


Abstract
Array-based technologies have been used to detect chromosomal copy number changes (aneuploidies) in the human genome. Recent studies identified numerous copy number variants (CNV) and some are common polymorphisms that may contribute to disease susceptibility. We developed, and experimentally validated, a novel computational framework (QuantiSNP) for detecting regions of copy number variation from BeadArray™ SNP genotyping data using an Objective Bayes Hidden-Markov Model (OB-HMM). Objective Bayes measures are used to set certain hyperparameters in the priors using a novel re-sampling framework to calibrate the model to a fixed Type I (false positive) error rate. Other parameters are set via maximum marginal likelihood to prior training data of known structure. QuantiSNP provides probabilistic quantification of state classifications and significantly improves the accuracy of segmental aneuploidy identification and mapping, relative to existing analytical tools (Beadstudio, Illumina), as demonstrated by validation of breakpoint boundaries. QuantiSNP identified both novel and validated CNVs. QuantiSNP was developed using BeadArray™ SNP data but it can be adapted to other platforms and we believe that the OB-HMM framework has widespread applicability in genomic research. In conclusion, QuantiSNP is a novel algorithm for high-resolution CNV/aneuploidy detection with application to clinical genetics, cancer and disease association studies.
INTRODUCTION
Several human diseases are associated with chromosomal abnormalities including germline alterations leading to developmental defects and somatic alterations leading to cancer. Originally, the diagnosis of such defects has been carried out by cytogenetic karyotype analysis using chromosome banding techniques, more recently, molecular cytogenetic analysis has been developed with advances in fluorescence in situ hybridization (FISH) based technology allowing even more refined identification of the chromosomal defects underlying the specific phenotypes. Characterization of the defects at the molecular level using classic molecular biology approaches (such as PCR, cloning, sequencing or Southern blotting hybridization) can be laborious and time consuming. Recent developments in microarray technology have allowed the study of some chromosomal aberrations with a relatively easy and high-throughput molecular biology hybridization-based approach (for review see (1)). This new approach has been called ‘molecular karyotyping’, or ‘segmental aneuploidy profiling’, a descriptive term that is in line with the lack of structural information in the data generated using microarray platforms (2). Several oligonucleotide array platforms originally developed for genotyping have also been used for copy number analysis (3–9) and oligonucleotide arrays have been specifically designed for comparative genome hybridization (CGH) applications (10,11). More recently, tiling array strategies have been successfully applied to detect copy number alterations on chromosome 22 (12). Tiling arrays offer full regional coverage and very accurate mapping however, at present multiple arrays are needed to accommodate the whole genome. There is increasing interest in the ability of SNP array platforms to detect copy number variants (CNVs), as this approach allows simultaneous profiling of copy number polymorphisms (CNPs) and SNPs, leading to a better characterization of the genetic alterations under investigation. Some of the advantages of this approach for the detection of chromosomal abnormalities have been shown for the GeneChip® technology (Affymetrix, Santa Clara, CA, USA), using 10, 100 and 500 K platforms, and a variety of statistical analysis and visualization tools have been developed for these platforms (3,4,6–9,13–17). An alternative to GeneChip® is provided by Illumina's BeadArray™ technology for high-throughput SNP genotyping, where allele-specific hybridization is coupled with primer extension (Infinium® assay) (18). The technology was further developed to use allele-specific single-base extension in a two colour labelling method (19). This modification allows the generation of more genotypes from each array, when compared to the single colour system, as a single-bead type is sufficient to represent one SNP. Furthermore, this high-throughput method couples hybridization and primer extension, thus achieving higher specificity. It has been recently demonstrated that the bead array platform using the Infinium® assay is able to detect copy number alterations (20).
