both mutant and wild-type littermates produced a similar number of proliferating endothelial cells (mutant proliferation was 94 ± 4% of wild-type index, n = 3; ∼50 BrdU/PECAM double-positive cells per millimeter squared were found in the subventricular plexus, and 25 double-positive cells per millimeter squared in vessels on the pial face). Furthermore, as mutant hindbrains contained fewer vessel branches, the number of BrdU-labeled endothelial cells per vessel cross section increased, confirming that there was a tendency for nascent cells to be incorporated into existing vessels (Fig. 2, cf. K and L).
The phenotype of embryos expressing VEGF120 only is distinct from that of embryos lacking either PDGF-B or PDFGR-β (Hellstrom et al. 2001), in which central nervous system vessels are larger at 11.5 dpc, but appear normal at 10.5 dpc. Here, the increase in vessel caliber is due to endothelial hyperproliferation, likely caused by the absence of an inhibitory signal following the lack of pericyte recruitment. Moreover, unlike the vessels of embryos lacking heparin-binding VEGF-A, those of PDGF-B and PDFGR-β knockouts are not impaired in branching (Hellstrom et al. 2001; C. Ruhrberg and D.T. Shima, unpubl.).
In summary, our results establish that (1) VEGF120 and VEGF164 have a similar proliferative signaling capacity both in vitro and in vivo, and (2) the vascular patterning defects of mice expressing solely soluble VEGF-A are due to an altered distribution of endothelial cells rather than to their impaired proliferation. We conclude that VEGF120 fully supports endothelial growth in vivo, but it promotes the incorporation of new endothelial cells into existing capillaries, rather than into new branches, in the absence of heparin-binding VEGF-A.
Altered distribution of VEGF-A protein in the absence of its heparin-binding isoforms
Mechanistically, the formation of a new branch point requires the selection of a bud site and the directed extension of the growing bud toward the target. For example, in the developing lung, the distal mesenchyme expresses Fgf10 to allure growing lung buds, whereas Drosophila airways branch when small clusters of cells around the tracheal sacs secrete branchless FGF to attract migrating tracheal cells (Sutherland et al. 1996; Weaver et al. 2000). As heparin-binding VEGF-A isoforms have the potential (1) to become localized in the extracellular matrix of cultured cells by binding to HSPGs and (2) to stimulate the motility and proliferation of endothelial cells in vitro, it is conceivable that extracellular matrix-bound VEGF-A deposits could induce and guide the extension of new branches. We therefore asked if secreted VEGF-A was abnormally localized in VEGF120/120 embryos, which completely lack heparin-binding VEGF-A isoforms.
At 10.5 dpc, the VEGF-A gene was expressed at high levels near the hindbrain midline, presumably to attract new branches from the laterally expanding subventricular plexus and the perpendicularly invading perineural plexus (Fig. 3A,B; see also Fig. 2B). To visualize the distribution of the secreted VEGF-A protein pool, we immunostained nonpermeabilized hindbrains with an antiserum that recognizes all VEGF-A isoforms (see Materials and Methods). Immunostaining revealed that secreted VEGF-A protein accumulated in patches surrounding the sites of its production, forming a steep concentration gradient that tapered off toward the front of the growing subventricular plexus (Fig. 3B,C). In the absence of heparin-binding isoforms, VEGF-A protein no longer formed a steep concentration gradient but instead appeared more widely dispersed (Fig. 3, cf. C and D, E and F; see also 11.25-dpc appearance in Fig. 6A,B, below). These data demonstrate a distinct difference in the localization of heparin-binding VEGF-A isoforms compared with VEGF120.
Figure 3.

Altered distribution of secreted VEGF-A protein in the absence of heparin-binding isoforms. (A) VEGF-A gene expression near the hindbrain midline at 10.5 dpc, monitored with a VEGF-A lacZ reporter. (B) At 10.5 dpc, extracellular VEGF-A protein (red) concentrates near the midline, and the PECAM-positive microvessel network (green) is closing in on the VEGF-positive area. (C,D) Comparison of VEGF-A protein distribution in the midline region of wt/wt (C) and 120/120 (D) hindbrains at 10.5 dpc. (E,F) Pseudo-coloring of C,D to highlight regions of highest (red) and lowest (blue) staining intensity. The midline is labeled with an asterisk. Bar, 50 μm.
Figure 6.
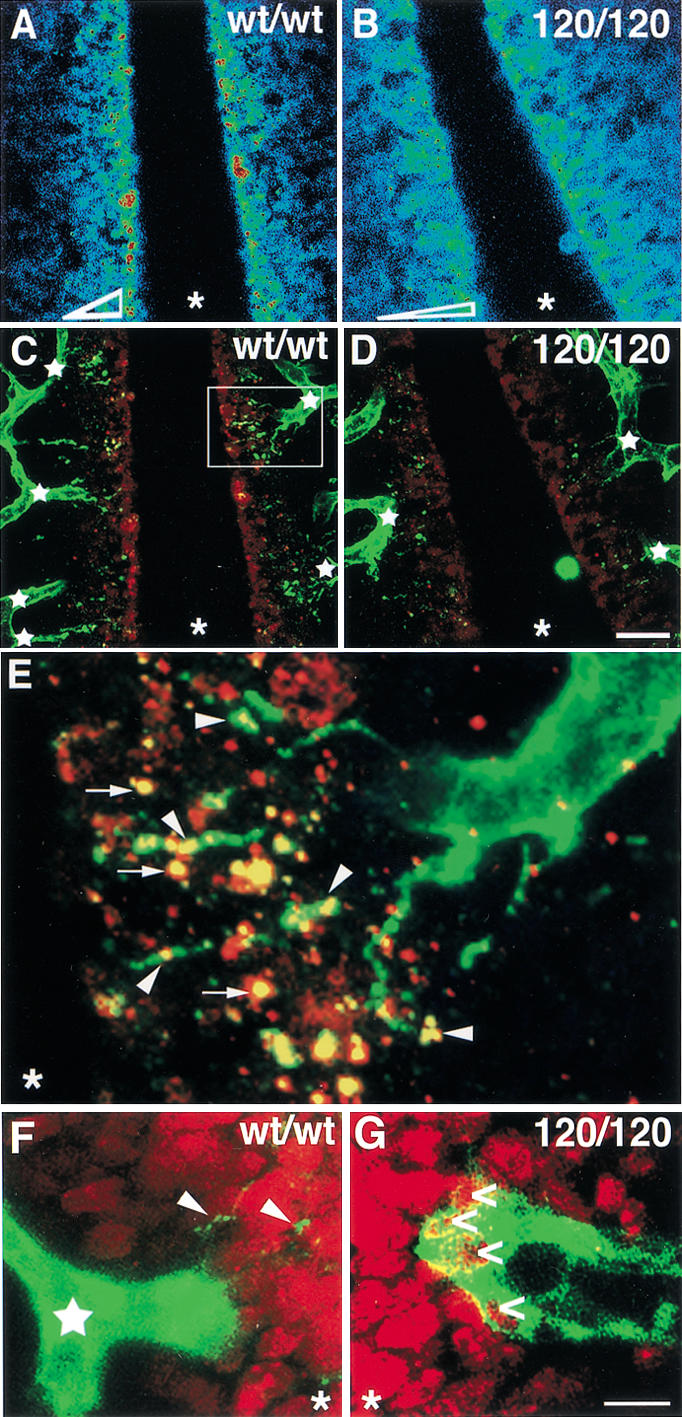
Filopodia extension toward VEGF-A deposits is impaired in the absence of heparin-binding isoforms. (A–D) Distribution of extracellular VEGF-A protein near the midline region of an 11.25-dpc wild type (A,C) and 120/120 (B,D) hindbrain. (A,B) Highest VEGF-A staining intensities are shown in red, lowest in blue; the shape of the VEGF-A gradient is shown at the bottom of the two panels. (C,D) PECAM-positive microvessels (green) extend many new branches (junctions labeled by white stars) toward the midline region in wt/wt (C), but not in 120/120 (D) hindbrains, in which, instead, the vessel network often terminates in closed loops. The microvessels closest to the midline extend multiple filopodia toward extracellular VEGF-A patches (red) in wt/wt hindbrains (C), but make only few filopodia in 120/120 hindbrains (D). (E) The area boxed in C, magnified 5×. A microvessel sprout extends filopodia (green) to contact VEGF-A patches (red); regions of overlap are indicated in yellow. (F,G) PECAM-positive vessel tips (green) near the midline region in wt/wt (F) and 120/120 (G) hindbrains; BrdU-positive (red) proliferating neuronal nuclei (in F,G) or double-positive endothelial nuclei (in G; open arrows). Some filopodia extending from the wild-type tip cell are indicated with arrowheads (F). The midline position in all panels is labeled with an asterisk. Bars: A–D, 20 μm; F,G, 100 μm.
