


Abstract
Nucleotide repair genes are not generally altered in sporadic solid tumors. However, point mutations are found scattered throughout the genome of cancer cells indicating that the repair pathways are dysfunctional. To address this point, in this work we focus on the expression pathways rather than in the DNA structure of repair genes related to either genome stability or essential metabolic functions. We present here a novel statistical analysis comparing ten gene expression pathways in human normal and cancer cells using serial analysis of gene expression (SAGE) data. We find that in cancer cells nucleotide-excision repair (NER) and apoptosis are the most impaired pathways and have a highly altered diversity of gene expression profile when compared to normal cells. We propose that genome point mutations in sporadic tumors can be explained by a structurally conserved NER with a functional disorder generated from its entanglement with the apoptosis gene network.
INTRODUCTION
Cancer cells have large and small abnormalities in their genetic material: additional or missing chromosomes, mutated genes and other types of alterations. The lost of genome stability pathways is associated with genetic deterioration of cancer cells and is one of the most important aspects of carcinogenesis. In fact, mutations in mismatch repair (MMR), nucleotide-excision repair (NER), base-excision repair (BER) and recombinational repair genes have been causally implicated in the acquisition of a genome instability phenotype (1).
Genome instability in solid tumors originates from either somatic mutations (observed in the majority of sporadic cancers) or germline mutations (associated to rare hereditary cancer syndromes). Considering the list of repair genes presented in Cancer Gene Census (2), germline mutations can be observed in NER, BER and MMR, while somatic mutations are described only in recombinational repair (homologous recombination and non-homologous end joining). On the other hand, mutations in apoptotic genes are recurrently observed in both types of solid tumors as listed in the census.
The genotype signature of the malfunctioning of these stability gene networks is 2-fold: aneuploidy (e.g. translocations, gain or loss of entire or large parts of chromosomes) and/or random point mutations (e.g. nucleotide changes randomly distributed throughout the genome) (3). The omnipresence of random point mutations in sporadic solid tumors (4) and the recurrent absence of mutations in nucleotide repair genes (2) suggest a functional deficiency in these stability pathways without structural alterations in the related DNA sequence.
There are different views explaining how a cell loses genome stability and acquires a cancerous phenotype (5,6). In one proposed scenario, large chromosomal changes are required for triggering the onset of cancer, such as varying the number of whole chromosomes or cutting and/or pasting their fragments among different chromosomes. Then either the expression of unbalanced gene dosage (7) and/or alterations in mitotic check points (8) can, under adequate conditions, give place to a cancer. An alternative idea proposes that cancer cells have a ‘mutator phenotype’ that favors the acquisition of point mutations, which eventually affect tumor suppressors or oncogenes yielding to cancer (9). Supporting this idea, a list of mutated genes found in human colorectal and breast cancer covering several gene functions shows that point mutations are the most common alterations found throughout the genome of cancer cells (over 87%) (10).
The two scenarios are qualitatively possible, since both offer explanations to the typical chromosome configurations and nucleotide alterations of a cancer cell. In order to discriminate between different scenarios, studies of chromosome and gene structures should be complemented by quantitative analysis of gene expression of cancer cells. As a contribution in this direction, we present here a pioneer, comprehensive statistical analysis of 10 gene expression pathways in normal and cancer cells using serial analysis of gene expression (SAGE) data from the public gene expression resource (SAGE Genie) (11) available at Cancer Genome Anatomy Project (CGAP) (12).
MATERIALS AND METHODS
Data selection
Human cancer and normal tissue SAGE libraries are retrieved using SAGE Library Finder tool at SAGE Genie website (http://cgap.nci.nih.gov/SAGE) based on the search criteria: tag length (short 10 bp), tissue preparation [bulk, short-term culture (STC), antibody purified (ABP), microscope dissected (MCD) or cell line] and tissue histology (cancer or normal). The final list is presented in Supplementary Tables S1 to S4 and contains only cancer libraries that had at least one normal equivalent tissue library, and vice-versa, matching both the search criteria. The list of SAGE libraries is also retrieved for tag-to-gene corresponding libraries using SAGE Absolute Level Lister (SALL) tool at SAGE Genie website. This tool links SAGE unique tags to genes via UniGene cluster IDs (e.g. it packs into one file Tag Sequence, Tag Frequency, UniGene Cluster ID and Gene Symbol). SALL database retrieval was conducted in June 2006 (UniGene Build #191 and #192). Therefore, according to the search criteria, we retrieved the largest human SAGE collection up to date at NCI's Cancer Genome Anatomy Project for the analysis presented here.