Taken together, GeneChip® and BeadArray™ provide the two most widely used SNP chip platforms at the time of writing. We have developed a highly tailored Objective Bayes Hidden-Markov Model (OB-HMM) to automatically infer regions of segmental aneuploidy (copy number variation) from BeadArray™ genotyping data (QuantiSNP). We demonstrate that the Objective Bayes paradigm provides a powerful framework for model building as it affords the benefits of Bayesian marginal probability calculus (information processing) while allowing calibrated hyperparameters in the priors which ensure certain long-running (frequentist) coverage properties (for a general discussion and references on Objective Bayes, see (21,22)). In the context of our work we report on the development of a re-sampling data-driven strategy to automatically set certain prior parameters given a user defined, frequentist, false positive rate. All other parameters are set via maximum marginal likelihood matched to prior training data with known structure. In this way the OB-HMM framework allows for a formal power analysis to be undertaken. Characterization of the power of the method is vitally important in experimental design when sample sizes and end costs are being evaluated. It is also important a posteriori in qualifying the risks and costs associated with subsequent validation studies for CNVs detected by the model.
To test the algorithm, our results were compared to the mapping obtained using other cytogenetics and/or molecular genetics technologies. We showed that our method is able to produce accurate copy number detection and high-resolution breakpoint identification. The advantages of our approach are presented and discussed in comparison to the only other current software, BeadStudio LOH+ (Illumina). We believe the OB-HMM method is highly suited to the analysis of high-throughput genomic data when one of the hidden states has special status as a ‘null’ or normal state. In this case, the OB-HMM allows for setting of parameters which ensure certain frequentist coverage properties for excursions of the model out of the null state, while benefiting from Bayesian marginal inference. To our knowledge, we are the first to consider OB-HMM for genomic data analysis, and we believe the framework we have developed is well suited to many other genomic data types, including other SNP array platforms and array CGH. In previous work, several other authors have considered conventional HMM-based statistical methods to detect copy number changes using array CGH (23,24) and GeneChip® SNP array data (15,16,25,26). In addition, we present novel extensions including: the ability to combine data from several platforms of differing resolution (combining the Human-1 and HumanHap300 arrays in this case) and the ability to infer CNVs across several samples, which allows for increased precision to detect common regions of CNVs when analysing several individuals.
MATERIALS AND METHODS
Sample validation and whole genome genotyping
Fifteen samples with different cytogenetics alterations and three normal controls were used (Table 1). All experiments were performed according to the principle expressed in the Declaration of Helsinki. See Supplementary Data, Materials and Methods S1 for a detailed description of DNA extraction, high-throughput SNP genotyping using Sentrix® Human-1 Genotyping and Sentrix® HumanHap300 (Illumina, San Diego, USA) and a description of experimental validation.
Table 1.
Sample description
| Molecular cytogenetics | Reference | |||||||
|---|---|---|---|---|---|---|---|---|
| Sample ID | Chromosomal alterations | Chr. | Method | Normal marker (tel) | Del/Dup marker (tel) | Del/Dup marker (cen) | Normal marker (cen) | |
| 1 | Normal | NA | NA | NA | NA | NA | NA | NA |
| 2 | Normal | NA | NA | NA | NA | NA | NA | NA |
| 3 | Normal | NA | NA | NA | NA | NA | NA | NA |
| 4 | Deletion | 6p | FISH | NA | NA | 5 957 425 | 6 082 107 | (39) |
| 5 | Deletion | 6p | FISH | NA | NA | 6 265 901 | 7 052 829 | (36) |
| 6 | Deletion | 6p | FISH | NA | NA | 6 429 538 | 7 672 009 | (39) |
| 7 | Deletion | 6p | FISH | NA | NA | 4 157 742 | 6 939 085 | (38) |
| 8 | Deletion | 6p | FISH | NA | NA | 9 682 865 | 9 950 880 | (39) |
| 9 | Deletion | 6p | FISH | NA | NA | 6 739 542 | 7 304 962 | (40) |
| 10 | Duplication | 6p | FISH | NA | NA | 15 111 309 | 20 066 682 | NA |
| 11 | Translocation | 6p,7q | FISH | NA | NA | NA | NA | (37) |
| 12 | Translocation | 6p,9p | FISH | NA | NA | NA | NA | (35) |
| 13 | Translocation | 6p,9q | FISH | NA | NA | NA | NA | (37) |
| Molecular genetics | ||||||||
| 14 | Deletion | Xp | Sequencing | From 31 589 077 (31 589 080) to 31 743 409 (31 743 412) | NA | |||
| 15 | Duplication | 17p | MLPA | 13 445 969 | 14 051 072 | 15 148 195 | 15 548 103 | NA |
| 16 | Duplication | 6p | MLPA | 41 255 724 | 43 608 796 | 47 024 373 | 51 272 159 | NA |
| 17 | Deletion | 5q | PCR | Homozygous deletion of exon 7 and 8 of the SMN1 gene | NA | |||
| 18 | Deletion | 3p | MLPA | 342 746 | 10 051 146 | 10 166 632 | 10 194 541 | NA |
Summary of samples and chromosomal alteration as characterized with different classic technologies. (All positions are in bp on Build35; May 2004 Assembly.) NA—not applicable.