Heparin-binding VEGF-A is required for the directed extension of filopodia from branching microvessel networks
To examine the consequences of an altered VEGF-A gradient on vessel pattern, we examined the morphology and behavior of brain endothelial cells at the leading edge of the laterally expanding subventricular plexus in the presence and absence of heparin-binding VEGF-A. Normally, endothelial cells at the tips of growing vessel branches extended numerous long filopodial processes toward the midline (Fig. 4A). Over time, as the distance between the advancing vessel front and the midline decreased, these filopodial processes shortened (Fig. 4, cf. A and C). The number of laterally extending filopodia was greatly diminished in the hindbrains of embryos lacking heparin-binding VEGF-A (Fig. 4, cf. A and B, C and D, F and H). Rather than polarizing toward the midline, most mutant vessels ended in large distended tips (Fig. 4B,D, stars) or terminated in loops (Fig. 4D; see also Fig. 6, below). Similar to the laterally expanding subventricular plexus, perpendicularly growing vessels normally extended many filopodia toward the midline region, and they appeared as dots on the ventricular side of the flat-mounted brain. Branching mutants extended fewer perpendicular filopodia, and they failed to correctly target the midline region (Fig. 4, cf. C and D, E and G).
Figure 4.

Impaired filopodia extension at the leading front of growing microvessel networks in the absence of heparin-binding VEGF-A. (A–H) PECAM-positive microvessels with laterally (arrowheads) and perpendicularly (arrows) extending filopodia in wt/wt (A,C,E,F) and 120/120 (B,D,G,H) hindbrains at 10.75 (A,B) and 11.25 (C–H) dpc; the midline is situated off the left side of the image in A and B and is indicated with a red line in C and D; examples of fusing microvessels are indicated with open arrowheads; examples of dilated or bulbous vessel ends in the 120/120 hindbrains are indicated with stars; examples of abnormally localized perpendicular filopodia in D are circled; and the corresponding region devoid of filopodia in C is indicated with a hash. (E,F) The lateral (E) and perpendicular (F) filopodial extensions in the areas boxed in C, magnified 2.5×. (G,H) The lateral (G) and perpendicular (H) filopodial extensions in the areas boxed in D, magnified 2.5×. Bars: A,B, 10 μm; C,D, 20 μm.
Filopodia extension and branching of microvessels is directed to extracellular VEGF-A deposits
To examine the likelihood of a general role for filopodial extensions in vessel branching, we turned our attention to the retina, which develops a vascular supply postnatally, and, similar to the embryonic hindbrain, can be flat-mounted to visualize the entire vascular network (Stone and Dreher 1987). Importantly, the expansion of the vascular network appears to follow the movement of VEGF-A secreting astrocytes, which leave the optic nerve head around the same time as endothelial cells but spread ahead of the vessels to form a dense meshwork (Fig. 5D–F; Chang-Ling and Stone 1992).
Figure 5.

Filopodia extension from retinal cells. (A) Endothelial cells at the front of the growing vascular network (green) extend numerous filopodia into their surrounds and toward each other (arrowheads). (B,C) Endothelial filopodia (green, A) are enriched in actin (red, B); regions of overlapping staining appear yellow (C). (D–H) Endothelial cells at the front of the growing vascular network (green, D) track GFAP-positive astrocytes (red, E); some regions in which endothelial filopodia follow astrocyte processes are indicated with arrowheads (D–F). (G,H) An area shown in D and F, in which endothelial filopodia (G) follow astrocyte processes (H), magnified 2×; an undulating filopodium that has not lined up with astrocyte processes is indicated with an arrow (G,H). (A–H) Isolectin B4-positive blood vessels are shown in green, GFAP-positive astrocytes in red; isolectin B4-positive microglial cells in D, F, G, and H are labeled with stars. (I) Endothelial cells at the front of the growing vascular network extend filopodia (arrowheads) toward VEGF-A-expressing astrocytes (asterisks); isolectin B4-positive blood vessels are shown in brown; VEGF-A mRNA expression is shown in purple. Bars: A–C, 8.5 μm; D–F, 20 μm; G,H, 10 μm; I, 25 μm.
As observed in the developing hindbrain, endothelial cells at the leading edge of the branching retinal vasculature extended many thin processes in the direction of vascular expansion (Fig. 5A,D,G). These filopodia were enriched in actin (Fig. 5B,C) and usually aligned with astrocyte processes to form parallel bundles (Fig. 5, cf. D and E, G and H, arrowheads). It seemed likely that VEGF-A directed filopodial behavior, because the tip cells of growing vessel branches extended filopodia toward single VEGF-A-expressing astrocytes as if to make contact (Fig. 5I).
A spatial relationship among secreted VEGF-A protein, filopodia, and branch formation was also apparent in the hindbrain (Fig. 6). By 11.25 dpc, microvessels at the front of the expanding subventricular plexus had normally initiated branch formation to vascularize the midline region (Fig. 6C, stars). The tips of these nascent branches were decorated with filopodia that extended toward the midline, where they contacted extracellular VEGF-A deposits (Fig. 6E). Concomitant with the failure to form a normal VEGF-A concentration gradient (Fig. 3, cf. D and E, Fig. 6, cf. A and B), microvessels in VEGF120/120 embryos initiated fewer new branch sites, and the vessel network appeared to terminate in closed loops, with reduced filopodial extension toward the midline (Fig. 6, cf. C and D). Moreover, the observation that VEGF-A in the mutants was more broadly distributed away from the midline region toward the established plexus (Fig. 6, cf. A,B and C,D) provides an explanation for the sporadic appearance of ectopic filopodia (circled in Fig. 4D).