Mathematical definitions and analysis of SAGE libraries
In the SAGE database, a SAGE library corresponds to one tumor sample exam, which is made from mRNA extracts from different tissue preparations (bulk, short-term culture, antibody purified, microscope dissected or cell line) and histology (cancer or normal), as described in detail by Lash et al. (13). One such library gives the amount of every detected transcript in the sample, each one being labeled by a 10-letter tag, corresponding to 10 bases close to the poly-A tail, whose length is long enough to discriminate every possible transcript. Transcripts related to different gene networks may be grouped and used to quantify and characterize their expression activity. Here we analyze both the amount of transcript production and its diversity in ten gene pathways, chosen due to either their recognized relation with genome stability (apoptosis, chromosome stability, mismatch repair, nucleotide-excision repair, base-excision repair and recombinational repair) or, as a control group, due to their essential life-supporting activities (ribosome, ATP synthase, electron transport chain and glycolysis). The tumor types were selected such that they present a library of normal cells, to be used as control. The complete list of SAGE libraries and details about database search are available in the Supporting Online Material.
To obtain a quantitative expression of sample distribution of SAGE tags, we have measured the information content of SAGE libraries using Shannon Information Theory (14–18) defined as follows. Consider n as the number of all selected SAGE libraries of a given tumor type. Each library of this set is labeled by α(α = 1, … , n) and has Nα tags, among Mα possible ones, that is, possible transcripts. For a given SAGE library in this set, we can define s(i,α) as being the number of transcripts (tags) of a given type i, (i = 1, … , Mα), whose sum for a given α adds up to Nα. The probability p(i,α) that, among the Nα tags of the α-library, a randomly chosen transcript is of the type i is written as
| 1 |
such that ∑ip(i, α) = 1. The normalized Shannon information function Hα is defined as
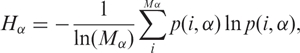 |
2 |
where we have divided all terms by the factor ln(Mα) in order to normalize the quantities, guaranteeing that 0 ≤ Hα ≤ 1. The idea is to compare among samples of different tissues that may present different numbers of Mα possibilities (e.g. different numbers of possible transcripts). While Nα reflects gene expression activity (the amount of tags in the αth library), Hα reflects the spread of the distribution s(i, α), i.e. it measures the diversity that exists in the αth library.
Finally, in order to normalize the quantities by sets of tags, taking as reference normal tissue histology, we define the relative diversity hα for any given set of genes as
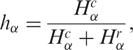 |
3 |
where 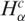 and
and 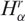 are, respectively, the diversity of cancer and normal SAGE libraries. Observe that 0 ≤ hα ≤ 1, and hα < ½ implies
are, respectively, the diversity of cancer and normal SAGE libraries. Observe that 0 ≤ hα ≤ 1, and hα < ½ implies 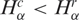 , that is, the transcript distribution in the αth library is narrower in cancer cells than in the normal tissue, while hα > ½ represents the inverse case. In analogy, the relative gene expression activity nα of the α library is defined as
, that is, the transcript distribution in the αth library is narrower in cancer cells than in the normal tissue, while hα > ½ represents the inverse case. In analogy, the relative gene expression activity nα of the α library is defined as
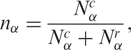 |
4 |
where 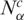 and
and 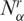 are, respectively, the gene expression activity of cancer and normal tissue (i.e. number of SAGE tags). Again, 0 ≤ nα ≤ 1, and nα < ½ implies
are, respectively, the gene expression activity of cancer and normal tissue (i.e. number of SAGE tags). Again, 0 ≤ nα ≤ 1, and nα < ½ implies 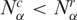 , that is, in this library the cancer cells have lower gene activity, producing less transcripts than the normal case (e.g. Supplementary Figures S1 and S2).
, that is, in this library the cancer cells have lower gene activity, producing less transcripts than the normal case (e.g. Supplementary Figures S1 and S2).
Diversity of gene expression pathways
To estimate the diversity of expression pathways related to genome stability, we carry out the following steps: (i) define gene expression pathways of interest; (ii) identify groups of genes that best represents each pathway; (iii) identify the best SAGE tags of these genes—among all possible tags—presented in the collection of tags of each SAGE library; (iv) arrange the SAGE tags into a separate file—one for each pathway; (v) verify the agreement of the original database with subset files; (vi) build a curated database; and (vii) estimate the degree of diversity of pooled SAGE tags, as defined in mathematical definitions section.