Statistical model
QuantiSNP: an Objective Bayes Hidden-Markov Model
QuantiSNP uses an OB-HMM to infer copy number variation and in the model the hidden states denote the (unknown) copy number at each SNP. The states are inferred using BeadArray™ genotyping data—in terms of log R ratios and B allele frequencies—for each SNP (Figure 1).
Figure 1.

Chromosome-wide data. Log R ratio values (top) and B allele frequencies (bottom) plotted for each SNP from one individual on chromosome 6. A deletion on the p-arm can be identified by the shift in the log R downwards and the loss-of-heterozygosity indicated by the disappearance of heterozygous state (0.5) in the B allele frequencies (as indicated by the arrows).
Transition probabilities
Table 2 lists the hidden states used in our HMM. Note that we divide the normal (diploid) state into homozygote and heterozygote sub-states to take into account regions of homozygosity since the frequency of homozygotes in heterozygous regions (2/3) differs from that in homozygous regions (1/2). We use an exponential function to define an a priori probability that some genetic event (hidden state change) occurs between adjacent SNP loci a distance d apart,
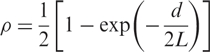 |
1 |
where L is a characteristic length which could either be inferred directly from the data, or adjusted to calibrate the model to a given false positive rate in an objective fashion (see below). The transition matrix of hidden states between adjacent SNPs i, j is given by:
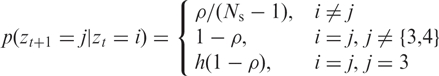 |
2 |
where h is the rate of heterozygosity which we set as 1/3 (chosen based on the mean AB frequencies given in the BeadStudio Manual), and Ns is the number of hidden states.
Table 2.
Hidden states, associated copy numbers and biological interpretation
| Hidden state, z | Copy number, c(z) | Number of genotypes, K(z) | Description |
|---|---|---|---|
| 1 | 0 | 0 | Full deletion |
| 2 | 1 | 1 | Single copy deletion |
| 3 | 2 | 3 | Normal (heterozygote) |
| 4 | 2 | 2 | Normal (homozygote) |
| 5 | 3 | 4 | Single copy duplication |
| 6 | 4 | 5 | Double copy duplication |
We associate each hidden state z with a given copy number c(z) and genotype number K(z). For each copy number there can be a number of genotypes, for example, for copy number 3 there can be one of four genotypes {AAA, AAB, ABB, BBB}. The genotype number gives the number of components in the mixture distribution of B allele frequencies for that state. We have split the diploid (copy number 2) into heterozygous and homozygous sub-states {3,4} to take into account naturally occurring regions of homozygosity without allelic loss.
Emission probabilities
Let r denote the log R ratio and b the B allele frequencies. These values are assumed to be independent given the hidden state z and all other model parameters θ. The emission probabilities are defined as a mixture of Gaussian and uniform distributions,
| 3 |
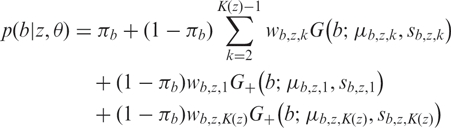 |
4 |
where Rmax = 6 is defined by the lowest observed value for the intensity R. The uniform distribution in each case acts as a non-informative state for capturing outliers in the data. As the B allele frequencies are in the range 0 < b < 1, we use half-normal distributions (G+) for the homozygous genotypes with fixed location parameters (0 and 1, respectively). The EM updating of the variance parameters is then the same as with the full normal distribution.