Concomitant with the redistribution of VEGF-A protein in the mutant hindbrains, we observed a reorganization of the proliferation pattern within the subventricular plexus. Endothelial proliferation in the subventricular zone was normally restricted to the established plexus, and the polarized tip cells lacked BrdU-positive nuclei (Fig. 6F; data not shown). In contrast, the tips of mutant vessels were usually enlarged and contained several positive nuclei (Fig. 6G). Taken together, the redistribution of secreted VEGF-A toward the growing vessel front explained why existing vessels did not branch appropriately but expanded in situ: Rather than polarizing and extending toward a spatially restricted growth stimulus, vessel tips were triggered to proliferate by the high levels of VEGF-A signal now present in their existing location.
Abnormal vessel branching in embryos that solely express heparin-binding VEGF-A
To further test the hypothesis that the balance of heparin-binding versus soluble VEGF-A controls vascular patterning, we examined mice that express solely VEGF188, the isoform that is completely sequestered on secretion on the cell surface and within the ECM of the producing cell (Park et al. 1993). These mice were created by replacing the genomic region encoding alternatively spliced exons 6 and 7 of the VEGF-A gene with a cDNA fragment of VEGF188 to direct all splicing toward production of the VEGF188 mRNA (Stalmans and Ng 2002). Analysis of postnatal retinal vascularization in these animals had previously uncovered gross vessel patterning defects, including impaired arterial vessel development, which likely occurs as a result of defective hyaloid remodeling (Stalmans and Ng 2002).
Analysis of branching morphogenesis in the neural tube revealed that rather than promoting an increase in vessel lumen size as observed in VEGF120/120 mice, VEGF188 induced the formation of ectopic and abnormally thin vessel branches during brain and retina vascularization (Fig. 7; data not shown). This defect was best visualized on the pial side of the neural tube, where excess numbers of microvessels invaded the area adjacent to the hindbrain midline, and many microvessel sprouts that had extended from the perineural plexus into the hindbrain branched prematurely, that is, before they had reached the subventricular zone (Fig. 7, cf. C and D; see also Fig. 2A–C).
Figure 7.

VEGF-A isoform and Nrp-1 mutants exhibit distinct embryonic vessel defects. VEGF188 promotes ectopic filopodia extension (A,B) and excess branching (C,D) during brain vascularization. Blood vessels in the left wt/wt (A,C) or right 188/188 (B,D) hindbrain are shown at 10.75 (A,B) and 13.5 (C,D) dpc. The direction of filopodia extension is indicated with arrows in A,B. Clusters of perpendicularly extending filopodia near the midline are labeled with stars in B. Vessels near the midline (indicated with an asterisk) are labeled with black arrowheads in C and D; examples of ectopic branching with single arrows in D; and long thin vessel stretches with a double arrow in D. (E,F) Ectopic expression of the VEGF188 isoform (E) rescues vascular defects in hindbrains expressing VEGF120, but lacking VEGF164 (F). (G,H) Vascular networks covering the somites of littermate embryos producing (G) or lacking (H) heparin-binding VEGF-A at 10.5 dpc; an exemplary vessel stretch devoid of branch points is labeled with black arrowheads. (I,J) Vascular networks covering the somites of littermate embryos producing (I) or lacking (J) Nrp-1 at 10.5 dpc. (K) Working model for the role of VEGF-A isoforms in vascular patterning. The balanced production of soluble VEGF120 versus heparin-binding VEGF-A (HB-VEGF) determines the shape of the VEGF-A gradient around producing cells and thereby controls the decision of nearby vessels to invest growth into new branches (right) or luminal caliber (left). Accordingly, vessel size and branching pattern are dramatically different in the 120/120 and 188/188 midbrain at 13.5 dpc. Blood vessels were visualized by immunohistochemistry using an antibody to PECAM. Bars: A,B, 10 μm; C,D, 200 μm; E,F, 100 μm; G,H, 200 μm; I,J, 200 μm; K, 25 μm.
Vessels in hindbrains expressing solely VEGF188 extended numerous filopodia, but they were organized abnormally. Although wild-type embryos had directed only few perpendicular filopodia into the midline region by 10.5 dpc (see above; Fig. 4A), littermates producing only heparin-binding VEGF-A accumulated many more filopodia in close proximity to the midline; here, they prefigured the sites at which ectopic microvessels were to be found at 13.5 dpc (Fig. 7, cf. B and D). Moreover, many vessel sprouts extended excess filopodia in multiple directions, for example, both toward the midline and away from it toward existing vessels (Fig. 7B). Thus, it appears that loss of heparin-binding or soluble VEGF-A results in opposing vascular phenotypes, in which vessel size and branching frequency were inversely linked and branching defects were preceded by altered filopodia behavior.
Vascular branching defects in VEGF-A isoform-specific embryos cannot be explained by loss of VEGF164/Nrp-1 signaling
Although mice producing solely either soluble (VEGF120/120) or heparin-binding (VEGF188/188) VEGF-A exhibit vascular defects and severely reduced survival (Carmeliet et al. 1999; Stalmans and Ng 2002), VEGF120/188 double heterozygotes are viable with no overt vascular branching abnormalities (Fig. 7E,F; data not shown). This observation suggests that the combinatorial expression of soluble VEGF120 with a heparin-binding VEGF-A isoform (i.e., VEGF188) is sufficient to drive the formation of a normal branching network. More importantly, as these double heterozygotes specifically lack VEGF164, it seems unlikely that the observed branching defects in VEGF120/120 or VEGF188/188 mice were related to the loss of VEGF164-dependent signaling via the neuropilin-1 (Nrp-1) receptor (Miao and Klagsbrun 2000). To further exclude this possibility, we examined Nrp-1-deficient mouse embryos (Nrp-1−/−) for vascular patterning defects (Kawasaki et al. 1999). Because Nrp-1−/−embryos are severely impaired in the vascularization of the neural tube and do not form a subventricular vascular plexus (Kawasaki et al. 1999; data not shown), we compared their somite vessels to those of VEGF120/120 mutants but found little similarity in the observed vascular pattern (Fig. 7G–J). Whereas many vessels extended throughout the length of somites without producing lateral branches when heparin-binding VEGF-A was lacking, somite vessels branched frequently in the absence of Nrp-1 (Fig. 7, cf. H and J). Thus, it appears that Nrp-1 fulfills essential functions during the vascularization of specific tissues, but that loss of VEGF164 signaling through the Nrp-1 receptor is not responsible for the widespread vascular branching defects in VEGF-A isoform-specific mice.
We conclude that heparin-binding VEGF-A isoforms provide spatially restricted cues for vascular pattern formation, and we suggest that the choice to increase branching complexity versus luminal diameter during angiogenic growth is controlled by the balance of heparin-binding versus soluble VEGF-A isoforms (Fig. 7K).
Discussion
In this study we have shown that altering the relative ratio of VEGF-A isoforms directly and discretely shapes vessel branching morphogenesis during angiogenesis in vivo. Microvessels from mouse embryos expressing decreased levels of heparin-binding VEGF-A isoforms (VEGFwt/120), or solely the non-heparin-binding form VEGF120 (VEGF120/120), exhibit a dose-dependent decrease in branch formation compared with those of wild-type embryos. In contrast, animals expressing solely the heparin-binding VEGF188 isoform (VEGF188/188) exhibit an opposing phenotype, with ectopic branching structures in the form of long and thin microvessels. Intriguingly, these branching and vessel morphogenesis defects were rescued when embryos were engineered to express both the most diffusible and the most avid heparin-binding isoform (VEGF120/188) in the absence of VEGF164.