We focused this study in six genomic stability pathways (apoptosis, chromosome stability, mismatch repair, nucleotide-excision repair, base-excision repair and recombinational repair). Here, the group of genes representing each gene expression pathway is considered as a group of UniGene Cluster IDs. The lists of selected genes and pathways are presented in Supplementary Tables S9–S14, including references used for selection. In order to link genes and SAGE tags, we used the UniGene number as common identifier. Next, we checked libraries looking for UniGene number duplication. This process reveals that, in the original database retrieved from the SAGE Genie website, several pooled tags (Unique Tags) present the same UniGene cluster ID and, therefore, they are pooled as single UniGene number. The curated libraries are then used to build the subset files of tags—one for each gene expression pathway. These files are used in the final step to estimate the diversity of gene expression. The curated database and a Microsoft Excel™ spreadsheet that automatically calculates diversity scores for multiple pathways are available upon request.
We have considered several internal controls in order to use as invariant references among cancer and normal SAGE libraries. The idea is to estimate the diversity of a gene expression pathway that produces co-expressed genes, ideally always in the same proportion, independently of tissue type or histology. For this purpose, we consider the following criteria for selection: (i) gene products should be present in stoichiometric amounts because they are part of the same stable complex and/or are functionally associated at the molecular level (19); (ii) the candidate pathway must occur in all cell types because they are necessary for the cell survival and/or are implicated in basal cell metabolism (20); (iii) the pathway must be involved in core, conserved biological functions (21). Among likely candidates, we evaluated four co-expressed gene groups (named here by its final products): (i) ribosome (e.g. ribosomal proteins); (ii) ATP synthase; (iii) electron transport chain; and (iv) glycolysis. The lists of selected control pathways are presented in Supplementary Tables S5–S8, including references used for selection.
Pairwise data of cancer versus normal
After estimating SAGE library parameters for each expression pathway, as defined above, the values of cancer libraries are compared to normal ones. The pairwise comparisons produce two distribution types of matched pairs. One related to SAGE library properly (overall SAGE tags); other related to gene expression pathways (a subset of SAGE tags). The pairwise comparisons are then plotted in order to examine either the entire data distributions or by tissue types individually. The number of pairwise libraries—cancer versus normal—is limited by the number of SAGE libraries available in SAGE Genie website up to date (http://cgap.nci.nih.gov/SAGE). Therefore, each cancer library is paired with each normal library of the same tissue type, as presented in Supplementary Tables S15–S16, providing 493 pairwise comparisons—368 for solid tumors (ST) and 125 for cell lines(CL): brain (ST = 192; CL = 20); breast (ST = 75; CL = 45); cerebellum (ST = 40; CL = 8); stomach (ST = 28); prostate (ST = 12; CL = 32); colon (ST = 4; CL = 8); pancreas (ST = 4; CL = 8); skin (ST = 3); ovary (ST = 3; CL = 2); liver (ST = 2; CL = 2); lung (ST = 2); kidney (ST = 1); peritoneum (ST = 1); thyroid (ST = 1). Indeed, there are only 492 pairwise libraries in SAGE tag-to-gene analysis because the skin library ‘SAGE_Skin_melanoma_B_DB3’ was not integrated with SAGE Absolute Level Lister (SALL) tool at the period of our study.
Analysis of protein/gene interaction networks
The protein–protein interaction network associating genes of the six genome stability pathways is generated using the database STRING (‘search tool for the retrieval of interacting genes/proteins’) (22,23) with input options ‘databases’, ‘experiments’ and 70% confidence level. In order to identify each gene in the database, we used both HUGO ID (24) and Ensembl Peptide ID (25) (Supplementary Table S17). Alternatively, the amino acid sequence of a given protein is supplied to identify the corresponding entry. The results from the search are saved in data files ‘tab-delimited text fields’ describing edge relationships and then handled in Medusa application (26) (i.e. optimized software for accessing protein interaction data from STRING). Pathways are discriminated by different colors and data are crossed with Cancer Gene Census (2) in order to indicate genes whose somatic mutations have been reported to be causally implicated in human cancer. The complete file matching entry IDs, data interactions and mutated genes are available upon request. Finally, graphs are exported to postscript files to have figure quality improved and edited in CorelDraw® graphic design tools (Corel Corp., Ottawa, Canada).