Hierarchical prior specification
We use standard normal-gamma conjugate priors for the emission model parameters which allows for efficient analytic integration in posterior calculations,
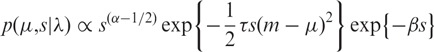 |
5 |
where τ,α,β,m are set ‘objectively’ (see below).
A Dirichlet prior is used for the B allele frequency mixture weights,
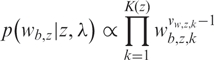 |
6 |
where we set a strong prior on equal weights vw,z,k = 10 000, since we expect the relative frequencies of each genotype to be approximately equal although some departure is allowable if there is strong evidence from the data. This also prevents mixture component weights from collapsing to zero which would cause ambiguity, for example, in the normal state there should be three components, if one component were to disappear, there would be no difference in the emission distribution from a deleted state which has two.
Beta priors are used for the outlier rates,
| 7 |
| 8 |
where we set αr = αb = βr = βb = 1 to give a uniform distribution.
Objective learning, expectation maximization(EM) and the Viterbi algorithm
Our model will be calibrated to a user-defined specificity (false positive) rate of excursions out of the normal (copy number = 2) state, however, we wish to restrict the number of prior parameters which need to be tuned in this manner. Hence, we choose to estimate most of the hyperparameters, λ = {τ, α, β, m}, via maximum marginal likelihood techniques to a reference dataset obtained from chromosome X multiple copy cell lines (20),
| 9 |
with the remaining (user specified) free parameter L in Equation (1) to be calibrated against Type I error, as described below.
Given the setting of the hyperparameters, we then use an EM algorithm to find maximum marginal a posteriori estimates for the parameters of the emission distributions.
| 10 |
The Viterbi algorithm can then be used to compute the sequence of hidden states with highest probability given the MAP parameter estimates of the emission model parameters and hyperparameter,
| 11 |
Posterior measures
Aberration events are defined as excursions of the sequence 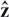 out of the normal states. For each aberration event given by the Viterbi algorithm, which spans a region from SNP i to j with copy number k, we associate with that event a Bayes Factor BF given by,
out of the normal states. For each aberration event given by the Viterbi algorithm, which spans a region from SNP i to j with copy number k, we associate with that event a Bayes Factor BF given by,
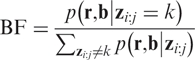 |
12 |
This posterior measure compares the evidence for the region zi:j being in hidden state k in comparison to all other sequences in which no part of this region is in this hidden state. The greater the value of BF, the more confidence we have in the event being of significance. We ignore all called events whose ratio is below a user-defined threshold.
Calibration to type I error
In order to provide calibration of our model to a fixed Type I (frequentist) error rate, we generated 100 pseudo-normal datasets for both the Human-1 and HumanHap300 SNP coverage. This was achieved by randomly sampling log R ratio and B allele frequency values from an individual assayed using the Human-1 and HumanHap300 platforms. These normal datasets allow us to quantify the false positive rates of the algorithms and calibrate the method to a user-specified rate.
We then applied QuantiSNP to each dataset to detect chromosomal aberrations. As the data is generated from samples of a normal individual, any detected aberrations will be false positive events. For various settings of the algorithm (different L and Bayes Factor thresholds), we then counted the number of false positive events. In this manner we are able to automatically define a Bayes Factor threshold and prior setting,  , which maximizes power for a given Type I error rate. (Further details in Figure 2.)
, which maximizes power for a given Type I error rate. (Further details in Figure 2.)
Figure 2.

(a) and (b) QuantiSNP is able to detect as many as 3–4 SNPs in simulated 5 SNP aberration region but only if we accept false calls rates of around 10 in 100 000 SNPs. However, in (c) and (d), when the length of the event increases to 10 SNPs, QuantiSNP successfully detects nearly all affected SNPs in the deletion and duplication events even at very stringent false call rates of less than 1 in 100 000 SNPs. In all cases, the localization of the true boundary is good, with less than one extra SNP called outside of the true aberrant region.