The cellular basis of branching defects in animals expressing solely VEGF120 is not caused by significant differences in the number of dividing endothelial cells during angiogenic invasion, strongly suggesting that the VEGF-A isoforms have similar capacities to drive endothelial cell proliferation in vivo. Furthermore, analyses of embryos deficient in Nrp-1 or solely the VEGF164 isoform provide evidence that the observed branching defects could not be attributed to VEGF164-specific signaling through the Nrp-1 receptor. Because embryos expressing either no VEGF164 (VEGF120/188; this study) or solely VEGF164 (VEGF164/164; C. Ruhrberg and D.T. Shima, unpubl.) exhibit normal vessel networks, we also suggest that isoform-specific signaling of any type is unlikely to be an absolute requirement to specify vascular branching patterns.
Instead, our analysis of VEGF-A protein distribution in relation to the branching endothelial network strongly supports the idea that the branching deficits in animals lacking heparin-binding VEGF-A are caused by an insufficiency of VEGF120 to interact with the extracellular matrix on secretion. We show that in the neural tube, secreted VEGF-A (predominantly VEGF164; see Fig. 1) normally establishes a steep concentration gradient proximal to the midline and attracts the tips of filopodia that emanate from vessels nearby. However, in VEGF120/120 animals, secreted VEGF120 assumes a shallow concentration gradient, with a shifted distribution that reaches the front of the branching plexus, where it reduces the polarity of tip cells and increases their proliferation rate. We conclude that the coordinate expression of VEGF-A isoforms shapes the spatial distribution of VEGF-A protein within tissues and thereby provides a mechanism for eliciting the discrete types of endothelial cell behavior required for the formation of branching networks during embryonic development.
Localizing secreted signals during vascular morphogenesis
Developmental studies have underscored that both the localized expression of intercellular signaling molecules and their spatially restricted distribution in the extracellular matrix regulate pattern formation and tissue morphogenesis in several model systems, including the vertebrate lung and the tracheal system of Drosophila (Lawrence and Struhl 1996; Teleman et al. 2001). Similarly, our study suggests that the spatial restriction of the action of VEGF-A is a critical requirement during vascular pattern formation and relies on its precise deposition in the extracellular space. Studies on the founding member of the heparin-binding growth factor family, basic fibroblast growth factor (bFGF), provided the first indication that heparin-binding activity equated with the ability for storage of intercellular signaling molecules in the ECM following their secretion (Vlodavsky et al. 1987). Besides VEGF-A and bFGF, many other intercellular signaling molecules with vascular effects, in particular other members of the VEGF and PDGF families, are now known to be alternatively spliced or proteolytically processed to regulate their ability to interact with proteoglycans and/or the ECM (Eriksson and Alitalo 1999; Betsholtz et al. 2001). Although molecular details about these growth factor-ECM interactions are largely lacking, the glycosylphosphatidylinositol-anchored cell surface HSPG glypican-1 is an interesting candidate, because it associates with secreted VEGF165, but not VEGF121 in vitro (Gengrinovitch et al. 1999).
The release of VEGF-A from HSPGs through the activity of proteolytic enzymes such as matrix metalloproteinases (MMPs) or heparinases could represent an important means of regulating ligand activation during vascular morphogenesis (Houck et al. 1992). In support of this notion, it is thought the angiogenic switch in models of stepwise carcinogenesis is provided by the up-regulation of MMP-9 activity and the subsequent release of active VEGF-A from a dormant state within the tumor extracellular matrix (Bergers et al. 2000). However, the examination of embryos deficient in either MMP-2 or MMP-9, two proteases implicated in angiogenesis and VEGF-A biology, does not support a role for either protein in the establishment of vascular branching patterns (C. Ruhrberg, D.T. Shima, N. Ortega, Z. Werb, and S. Itohara, unpubl.).
In analogy to the need for complex gene regulatory mechanisms during branching within the lung and trachea, it is likely that regulating the localized expression of the VEGF-A gene plays an important part in determining blood vessel branching patterns. Hypoxia is a key factor controlling VEGF-A expression during postnatal neovascularization (Dor et al. 2001), yet its contribution to VEGF-A gene regulation and vascularization in the embryo is still a matter of debate, partly because of the difficulties in manipulating and measuring local oxygen concentrations in utero. However, focal oxygen deficits at the avascular front of a growing vessel network might trigger localized VEGF-A expression to stimulate new branch formation from existing vessels, in much the same way as hypoxia is thought to induce branchless expression to direct the extension of terminal branches of tracheal airways in the Drosophila embryo (Jarecki et al. 1999). The arrival of a new capillary would increase the local oxygen concentration and subsequently lead to a down-regulation of VEGF-A expression. The reiteration of this VEGF-A gene induction and repression cycle could regulate expansion of the branching vasculature to coordinate vascular network density with the metabolic needs of growing embryonic organs.
In addition to the control of VEGF-A mRNA abundance, VEGF-A splicing is regulated in a complex manner during embryogenesis to yield stage- and tissue-specific ratios of the various isoforms (Ng et al. 2001). Even subtle alterations in the VEGF isoform ratio during embryonic development are likely to have a significant biological outcome, because a reversal of the relative ratios of VEGF120 to VEGF164 (VEGF120/wt mice; the present study) or the increase of VEGF188 at the expense of VEGF164 (VEGF188/wt; C. Ruhrberg and D.T. Shima, unpubl.) both cause alterations in vascular branching pattern. These observations suggest that splicing of the VEGF-A gene could represent a major control point for decisions about vascular pattern formation, and they underscore the importance of understanding this aspect of VEGF-A gene regulation.
Finally, our identification of VEGF-A as a localized stimulatory cue for vascular branching, and the availability of mouse mutants specifically deficient in the branching process, provides a basis to identify additional components involved in vascular branching morphogenesis. In particular, it will be important to look for negative regulators, which, like Bmp-4 in the mammalian lung or sprouty in the Drosophila trachea, restrict branch initiation and elongation to selected sites along a tubule (Placzek and Skaer 1999). Interestingly, overexpression of mammalian sprouty homologs inhibits endothelial cell proliferation in vitro and, in the mouse embryo, leads to defective vessel formation; it will be interesting to determine if this latter defect is related to branch formation (Impagnatiello et al. 2001; Lee et al. 2001).
Cellular responses to branching cues
Little is known about the cellular events that accompany branching. Insight into the endothelial response to VEGF-A signals during branching is likely to come from an analysis of the mechanisms and consequences of filopodia extension, because their induction represents the earliest observed behavioral change associated with branch formation. Given the role of filopodial protrusions in other model systems of development, it would be plausible to implicate them as sensors of positional information, which is provided in the form of localized VEGF-A deposits. For example, a sensor role has been proposed for the filopodia that are extended by Drosophila imaginal disc cells toward morphogens in the disc organizing center, by sea urchin primary mesenchymal cells toward signals in the overlying ectoderm, and by mammalian neuronal growth cones and muscle cells toward each other during synapse formation (Ramirez-Weber and Kornberg 2000). Detection of extracellular VEGF-A deposits by filopodia and transduction of this positional signal into a branching response—which includes endothelial cell polarization, elongation, and migration—presumably involves VEGF-A cell surface receptors, such as the VEGF receptor tyrosine kinases KDR/Flk-1 and Flt-1. More speculatively, our observations on the intimate association of extracellular VEGF-A deposits and filopodia raise the possibility that filopodial protrusions could facilitate the uptake and/or degradation of extracellular VEGF-A to down-regulate the growth stimulatory signal once a new branch has reached its target. To further understand the role of filopodia and the precise cellular behaviors required for branching, we are currently developing an organ culture model of microvessel branching growth, in which endothelial cells at the tips of branches extend filopodia toward, and guide branching growth to VEGF-expressing cells (M. Golding, C. Ruhrberg, and D.T. Shima, unpubl.).