Statistical analysis
Under the null hypothesis, the stochastic contrast among k expression pathways is given by hαA = hαB = · = hαK. Although the distributions do not seriously deviate from normality, as observed in Figures 1 and 2, with equal sample sizes among pathways, the data failed to meet the assumptions of ANOVA for normality and homogeneity of variance and, thus, we used Kruskal–Wallis one-way analysis of variance followed by Mann–Whitney test for comparisons. The tests were performed in SPSS nonparametric statistical package (SPSS for Windows, release 14.0.0. SPSS Inc., Chicago, IL). Values are expressed as mean ± SEM. Significance is considered at P < 0.05.
Figure 1.

Distributions 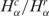 and
and 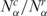 for different tissue types. (A) Nonparametric distribution of matched pairs of solid tumor libraries. The length of the black horizontal stripes correspond to the number of libraries presenting ratios of and that are larger than one, while the gray stripes to the left correspond to the number of libraries with ratios less than one. The number of matched pairs is indicated for each tissue type. (B) Cell lines, as described in A. (C) Histogram distribution of matched pairs of libraries compared by the number of SAGE tags. Inset shows diversity Hα as a function of the number Nα for cancer and normal libraries. (D) Histogram distribution of matched pairs of libraries compared to diversity of SAGE tags. The percentage of matched pairs with
for different tissue types. (A) Nonparametric distribution of matched pairs of solid tumor libraries. The length of the black horizontal stripes correspond to the number of libraries presenting ratios of and that are larger than one, while the gray stripes to the left correspond to the number of libraries with ratios less than one. The number of matched pairs is indicated for each tissue type. (B) Cell lines, as described in A. (C) Histogram distribution of matched pairs of libraries compared by the number of SAGE tags. Inset shows diversity Hα as a function of the number Nα for cancer and normal libraries. (D) Histogram distribution of matched pairs of libraries compared to diversity of SAGE tags. The percentage of matched pairs with 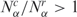 and
and 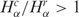 are indicated. The list of SAGE libraries is available in Supplementary Tables S1–S4.
are indicated. The list of SAGE libraries is available in Supplementary Tables S1–S4.
Figure 2.

Distributions of and for different gene expression pathways. (A) and (B) Gene expression pathways involved in core cell functions. The group of genes of each pathway is presented in Supplementary Tables S5–S8, defined accordingly to Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways (35). (C) and (D) Gene expression pathways involved in genome stability functions. The group of genes of each pathway is presented in Supplementary Tables S9–S14, defined according to several references (35–38). The percentage of matched pairs with 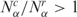 or
or 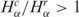 is indicated in all distributions. Another supplemental file listing all genes involved in core biological function and genome stability pathways are provided in spreadsheet format (Supplementary Tables S17 and S18). These data are also presented in log-log scatter plots (Supplementary Figures S8 and S9) and a correlation analysis between SAGE tag and SAGE tag-to-gene data is presented in Supplementary Figure S10.
is indicated in all distributions. Another supplemental file listing all genes involved in core biological function and genome stability pathways are provided in spreadsheet format (Supplementary Tables S17 and S18). These data are also presented in log-log scatter plots (Supplementary Figures S8 and S9) and a correlation analysis between SAGE tag and SAGE tag-to-gene data is presented in Supplementary Figure S10.
RESULTS AND DISCUSSION
In what follows we present the results concerning the above defined quantities for different tumor types. First, we compare the gene expression activity and diversity of each cancer SAGE library from several tissue types with its respective normal case (Figure 1A and B). The great majority of cancer tissues showed an increased gene expression activity and diversity, since in almost all cases the majority of libraries present and 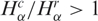 . The resulting global histogram distributions of cancer-normal pairwise libraries are then plotted in Figure 1C and D, showing that the number and diversity of tags in cancer libraries are higher than normal tissue in 56.6 and 72.8% of the cases.
. The resulting global histogram distributions of cancer-normal pairwise libraries are then plotted in Figure 1C and D, showing that the number and diversity of tags in cancer libraries are higher than normal tissue in 56.6 and 72.8% of the cases.