Joint inference on multiple samples
We can extend QuantiSNP to analyse multiple samples, either the same individual assayed several times or single samples from multiple individuals, and use the data jointly to update our transition matrix. If the regions of CNVs are common across samples, joint inference allows for borrowing of strength and improved resolution to detect CNV boundaries.
To allow for joint inference we place a Dirichlet prior on the transition matrix at each SNP, centred on the expected values given by Equation (2), and with precision K,
| 13 |
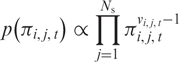 |
14 |
where vi, j, t = Kρ and ρ is given by Equation (2).
To test the effectiveness of joint updating of the transition matrices we artificially generated data for 1000 assays of a 500 SNP long chromosome containing a single 5 SNP aberrant region. We then applied QuantiSNP independently to each individual assay and then QuantiSNP, in its multi-sample analysis mode (we manually set K = 100), to the entire dataset. This procedure was repeated 100 times. We then assessed the performance by counting the number of deleted SNPs that were correctly called by the single- and multi-sample analysis modes and computing the average over the 100 iterations. Figure 3 shows the improved detection of a small deletion and duplication region shared by 1000 individuals.
Figure 3.

Multi-sample detection rates. Comparison of single-sample (red) and multi-sample analysis (blue) performance in (a,c) duplications and (b,d) deletions. In (a) and (b), the multi-sample analysis has greatly improved the detection capability of QuantiSNP for a 5 SNP duplication and deletion event respectively by increasing the number of SNPs called aberrant. In (c) and (d), the multi-sample analysis reduces the number of SNPs that are falsely called as aberrant towards zero.
RESULTS
QuantiSNP: an Objective Bayes Hidden-Markov Model for copy number variation detection
We have developed an OB-HMM approach for detecting copy number variation from BeadArray™ data (for details see the Materials and Methods section). In the model, hidden states denote the (unknown) copy number at each SNP. The states are inferred using the BeadArray™ genotyping data which comprises two signals at each SNP: (1) Log R ratios which are a measure of the magnitude of the combined fluorescent intensity signals from both sets of probes and (2) B allele frequencies which represent the relative ratio of the fluorescent signals from one allelic probe to the other. Figure 1 shows the log R ratios and B allele frequencies for one individual across chromosome 6 which includes a deletion.
The parameters of our model are learnt from the data using an EM algorithm (27) and, given these parameters, the maximum likelihood sequence of hidden states are inferred using the Viterbi algorithm (28). In our analysis, we apply the EM and Viterbi algorithms to one chromosome at a time. Identifiability of the states is maintained via our non-symmetric prior distribution structure for the B allele frequencies. The number of mixture components is conditioned on the hidden states and therefore arbitrary re-labelling is not possible. We assign a Bayes Factor to each region of copy number variation detected. This provides a probability measure of the strength of evidence from the data for the presence of a copy number variant in a region versus the null hypothesis that there is no variant. The greater the value of the Bayes Factor, the stronger the evidence for the existence of a copy number variant.
In our model, we have used fully conjugate prior distributions throughout enabling efficient analytical integration to be performed for posterior calculations. Estimates for hyperparameters in prior distributions were obtained by learning from reference datasets of chromosome X multiple copy cell lines (20), for which the copy numbers are known, using maximum marginal likelihood inference. For the remaining model parameters, we have chosen to set these ‘objectively’ in order to calibrate our model to user-specified false positive error rates.
In Bayesian inference, prior probability models are developed for unknown parameters and these prior beliefs are then updated in light of new data, using Bayes’ Rule, to give posterior probability distributions for the parameters. In a subjective Bayesian approach, prior distributions are elicited using expert knowledge or personal beliefs, and the Bayesian framework provides a powerful means by which to incorporate such information into an inference problem. In instances where little or no substantive prior knowledge is available, the Objective Bayes approach provides a principled method to set parameters of the priors; such that the resulting Bayesian procedures possess good long-run frequency properties (29) (for general discussion of Objective Bayes see (21,22)).