Implications for developmental, pathological, and therapeutical vascular morphogenesis
In this study we have identified microvessel size and branching frequency as morphogenetic features that are influenced differentially by heparin-binding and soluble VEGF-A isoforms during organogenesis. Several observations from previous work on the VEGF-A isoforms, both during developmental angiogenesis and pathological neovascularization in the adult, are consistent with our findings and, taken together, they begin to explain how VEGF-A exerts some of its effects during vascular development (see model, Fig. 7K). For example, we would predict that VEGF120 plays a major role as a long-range attractant used to initiate the early stages of vessel pattern, and that the heparin-binding forms of VEGF-A are more critical during the establishment of an intricate vessel architecture within target organs. In support of this idea, the relative levels of VEGF120 progressively decrease as embryonic development advances, with a concomitant increase of heparin-binding VEGF-A (Ng et al. 2001). Studies on vasculogenesis in Xenopus support an early role for VEGF120 in establishing the gross vascular pattern, in which the VEGF120 ortholog acts as a locally produced, but diffusible, long-range chemoattractant for lateral mesoderm angioblasts that contribute to dorsal aorta formation (Cleaver and Krieg 1998). Similarly, VEGF120 is capable of initiating the formation of the large axial vessels and the heart during mouse development (this study). However, during later stages of vascular development, when the fine patterning and positioning of vessels becomes critical for proper organogenesis, VEGF120 alone is insufficient: VEGF120/120 mice exhibit discrete defects in the postnatal assembly of the myocardial vasculature, the kidney glomerulus, the lung epithelial-vascular interface, and the retinal vessel array (Carmeliet et al. 1999; Ng et al. 2001; Mattot et al. 2002; Stalmans and Ng 2002).
Might the VEGF-A isoforms also play a role during the growth and patterning of vessels associated with pathological situations? Interestingly, vessels within tumors that express single VEGF-A isoforms share some morphological features with those in our single isoform-expressing embryos (Grunstein et al. 2000). Moreover, high level ectopic expression of VEGF-A in adult mice leads to the formation of disorganized networks, sinusoidal vessels, and convoluted tuft-like structures (akin to VEGF120/120 branching abnormalities; see Figs. 2H, 3I), rather than a hierarchical vascular tree (Carmeliet 2000; Lee et al. 2000; Sundberg et al. 2001), most likely because a saturating growth signal is provided in the absence of proper positional information. It therefore seems likely that secreted angiogenic growth factors, such as the VEGF-A isoforms, do not just stimulate new vessel formation but also direct vascular pattern formation during neoangiogenesis. If true, proangiogenic therapies, aimed at creating new vessel networks (e.g., to rescue chronic ischemia), might benefit from the manipulation of VEGF-A isoform expression and the presentation of VEGF-A in a more natural extracellular context to provide appropriate patterning cues.
Materials and methods
Animals
Mice containing a deletion of exons 6 and 7 of the VEGF-A gene (Carmeliet et al. 1999) or producing VEGF188 as the only VEGF-A isoform (Stalmans and Ng 2002) were maintained in a Swiss/129Sv background and genotyped as described previously. To obtain stage-matched homozygous and wild-type littermates, heterozygote animals were mated to each other, and the morning of vaginal plug formation was counted as 0.5 dpc.
Mice lacking PDGFR-β, PDGF-B, or Nrp-1 have been previously described (Leveen et al. 1994; Soriano 1994; Kawasaki et al. 1999), as have been mice containing a LacZ reporter in the 3′UTR of one or both VEGF-A alleles (Miquerol et al. 1999) and mice carrying a LacZ gene under the control of the Tie2 promoter (Schlaeger et al. 1997).
RT-PCR and sequencing
Total RNA was reverse transcribed using Superscript II (GIBCO-BRL). Single-stranded cDNA was amplified using Taq polymerase (Perkin-Elmer) as recommended by the manufacturer, except that 10% dimethylsulfoxide was included in the reaction. Positive controls were provided by plasmid vectors containing cDNA inserts for VEGF120, VEGF164, and VEGF188 (a kind gift of Y.S. Ng and P. D'Amore, Harvard Medical School, Boston, MA). The following oligonucleotide pairs were used for PCR amplification: GAPDH-F (5′-GCTGAGTATGTCGTGGAGTC-3′)/GAPDH-R (5′-TTGGTGGTGCAGGATGCATT-3′) and VEGF-F (5′-ATGAACTTTCTGCTCTCTTGG-3′)/VEGF-R (5′-TCACCGCCTTGGCTTGTCACA-3′). PCR products amplified with VEGF-specific oligonucleotides were extracted from agarose gel slices and sequenced with VEGF-F and VEGF-R oligonucleotides as primers, using the dideoxy chain termination method with the ABI PRISM cycle sequencing kit (Perkin-Elmer).
Histology, immunostaining, and in situ hybridization
Visualization of β-galactosidase activity was performed as previously described (Schlaeger et al. 1997). Whole-mount in situ hybridization was performed as previously described (Strahle et al. 1994) except that tissues were digested with proteinase K for 20 min, and posthybridization washes were performed in TBST (10 mM Tris-HCl at pH 8, 150 mM NaCl, and 0.1% Tween-20; Sigma). A plasmid containing the VEGF-A cDNA (kindly provided by Kari Alitalo, Helsinki, Finland) was transcribed using RNA polymerase (Promega) and digoxigenin labeling mix (Roche) following the manufacturer's instructions. For immunostaining, samples were fixed in 4% formaldehyde in PBS for 2 h on ice and then permeabilized by washing twice in PBS containing 0.1% TritonX-100 (PBX), followed by a 5-min incubation in 50% methanol/PBX and then methanol. Samples were rehydrated, and endogenous peroxidase activity was quenched with 0.1% hydrogen peroxide in PBS for 30 min at room temperature. After washing twice in PBS, samples were incubated for 2 h at 4°C in 10% normal goat serum in PBX, and then incubated with primary antibodies in the same solution overnight at 4°C with gentle agitation. Samples were washed five times for 1 h each in PBX, incubated overnight at 4°C with secondary antibody in 10% normal goat or rabbit serum (depending on the secondary antibody used) in PBX, and washed as described for the primary antibody. Extracellular VEGF-A staining was performed immediately after fixation but without inclusion of methanol or TritonX-100 in the procedure.
The following primary antibodies were used: rat anti-mouse CD31/PECAM MEC13.3 (Pharmingen); rabbit anti-cow GFAP (glial fibrillary acidic protein; Dako); and rabbit affinity-purified anti human VEGF (Research Diagnostics Inc.), which recognizes all mouse VEGF-A isoforms by immunoblotting (data not shown). To test the specificity of the VEGF-A antiserum, immunostaining was performed after quenching with a 50-fold excess of recombinant VEGF164 (a gift of D. Krilleke, Cancer Research UK, London, U.K.) or an unrelated protein, α-SNAP (a gift of J. Mueller, Cancer Research UK, London, U.K.). Secondary antibodies used included rabbit anti-rat IgG, horseradish peroxidase (HRP)-conjugated (Dako); goat anti-rabbit IgG, Alexa543-conjugated (Molecular Probes); and goat anti-rat IgG, Alexa488-conjugated (Molecular Probes).