To evince the contribution from the different gene networks, which is not shown in the global histograms, in Figure 2 we present the same matched pairs of SAGE libraries, assessed for subsets of tags corresponding to different gene expression pathways. Comparing the histograms presented in Figure 2A and C, we conclude that apoptosis and NER pathways present a smaller number of cancer libraries with 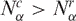 , indicating reduced gene expression in these pathways. When considering the diversity of tags (Figure 2B and D) the results are opposite, since apoptosis and NER present more cancer libraries with
, indicating reduced gene expression in these pathways. When considering the diversity of tags (Figure 2B and D) the results are opposite, since apoptosis and NER present more cancer libraries with 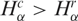 , indicating increased diversity in these gene expression pathways. Furthermore, observe the contrast between the diversity distributions for Ribosome and NER pathways: Ribosome histogram is narrowly peaked around 1, indicating that cancer and normal cells have almost identical transcript profiles for this highly conserved pathway, while NER diversity histogram presents a broad distribution, biased to In fact, except for NER, all other pathways presented a uniform gene activity change in the sense that the relative profiles are conserved. Moreover, NER presents a negative correlation between gene activity and diversity (log–log scatter plots of core cell functions and genomic stability pathways are presented in Supplementary Figures S3 and S4). This result indicates that NER gene activity decreases due to the reduction of normal gene expression peaks in cancer cells. In this way, the increased relative contribution of a broad profile background enhances diversity.
, indicating increased diversity in these gene expression pathways. Furthermore, observe the contrast between the diversity distributions for Ribosome and NER pathways: Ribosome histogram is narrowly peaked around 1, indicating that cancer and normal cells have almost identical transcript profiles for this highly conserved pathway, while NER diversity histogram presents a broad distribution, biased to In fact, except for NER, all other pathways presented a uniform gene activity change in the sense that the relative profiles are conserved. Moreover, NER presents a negative correlation between gene activity and diversity (log–log scatter plots of core cell functions and genomic stability pathways are presented in Supplementary Figures S3 and S4). This result indicates that NER gene activity decreases due to the reduction of normal gene expression peaks in cancer cells. In this way, the increased relative contribution of a broad profile background enhances diversity.
In order to consolidate these results and simultaneously compare all gene expression pathways, we present in Figure 3A the average values of the relative activity nα for each gene network. As we can observe, NER and apoptosis present the lowest amount of relative activity (P < 0.001), indicating again an altered state of gene expression. In contrast, NER has the highest relative diversity (P < 0.001) (Figure 3B), which corroborates that the low level of gene expression occurs together with changes on gene expression profile of this repair pathway. Since gene expression in cell lines could not reliably reflect the gene expression in bulk tissues, we also present in Supplementary Figures S5 and S6 an individual analysis, by tissue type and preparation. Overall, the results indicate that the conclusions drawn on these observations follow the same outcome of the pooled analysis, especially considering the most representative solid tumors in the sample (i.e. brain, breast, cerebellum and stomach).
Figure 3.

Stochastic contrasts among gene expression pathways according to diversity and number of SAGE tags. (A) Relative activity nα as defined in Equation (4). (B) Relative diversity hα as defined in Equation (3). The values are expressed as mean ± SEM (n = 492). Statistical analyses are carried out by Kruskal–Wallis one-way analysis of variance followed by Mann–Whitney test for comparisons. *Different from controls with P < 0.001; **different from others with P < 0.001.
In cancer cells programmed cell death mechanism is in general structurally impaired (27), what is coherent with the observed gene expression profile of apoptosis transcripts. However, NER is in general structurally intact in sporadic solid tumors, since no somatic mutations in NER genes have been reported to be causally implicated in oncogenesis (2). The observed transcript profile then suggests that NER-transactivation-dependent functions are affected in cancer cells.
As both apoptosis and NER networks are simultaneously affected, a causal correlation is plausible, considering that both networks are entangled. One scenario is suppression of NER transcription activity due to global alterations in cell-death control. A second alternative would be apoptosis and NER impairment caused by the malfunctioning of a gene, either due to failures in activation-dependent functions or gene mutations. A natural candidate in this last case is TP53 based on the wealth of experimental evidence that this gene plays a role in both apoptosis and NER networks (28). As an illustration of these two possibilities we present in Figure 4A a protein–protein interaction network associating genes of the six genome stability pathways investigated here. The graph is generated using database STRING (22) with input options ‘Experimental/Biochemical Data’ and ‘Association in Curated Databases,’ with 70% confidence interval, meaning in this graph that genes are linked whenever direct (physical) or indirect (functional) protein interaction is reported in curated databases. Figure 4A suggests that either scenario is possible. This graph indicates a strong interaction among all pathways with significant overlapping among different genes. Concerning apoptosis and NER, TP53 plays a key role, connecting both networks (Supplementary Figure S7). In fact, there are many reports in the literature pointing p53 affecting both dependent and independent transactivation NER functions, as well as affecting apoptosis (28–31). Also, it is reasonable to assume that damage in a specific gene function may affect its neighbors in the network, causing perturbations that may disrupt the whole network. The implication of these observations is that the vulnerability of NER and apoptosis could reside in the same core ‘node.’ Other scenarios are also possible, as defective genes independently acting on both networks, but then more genes should be simultaneously impaired in order to account for the results presented here.