For our problem, a Bayes procedure with good frequency properties is particularly attractive. In copy number variation, we are principally interested in excursions into and out of the normal diploid state (or haploid for sex chromosomes) and, it is therefore natural to express interest in a frequentist property, such as the false positive rate, which tells us the long-run frequency with which we would make incorrect CNV detections. In our model, the rate of excursions (and hence our false positive error rate) is controlled by a characteristic length parameter L and a threshold value BFthresh. The greater the characteristic length, the less likely we are to make excursions into and out of the null state, and hence fewer copy number variant events will be called. Furthermore, if an excursion is made, the rate at which we accept this copy number variant is further determined by the significance we attribute to it: we only accept a copy number variant if the Bayes Factor associated with the event is greater than a threshold value BFthresh.
The selection of appropriate values or prior distributions for these parameters is very difficult. Although Jeffreys (30) (a more recent discussion is given by (31)) provides a scale for the interpretation of the Bayes Factor, this scale merely provides a descriptive statement for ease of interpretability, rather than facilitating an actual calibration. In addition, despite recent successes in mapping copy number variation in humans (8,32), the high reported false negative rates in these experiments mean that the true length distribution of copy number variants remains unknown and prevents us from adopting semi-Markov type approaches (33,34) which could exploit such knowledge. By adopting the Objective Bayes paradigm, we now have an objective by which to choose appropriate parameter values, in this case, we select parameter values that calibrate our model to given false positive error rates.
We perform the calibration using a re-sampling data-driven strategy to generate pseudo null datasets (where we know that there is no copy number variation) on which we can apply our algorithms (details given in the Materials and Methods section). The rate at which copy number variant events were detected on these simulated null datasets then provides an empirical measure of the false positive rate for different values of L and BFthresh. In addition, we have similarly estimated the power of our procedure to detect events of various sizes at different false positive rates, by re-sampling BeadArray™ data from chromosome X multiple copy cell lines with known copy numbers 1–4.
This calibration analysis gives us the ability to perform a formal power analysis of our OB-HMM method (Figure 2). We believe this to be a fundamental reversal in the normal practice of developing CNV detection algorithms, where algorithms are first developed, then applied to the experimental sample, and false positive and false negative rates subsequently inferred via independent experimental validation of detected and non-detected CNV events. In our strategy, the characteristics of the algorithms are defined before application to the experimental sample, via the calibration analysis, offering the experimentalist the capability of being able to specify the desired false positive rate suitable for their experiment a priori.
We also adapted our OB-HMM method to perform analysis on multiple samples simultaneously. If regions of copy number variation are common across individuals (perhaps due to a shared disease phenotype), the use of the data jointly allows for borrowing of strength and improved detection of copy number variants. For example, a 5 SNP CNV which is typically undetectable by QuantiSNP at stringent false positive rates can be located accurately if the same CNV region is aberrant in 1000 individuals (Figure 3).
Sample collection and characterization
We used 18 samples, including three normal controls, that were characterized using different cytogenetics and molecular genetics technologies (Table 1). Nine samples have been previously characterized using FISH (35–40), one sample containing a duplication was characterized by FISH (Mirza et al., manuscript in preparation). Five samples (No. 14–18) were characterized using molecular genetics analysis (Supplementary Data, Materials and Methods S1 for details) and for these samples the study was conducted as a blind experiment.
Infinium genotyping and BeadStudio LOH+ data analysis
We generated high-density genotyping data for the 18 samples using both Sentrix® Human-1 Genotyping (∼1 09 000 SNPs) and Sentrix® Humanhap300 (∼3 17 000 SNPs) (Illumina, San Diego, USA). After scanning, the data were uploaded into BeadStudio and analysed using the BeadStudio LOH+ module with the default window size (1.1 Mb for Human-1 and 0.46 Mb for HumanHap300), the automatic bookmark system (Version 1.0) and a p-value cutoff <0.005. In BeadStudio (version 2.3.43), data from the same Infinium® assay can be combined, but at present this is not applicable to different types of arrays, such as Infinium® I and II. Therefore we performed a parallel analysis of the two array platforms (detailed output in Supplementary Table S1).