In some experiments, blood vessels were visualized using either 20 μg/mL horseradish peroxidase conjugated isolectin B4 (Sigma), or biotinylated isolectin B4 followed by fluorescent streptavidin conjugates (Molecular Probes). To visualize polymerized actin, rhodamine-conjugated phalloidin (Molecular Probes) was included during incubation with the streptavidin conjugates.
Samples incubated with HRP-conjugated antibodies were developed with diaminobenzidine/hydrogen peroxide (SigmaFast; Sigma); images were recorded using a MZ 125 microscope (Leica) equipped with a digital camera (CoolSNAP, RS Photometrics) and were processed using Openlab 2.2 software (Improvision Ltd.). Fluorescently labeled hindbrain samples were mounted using SlowFade (Molecular Probes), and images were recorded with an LSM510 laser scanning confocal microscope (Zeiss). Fluorescently labeled retinas were mounted in Mowiol (Mowiol 4–88, Hoechst) supplemented with 2.5% anti-bleaching agent DABCO (Sigma), and images were recorded with a Nikon E1000 fluorescence microscope equipped with a Coolpix 990 digital camera (Nikon) or with a LCS NT laser scanning confocal microscope (Leica). All digital images were processed using Adobe Photoshop 6.0 (Adobe Systems Inc.).
Quantification of vessel branch points and capillary profiles
For light microscopy, midbrains, hindbrains, and spinal cords of varying developmental stages were processed to visualize microvessel networks and then flat-mounted with the ventricular side facing upward. Littermate samples were photographed at the same magnification, and for each genotype, the average number of branch points was determined in three randomly chosen 500-μm2 regions; the error bar represents the standard deviation from the mean. For 10.75- or 11.25-dpc embryos, our analysis included only littermates that differed by no more than one somite; for 13.5-dpc embryos, we included only embryos with similar stages of limb development.
For electron microscopy, hindbrains of stage-matched 10.75-dpc littermates were fixed for 2 h in a fixative containing 2% formaldehyde, 2% glutaraldehyde, and 0.1 M cacodylate buffer (pH 7.5) and were processed for Epon embedding as previously described (Rabouille et al. 1995). The average number of endothelial cells per capillary cross-section was determined by counting the number of endothelial nuclei in random capillary cross-sections; the error bar represents the standard error of the mean. The P value was calculated using a nonparametric Mann-Whitney Test.
HUVEC proliferation assay
After starvation for 5 h in endothelial growth medium (Invitrogen) containing 0.1% FBS, human umbilical vein endothelial cell (HUVECs) were exposed for 96 h to various concentrations of bFGF (R&D Systems), VEGF121 (R&D Systems), or VEGF165 (Peprotech Ltd); cell growth was quantitated with a fluorimetric DNA assay as described previously (Rao and Otto 1992)
BrdU incorporation into proliferating endothelium
Timed pregnant females were injected intraperitoneally with 0.2 mL of 10 mg/mL BrdU (Sigma) in PBS and sacrificed after 1 h. Dissected embryonic hindbrains were processed for immunostaining as described above, with the exception that they were incubated for 30 min in 4 N HCl followed by neutralization in 100 mM sodium tetraborate for 5 min before the staining procedure. Blood vessels were visualized with biotinylated isolectin B4 followed by FITC-conjugated streptavidin, whereas BrdU was detected using a mouse anti-BrdU antibody (Becton Dickinson).
Acknowledgments
We acknowledge the following with gratitude: Yin-Shan Ng, Patricia D'Amore, Peter Carmeliet, Andras Nagy, and Urban Deutsch for providing mouse strains; Natalie Ortega, Zena Werb, and Shigeyoshi Itohara for providing MMP-9 and MMP-2 null mouse embryos; Dominik Krilleke for help with genotyping; the Biological Resources Unit of Cancer Research UK (CRUK) for animal care and assistance with experimentation; Julian Lewis, Andras Nagy, and Marcus Fruttiger for helpful discussions; Andrea Lundkvist for valuable discussion and help with retinal whole-mount immunohistochemistry; and Joyce Mueller and Katrin Deinhardt for critical reading of the manuscript. This study was funded by CRUK (D.T.S) and by grants from the Novo Nordisk Foundation, the Swedish Cancer Foundation, and Inga Britt and Arne Lundberg Foundation (to C.B.). H.G. is supported by an EMBO postdoctoral fellowship; C.R. by an MRC postdoctoral fellowship.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL d.shima@cancer.org.uk; FAX 44-20-7269-2880.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.242002.
References
- Abrahamson DR, Robert B, Hyink DP, St. John PL, Daniel TO. Origins and formation of microvasculature in the developing kidney. Kidney Int Suppl. 1998;67:S7–S11. doi: 10.1046/j.1523-1755.1998.06702.x. [DOI] [PubMed] [Google Scholar]
- Baeg GH, Perrimon N. Functional binding of secreted molecules to heparan sulfate proteoglycans in Drosophila. Curr Opin Cell Biol. 2000;12:575–580. doi: 10.1016/s0955-0674(00)00134-4. [DOI] [PubMed] [Google Scholar]
- Bär T. Patterns of vascularization in the developing cerebral cortex. CIBA Found Symp. 1983;100:20–36. doi: 10.1002/9780470720813.ch3. [DOI] [PubMed] [Google Scholar]
- Bellusci S, Grindley J, Emoto H, Itoh N, Hogan BL. Fibroblast growth factor 10 (FGF10) and branching morphogenesis in the embryonic mouse lung. Development. 1997;124:4867–4878. doi: 10.1242/dev.124.23.4867. [DOI] [PubMed] [Google Scholar]
- Bergers G, Brekken R, McMahon G, Vu TH, Itoh T, Tamaki K, Thorpe P, Itohara S, Werb Z, Hanahan D. Matrix metalloproteinase-9 triggers the angiogenic switch during carcinogenesis. Nat Cell Biol. 2000;2:737–744. doi: 10.1038/35036374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betsholtz C, Karlsson L, Lindahl P. Developmental roles of platelet-derived growth factors. Bioessays. 2001;23:494–507. doi: 10.1002/bies.1069. [DOI] [PubMed] [Google Scholar]