Figure 4.

Graph of interactions among genes involved in apoptosis and DNA repair pathways generated using database STRING (22) with input options ‘Experimental/Biochemical Data,’ ‘Association in Curated Databases,’ and 70% confidence level. (A) Different pathways are represented in different colors. Nodes with more than one color represent genes participating in more than one pathway. Square nodes represent genes whose somatic mutations have been reported to be causally implicated in oncogenesis (2). The group of genes of each pathway is presented in details in Supplementary Tables S9–S14, defined according to several references (35–38). Genes without known interactions with other genes are listed in the bottom left of the figure. (B) Connectivity k of interacting nodes, which shows the number of links that a given node has with other nodes. The color of a node indicates its pathway, as in (A); mutated genes are also indicated. Mismatch repair (MMR), nucleotide-excision repair (NER), base-excision repair (BER).
Furthermore, observing the network architecture and the organization of interactions in Figure 4A, one can see that NER topology suggests the existence of a functional module overlapping three pathways, i.e. NER, MMR and chromosome stability (module ρ). To quantify the interaction pattern among genes in the network we calculated the connectivity k, defined as the number of links that a given node has with other nodes (32). Figure 4B presents the nodes by increasing connectivity. There are two striking features in this figure. First, all ρ-nodes present high connectivity. Second, there are no mutations in high connectivity nodes. This may be indicating that the joint functions of the nodes in ρ are essential to turn the cell viable, what would play the role of a protection mechanism for the organism against proliferation of mutation-prone clones. Furthermore, TP53 appears as the mutated gene with highest k degree, what could be interpreted as a gene with high enough connectivity such that a mutation has a great effect in disrupting the cell apoptosis and repair system, but low enough connectivity such that the cell is still viable. Following this speculative point of view, mutations in more than one gene with lower connectivity should be required to disrupt both apoptosis and repair systems, that is, to cause cancer. This would hence explain why mutations in other genes than TP53 are less probable and why TP53 is not mutated in all tumors (10). In other words, it may happen that higher connectivity and higher mutation probability in cancer cells are correlated up to a connectivity threshold, when mutations render the cell unviable. This possibility is consistent with the growth failure and premature death described in at least three NER-deficient mouse models (33) and with the correlation between protein connectivity and indispensability described in yeast proteome (34).
In summary, the above statistical analysis indicates that, relative to normal tissues, cancer cells present (i) enhanced overall gene expression, indicating a higher transcriptional activity, (ii) decreased apoptosis and NER gene expression activity, (iii) conserved expression profiles for control gene pathways, (iv) high diversity in transcript profiles for NER, suggesting suppression of expression peaks, enhancing the relative background contribution. It is then possible that conditions that disable apoptosis, probably due to mutations in apoptotic genes, also affects NER-transactivation-dependent functions via p53. NER malfunctioning could then account for random point mutations scattered throughout the cancer cell genome. Furthermore, the analysis of network connectivity points to a highly connected module (ρ) involving genes from NER, MMR and chromosome stability where mutations are recurrently absent in cancer cells, but whose functions could be impaired by mutations in peripheral genes, linking this module with apoptosis.
A natural perspective of these findings is to extend the same approach for diagnostic purpose, for example, and thus test the robustness of our conclusions, which could also indicate whether this model can be applied to identification of other pathways involved in cancer progression.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
This work has been partially supported by Brazilian Agencies FAPERGS, CNPq and CAPES. We acknowledge KEGG, STRING and CGAP/SAGE databases for providing public access to its data. Funding to pay the Open Access publication charge was provided by CNPq (grant 140947/2006-0).
Conflict of interest statement. None declared.