- Bullock SL, Fletcher JM, Beddington RS, Wilson VA. Renal agenesis in mice homozygous for a gene trap mutation in the gene encoding heparan sulfate 2-sulfotransferase. Genes & Dev. 1998;12:1894–1906. doi: 10.1101/gad.12.12.1894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P. VEGF gene therapy: Stimulating angiogenesis or angioma-genesis? Nat Med. 2000;6:1102–1103. doi: 10.1038/80430. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, et al. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Ng YS, Nuyens D, Theilmeier G, Brusselmans K, Cornelissen I, Ehler E, Kakkar VV, Stalmans I, Mattot V, et al. Impaired myocardial angiogenesis and ischemic cardiomyopathy in mice lacking the vascular endothelial growth factor isoforms VEGF164 and VEGF188. Nat Med. 1999;5:495–502. doi: 10.1038/8379. [DOI] [PubMed] [Google Scholar]
- Chang-Ling T, Stone J. Factors determining the migration of astrocytes into the developing retina: Migration does not depend on intact axons or patent vessels. J Comp Neurol. 1992;303:375–386. doi: 10.1002/cne.903030304. [DOI] [PubMed] [Google Scholar]
- Cleaver O, Krieg PA. VEGF mediates angioblast migration during development of the dorsal aorta in Xenopus. Development. 1998;125:3905–3914. doi: 10.1242/dev.125.19.3905. [DOI] [PubMed] [Google Scholar]
- Davies J. Intracellular and extracellular regulation of ureteric bud morphogenesis. J Anat. 2001;198:257–264. doi: 10.1046/j.1469-7580.2000.19830257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Porat R, Keshet E. Vascular endothelial growth factor and vascular adjustments to perturbations in oxygen homeostasis. Am J Physiol Cell Physiol. 2001;280:C1367–C1374. doi: 10.1152/ajpcell.2001.280.6.C1367. [DOI] [PubMed] [Google Scholar]
- Eriksson U, Alitalo K. Structure, expression and receptor-binding properties of novel vascular endothelial growth factors. Curr Top Microbiol Immunol. 1999;237:41–57. doi: 10.1007/978-3-642-59953-8_3. [DOI] [PubMed] [Google Scholar]
- Esko JD, Lindahl U. Molecular diversity of heparan sulfate. J Clin Invest. 2001;108:169–173. doi: 10.1172/JCI13530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrara N, Alitalo K. Clinical applications of angiogenic growth factors and their inhibitors. Nat Med. 1999;5:1359–1364. doi: 10.1038/70928. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- Gebb SA, Shannon JM. Tissue interactions mediate early events in pulmonary vasculogenesis. Dev Dyn. 2000;217:159–169. doi: 10.1002/(SICI)1097-0177(200002)217:2<159::AID-DVDY3>3.0.CO;2-9. [DOI] [PubMed] [Google Scholar]
- Gengrinovitch S, Berman B, David G, Witte L, Neufeld G, Ron D. Glypican-1 is a VEGF165 binding proteoglycan that acts as an extracellular chaperone for VEGF165. J Biol Chem. 1999;274:10816–10822. doi: 10.1074/jbc.274.16.10816. [DOI] [PubMed] [Google Scholar]
- Gerber HP, Hillan KJ, Ryan AM, Kowalski J, Keller GA, Rangell L, Wright BD, Radtke F, Aguet M, Ferrara N. VEGF is required for growth and survival in neonatal mice. Development. 1999;126:1149–1159. doi: 10.1242/dev.126.6.1149. [DOI] [PubMed] [Google Scholar]
- Grunstein J, Masbad JJ, Hickey R, Giordano F, Johnson RS. Isoforms of vascular endothelial growth factor act in a coordinate fashion to recruit and expand tumor vasculature. Mol Cell Biol. 2000;20:7282–7291. doi: 10.1128/mcb.20.19.7282-7291.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellstrom M, Gerhardt H, Kalen M, Li X, Eriksson U, Wolburg H, Betsholtz C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J Cell Biol. 2001;153:543–553. doi: 10.1083/jcb.153.3.543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogan BL, Yingling JM. Epithelial/mesenchymal interactions and branching morphogenesis of the lung. Curr Opin Genet Dev. 1998;8:481–486. doi: 10.1016/s0959-437x(98)80121-4. [DOI] [PubMed] [Google Scholar]
- Houck KA, Leung DW, Rowland AM, Winer J, Ferrara N. Dual regulation of vascular endothelial growth factor bioavailability by genetic and proteolytic mechanisms. J Biol Chem. 1992;267:26031–26037. [PubMed] [Google Scholar]
- Impagnatiello MA, Weitzer S, Gannon G, Compagni A, Cotten M, Christofori G. Mammalian sprouty-1 and -2 are membrane-anchored phosphoprotein inhibitors of growth factor signaling in endothelial cells. J Cell Biol. 2001;152:1087–1098. doi: 10.1083/jcb.152.5.1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jarecki J, Johnson E, Krasnow MA. Oxygen regulation of airway branching in Drosophila is mediated by branchless FGF. Cell. 1999;99:211–220. doi: 10.1016/s0092-8674(00)81652-9. [DOI] [PubMed] [Google Scholar]
- Kawasaki T, Kitsukawa T, Bekku Y, Matsuda Y, Sanbo M, Yagi T, Fujisawa H. A requirement for neuropilin-1 in embryonic vessel formation. Development. 1999;126:4895–4902. doi: 10.1242/dev.126.21.4895. [DOI] [PubMed] [Google Scholar]
- Lawrence PA, Struhl G. Morphogens, compartments, and pattern: Lessons from Drosophila? Cell. 1996;85:951–961. doi: 10.1016/s0092-8674(00)81297-0. [DOI] [PubMed] [Google Scholar]
- Lee RJ, Springer ML, Blanco-Bose WE, Shaw R, Ursell PC, Blau HM. VEGF gene delivery to myocardium: Deleterious effects of unregulated expression. Circulation. 2000;102:898–901. doi: 10.1161/01.cir.102.8.898. [DOI] [PubMed] [Google Scholar]
- Lee SH, Schloss DJ, Jarvis L, Krasnow MA, Swain JL. Inhibition of angiogenesis by a mouse sprouty protein. J Biol Chem. 2001;276:4128–4133. doi: 10.1074/jbc.M006922200. [DOI] [PubMed] [Google Scholar]
- Leveen P, Pekny M, Gebre-Medhin S, Swolin B, Larsson E, Betsholtz C. Mice deficient for PDGF B show renal, cardiovascular, and hematological abnormalities. Genes & Dev. 1994;8:1875–1887. doi: 10.1101/gad.8.16.1875. [DOI] [PubMed] [Google Scholar]
- Lin X, Buff EM, Perrimon N, Michelson AM. Heparan sulfate proteoglycans are essential for FGF receptor signaling during Drosophila embryonic development. Development. 1999;126:3715–3723. doi: 10.1242/dev.126.17.3715. [DOI] [PubMed] [Google Scholar]
- Mattot V, Moons L, Lupu F, Chernavvsky D, Gomez RA, Collen D, Carmeliet P. Loss of the VEGF164 and VEGF188 isoforms impairs postnatal glomerular angiogenesis and renal arteriogenesis in mice. J Am Soc Nephrol. 2002;13:1548–1560. doi: 10.1097/01.asn.0000013925.19218.7b. [DOI] [PubMed] [Google Scholar]
- Metzger RJ, Krasnow MA. Genetic control of branching morphogenesis. Science. 1999;284:1635–1639. doi: 10.1126/science.284.5420.1635. [DOI] [PubMed] [Google Scholar]
- Miao HQ, Klagsbrun M. Neuropilin is a mediator of angiogenesis. Cancer Metastasis Rev. 2000;19:29–37. doi: 10.1023/a:1026579711033. [DOI] [PubMed] [Google Scholar]
- Miquerol L, Gertsenstein M, Harpal K, Rossant J, Nagy A. Multiple developmental roles of VEGF suggested by a LacZ-tagged allele. Dev Biol. 1999;212:307–322. doi: 10.1006/dbio.1999.9355. [DOI] [PubMed] [Google Scholar]