REFERENCES
- 1.Hoeijmakers JHJ. Genome maintenance mechanisms for preventing cancer. Nature. 2001;411:366–374. doi: 10.1038/35077232. [DOI] [PubMed] [Google Scholar]
- 2.Futreal PA, Coin L, Marshall M, Down T, Hubbard T, Wooster R, Rahman N, Stratton MR. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vogelstein B, Kinzler KW. Cancer genes and the pathways they control. Nat. Med. 2004;10:789–799. doi: 10.1038/nm1087. [DOI] [PubMed] [Google Scholar]
- 4.Venkatesan RN, Bielas JH, Loeb LA. Generation of mutator mutants during carcinogenesis. DNA Repair. 2006;5:294–302. doi: 10.1016/j.dnarep.2005.10.012. [DOI] [PubMed] [Google Scholar]
- 5.Marx J. Debate surges over the origins of genomic defects in cancer. Science. 2002;297:544–546. doi: 10.1126/science.297.5581.544. [DOI] [PubMed] [Google Scholar]
- 6.Merlo LMF, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat. Rev. Cancer. 2006;6:924–935. doi: 10.1038/nrc2013. [DOI] [PubMed] [Google Scholar]
- 7.Duesberg P, Li R, Fabarius A, Hehlmann R. The chromosomal basis of cancer. Cell. Oncol. 2005;27:293–318. doi: 10.1155/2005/951598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rajagopalan H, Nowak MA, Vogelstein B, Lengauer C. The significance of unstable chromosomes in colorectal cancer. Nat. Rev. Cancer. 2003;3:695–701. doi: 10.1038/nrc1165. [DOI] [PubMed] [Google Scholar]
- 9.Loeb LA, Loeb KR, Anderson JP. Multiple mutations and cancer. Proc. Natl. Acad. Sci. U.S.A. 2003;100:776–781. doi: 10.1073/pnas.0334858100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, Barber TD, Mandelker D, Leary RJ, Ptak J, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. doi: 10.1126/science.1133427. [DOI] [PubMed] [Google Scholar]
- 11.Boon K, Osorio EC, Greenhut SF, Schaefer CF, Shoemaker J, Polyak K, Morin PJ, Buetow KH, Strausberg RL, et al. An anatomy of normal and malignant gene expression. Proc. Natl. Acad. Sci. U.S.A. 2002;99:11287–11292. doi: 10.1073/pnas.152324199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wheeler DL, Church DM, Federhen S, Lash AE, Madden TL, Pontius JU, Schuler GD, Schriml LM, Sequeira E, et al. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003;31:28–33. doi: 10.1093/nar/gkg033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lash AE, Tolstoshev CM, Wagner L, Schuler GD, Strausberg RL, Riggins GJ, Altschul SF. SAGEmap: a public gene expression resource. Genome Res. 2000;10:1051–1060. doi: 10.1101/gr.10.7.1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shannon CE. A mathematical theory of communication. Bell Syst. Tech. J. 1948;27:379–423. [Google Scholar]
- 15.Kendal WS. The use of information theory to analyze genomic changes in neoplasia. Math. Biosc. 1990;100:143–159. doi: 10.1016/0025-5564(90)90038-z. [DOI] [PubMed] [Google Scholar]
- 16.Castro MAA, Onsten TTG, de Almeida RMC, Moreira JCF. Profiling cytogenetic diversity with entropy-based karyotypic analysis. J. Theor. Biol. 2005;234:487–495. doi: 10.1016/j.jtbi.2004.12.006. [DOI] [PubMed] [Google Scholar]
- 17.Gatenby RA, Frieden BR. Information dynamics in carcinogenesis and tumor growth. Mutat. Res. 2004;568:259–273. doi: 10.1016/j.mrfmmm.2004.04.018. [DOI] [PubMed] [Google Scholar]
- 18.Castro MAA, Onsten TGH, Moreira JCF, de Almeida RMC. Chromosome aberrations in solid tumors have a stochastic nature. Mutat. Res. 2006;600:150–164. doi: 10.1016/j.mrfmmm.2006.04.003. [DOI] [PubMed] [Google Scholar]
- 19.Teichmann SA, Babu MM. Conservation of gene co-regulation in prokaryotes and eukaryotes. Trends Biotechnol. 2002;20:407–410. doi: 10.1016/s0167-7799(02)02032-2. [DOI] [PubMed] [Google Scholar]
- 20.Thellin O, Zorzi W, Lakaye B, De Borman B, Coumans B, Hennen G, Grisar T, Igout A, Heinen E. Housekeeping genes as internal standards: use and limits. J. Biotechnol. 1999;75:291–295. doi: 10.1016/s0168-1656(99)00163-7. [DOI] [PubMed] [Google Scholar]