- Miquerol L, Langille BL, Nagy A. Embryonic development is disrupted by modest increases in vascular endothelial growth factor gene expression. Development. 2000;127:3941–3946. doi: 10.1242/dev.127.18.3941. [DOI] [PubMed] [Google Scholar]
- Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. Vascular endothelial growth factor (VEGF) and its receptors. FASEB J. 1999;13:9–22. [PubMed] [Google Scholar]
- Ng YS, Rohan R, Sunday ME, Demello DE, D'Amore PA. Differential expression of VEGF isoforms in mouse during development and in the adult. Dev Dyn. 2001;220:112–121. doi: 10.1002/1097-0177(2000)9999:9999<::AID-DVDY1093>3.0.CO;2-D. [DOI] [PubMed] [Google Scholar]
- Park JE, Keller GA, Ferrara N. The vascular endothelial growth factor (VEGF) isoforms: Differential deposition into the subepithelial extracellular matrix and bioactivity of extracellular matrix-bound VEGF. Mol Biol Cell. 1993;4:1317–1326. doi: 10.1091/mbc.4.12.1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrimon N, Bernfield M. Specificities of heparan sulphate proteoglycans in developmental processes. Nature. 2000;404:725–728. doi: 10.1038/35008000. [DOI] [PubMed] [Google Scholar]
- Placzek M, Skaer H. Airway patterning: A paradigm for restricted signalling. Curr Biol. 1999;9:R506–R510. doi: 10.1016/s0960-9822(99)80322-4. [DOI] [PubMed] [Google Scholar]
- Poltorak Z, Cohen T, Sivan R, Kandelis Y, Spira G, Vlodavsky I, Keshet E, Neufeld G. VEGF145: A secreted vascular endothelial growth factor isoform that binds to extracellular matrix. J Biol Chem. 1997;272:7151–7158. doi: 10.1074/jbc.272.11.7151. [DOI] [PubMed] [Google Scholar]
- Rabouille C, Hui N, Hunte F, Kieckbusch R, Berger EG, Warren G, Nilsson T. Mapping the distribution of Golgi enzymes involved in the construction of complex oligosaccharides. J Cell Sci. 1995;108:1617–1627. doi: 10.1242/jcs.108.4.1617. [DOI] [PubMed] [Google Scholar]
- Ramirez-Weber FA, Kornberg TB. Signaling reaches to new dimensions in Drosophila imaginal discs. Cell. 2000;103:189–192. doi: 10.1016/s0092-8674(00)00111-2. [DOI] [PubMed] [Google Scholar]
- Rao J, Otto WR. Fluorimetric DNA assay for cell growth estimation. Anal Biochem. 1992;207:186–192. doi: 10.1016/0003-2697(92)90521-8. [DOI] [PubMed] [Google Scholar]
- Rapraeger AC. Syndecan-regulated receptor signaling. J Cell Biol. 2000;149:995–998. doi: 10.1083/jcb.149.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risau W. Mechanisms of angiogenesis. Nature. 1997;386:671–674. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- Schlaeger TM, Bartunkova S, Lawitts JA, Teichmann G, Risau W, Deutsch U, Sato TN. Uniform vascular-endothelial-cell-specific gene expression in both embryonic and adult transgenic mice. Proc Natl Acad Sci. 1997;94:3058–3063. doi: 10.1073/pnas.94.7.3058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shima DT, Mailhos C. Vascular developmental biology: Getting nervous. Curr Op Genet Dev. 2000;10:536–542. doi: 10.1016/s0959-437x(00)00124-6. [DOI] [PubMed] [Google Scholar]
- Shima DT, Kuroki M, Deutsch U, Ng YS, Adamis AP, D'Amore PA. The mouse gene for vascular endothelial growth factor: Genomic structure, definition of the transcriptional unit, and characterization of transcriptional and post-transcriptional regulatory sequences. J Biol Chem. 1996;271:3877–3883. doi: 10.1074/jbc.271.7.3877. [DOI] [PubMed] [Google Scholar]
- Soker S, Takashima S, Miao HQ, Neufeld G, Klagsbrun M. Neuropilin-1 is expressed by endothelial and tumor cells as an isoform-specific receptor for vascular endothelial growth factor. Cell. 1998;92:735–745. doi: 10.1016/s0092-8674(00)81402-6. [DOI] [PubMed] [Google Scholar]
- Soriano P. Abnormal kidney development and hematological disorders in PDGF β-receptor mutant mice. Genes & Dev. 1994;8:1888–1896. doi: 10.1101/gad.8.16.1888. [DOI] [PubMed] [Google Scholar]
- Stalmans I, Ng Y-S. Arteriolar and venular patterning in retinas of mice selectively expressing VEGF isoforms. J Clin Invest. 2002;109:327–336. doi: 10.1172/JCI14362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone J, Dreher Z. Relationship between astrocytes, ganglion cells and vasculature of the retina. J Comp Neurol. 1987;255:35–47. doi: 10.1002/cne.902550104. [DOI] [PubMed] [Google Scholar]
- Strahle U, Blader P, Adam J, Ingham PW. A simple and efficient procedure for non-isotopic in situ hybridization to sectioned material. Trends Genet. 1994;10:75–76. doi: 10.1016/0168-9525(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Sundberg C, Nagy JA, Brown LF, Feng D, Eckelhoefer IA, Manseau EJ, Dvorak AM, Dvorak HF. Glomeruloid microvascular proliferation follows adenoviral vascular permeability factor/vascular endothelial growth factor-164 gene delivery. Am J Pathol. 2001;158:1145–1160. doi: 10.1016/S0002-9440(10)64062-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland D, Samakovlis C, Krasnow MA. branchless encodes a Drosophila FGF homolog that controls tracheal cell migration and the pattern of branching. Cell. 1996;87:1091–1101. doi: 10.1016/s0092-8674(00)81803-6. [DOI] [PubMed] [Google Scholar]
- Teleman AA, Strigini M, Cohen SM. Shaping morphogen gradients. Cell. 2001;105:559–562. doi: 10.1016/s0092-8674(01)00377-4. [DOI] [PubMed] [Google Scholar]
- Tischer E, Mitchell R, Hartman T, Silva M, Gospodarowicz D, Fiddes JC, Abraham JA. The human gene for vascular endothelial growth factor: Multiple protein forms are encoded through alternative exon splicing. J Biol Chem. 1991;266:11947–11954. [PubMed] [Google Scholar]
- Vlodavsky I, Folkman J, Sullivan R, Fridman R, Ishai-Michaeli R, Sasse J, Klagsburn M. Endothelial cell-derived basic fibroblast growth factor: Synthesis and deposition into subendothelial extracellular matrix. Proc Natl Acad Sci. 1987;84:2292–2296. doi: 10.1073/pnas.84.8.2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weaver M, Dunn NR, Hogan BL. Bmp4 and Fgf10 play opposing roles during lung bud morphogenesis. Development. 2000;127:2695–2704. doi: 10.1242/dev.127.12.2695. [DOI] [PubMed] [Google Scholar]
- Yayon A, Klagsbrun M, Esko JD, Leder P, Ornitz DM. Cell surface, heparin-like molecules are required for binding of basic fibroblast growth factor to its high affinity receptor. Cell. 1991;64:841–848. doi: 10.1016/0092-8674(91)90512-w. [DOI] [PubMed] [Google Scholar]