- 21.Stuart JM, Segal E, Koller D, Kim SK. A gene-coexpression network for global discovery of conserved genetic modules. Science. 2003;302:249–255. doi: 10.1126/science.1087447. [DOI] [PubMed] [Google Scholar]
- 22.von Mering C, Jensen LJ, Snel B, Hooper SD, Krupp M, Foglierini M, Jouffre N, Huynen MA, Bork P. STRING: known and predicted protein-protein associations, integrated and transferred across organisms. Nucleic Acids Res. 2005;33:D433–D437. doi: 10.1093/nar/gki005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Mering CV, Huynen M, Jaeggi D, Schmidt S, Bork P, Snel B. STRING: a database of predicted functional\associations between proteins. Nucleic Acids Res. 2003;31:258–261. doi: 10.1093/nar/gkg034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wain HM, Lush MJ, Ducluzeau F, Khodiyar VK, Povey S. Genew: the human gene nomenclature database, 2004 updates. Nucleic Acids Res. 2004;32:D255–D257. doi: 10.1093/nar/gkh072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Birney E, Andrews D, Caccamo M, Chen Y, Clarke L, Coates G, Cox T, Cunningham F, Curwen V, et al. Ensembl 2006. Nucleic Acids Res. 2006;34:D556–D561. doi: 10.1093/nar/gkj133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hooper SD, Bork P. Medusa: a simple tool for interaction graph analysis. Bioinformatics. 2005;21:4432–4433. doi: 10.1093/bioinformatics/bti696. [DOI] [PubMed] [Google Scholar]
- 27.Zhivotovsky B, Kroemer G. Apoptosis and genomic instability. Nat. Rev. Mol. Cell Biol. 2004;5:752–762. doi: 10.1038/nrm1443. [DOI] [PubMed] [Google Scholar]
- 28.Sengupta S, Harris CC. p53: Traffic cop at the crossroads of DNA repair and recombination. Nat. Rev. Mol. Cell Biol. 2005;6:44–55. doi: 10.1038/nrm1546. [DOI] [PubMed] [Google Scholar]
- 29.Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc. Natl. Acad. Sci. U.S.A. 1999;96:424–428. doi: 10.1073/pnas.96.2.424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc. Natl. Acad. Sci. U.S.A. 2002;99:12985–12990. doi: 10.1073/pnas.202485699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rubbi CP, Milner J. p53 is a chromatin accessibility factor for nucleotide excision repair of DNA damage. EMBO J. 2003;22:975–986. doi: 10.1093/emboj/cdg082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat. Rev. Genet. 2004;5:101–113. doi: 10.1038/nrg1272. [DOI] [PubMed] [Google Scholar]
- 33.Hoogervorst EM, van Steeg H, de Vries A. Nucleotide excision repair and p53-deficient mouse models in cancer research. Mutat. Res. 2005;574:3–21. doi: 10.1016/j.mrfmmm.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 34.Jeong H, Mason SP, Barabasi AL, Oltvai ZN. Lethality and centrality in protein networks. Nature. 2001;411:41–42. doi: 10.1038/35075138. [DOI] [PubMed] [Google Scholar]
- 35.Kanehisa M, Goto S, Hattori M, Aoki-Kinoshita KF, Itoh M, Kawashima S, Katayama T, Araki M, Hirakawa M. From genomics to chemical genomics: new developments in KEGG. Nucleic Acids Res. 2006;34:D354–D357. doi: 10.1093/nar/gkj102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang ZH, Cummins JM, Shen D, Cahill DP, Jallepalli PV, Wang TL, Parsons DW, Traverso G, Awad M, et al. Three classes of genes mutated in colorectal cancers with chromosomal instability. Cancer Res. 2004;64:2998–3001. doi: 10.1158/0008-5472.can-04-0587. [DOI] [PubMed] [Google Scholar]
- 37.Wood RD, Mitchell M, Lindahl T. Human DNA repair genes. Mutat. Res. 2005;577:275–283. doi: 10.1016/j.mrfmmm.2005.03.007. [DOI] [PubMed] [Google Scholar]
- 38.Jiricny J. The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell Biol. 2006;7:335–346. doi: 10.1038/nrm1907. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.