Normal controls
One normal control (No. 1) was run and analysed three times on both the Human-1 and HumanHap300 platforms: in both cases the genotyping data were highly concordant (>99%). Despite this, the BeadStudio LOH+ analysis suggested some discordant events among the three replicates (Supplementary Table S1). With QuantiSNP, we could combine the Human-1 and HumanHap300 datasets (which is not currently possible in BeadStudio). Using a log Bayes Factor of 30 and a characteristic length of 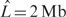 , we consistently identified two CNV events (one very small homozygous deletion on chromosome 1 and one duplication on chromosome 12) in all the three replicates of sample 1 using the combined dataset (Supplementary Table S2A). From our calibration study, we found that these settings corresponded to a false positive rate of less than one false CNV event call per 100,000 SNPs. This setting was chosen to be deliberately stringent in order to limit the number of CNV event calls made since we are unable to independently validate the existence or otherwise of large numbers of putative CNVs.
, we consistently identified two CNV events (one very small homozygous deletion on chromosome 1 and one duplication on chromosome 12) in all the three replicates of sample 1 using the combined dataset (Supplementary Table S2A). From our calibration study, we found that these settings corresponded to a false positive rate of less than one false CNV event call per 100,000 SNPs. This setting was chosen to be deliberately stringent in order to limit the number of CNV event calls made since we are unable to independently validate the existence or otherwise of large numbers of putative CNVs.
Clinical samples
Using BeadStudio LOH+ and HumanHap300 data, we identified 7/9 known deletions and 3/3 known duplications in either one or both of the array platforms in samples 4–18. As the study was conducted in a blind fashion after the analysis we realized that the sample 17 deletion (mapped by PCR) could not be identified, as there are no SNPs on the arrays that map to the deleted region on chromosome 5. In all samples, using HumanHap300 arrays, BeadStudio LOH+ discovered the validated event together with one or more unvalidated CNV events (Supplementary Table S1). Many of the additional events were mapped to chromosome X (52/105). At present, the sample sheet for the BeadStudio LOH+ module (version 2.3.41; Autobookmarking version 1.0) does not include a column to give information of the gender of the samples and therefore some of the X chromosome events may be solely due to differing copy numbers of X between genders. No new events around the translocation breakpoints were identified in cases 11,12 and 13.
QuantiSNP analysis was applied to each Human-1 and HumanHap300 datasets, and the combined dataset. Figure 4 shows two examples of the QuantiSNP analysis results output (for data visualization see Supplementary Figure S1). QuantiSNP identified 8/9 deletions (no SNP mapping on the deletion on chromosome 5 for sample 17) and 3/3 duplications in either one (1 case) or both (9 cases) array platforms, and always in the combined dataset (11 cases). In 7/11 of identified cases, (combined data analysis) the validated event was identified together with one or more unvalidated CNV events. Several of these additional events were found in more than one sample. A detailed analysis compared the additional CNVs with the Database of Genomic Variants (http://projects.tcag.ca/variation/—12th October 2006 Release) and 11/15 of these mapped to previously discovered CNV events (sample No.10 was excluded due to unusually high noise in the sample data). In Figure 5, we have plotted the average number of unvalidated CNV events detected in samples No. 2–9, 11–18 by QuantiSNP at different Bayes Factor thresholds (). This is not a true false call rate, as some of these events may be real CNVs, however, it is nonetheless a useful approximation. When compared to the false call rates derived from our simulation studies, we can see that there is good matching between the two boundaries for the Human-1 dataset. The comparison using the HumanHap300 data is less favourable, however, this is likely to be due to the different versions of the HumanHap300 used in the experiments (see Supplementary Data, Materials and Methods S1 for details). No new events around the translocation breakpoints were identified in cases 11, 12 and 13 in agreement with the BeadStudio LOH+ analysis described above. All QuantiSNP analysis was performed on a 3 GHz Pentium IV PC with 512 Mb.
Figure 4.

QuantiSNP Output. An example of output from QuantiSNP, shown are log R Ratio, B allele frequency, HMM copy number estimate and associated log Bayes Factor. (a) Sample No. 4 chromosome 6 deletion case; (b) Sample No.15 duplication on chromosome 17. In Supplementary Figure S1, the same data were visualized as a custom track in the UCSC Genome browser.
Figure 5.

Calibration of false call rates. Our false call rates obtained by simulation (red) fall within the bounds of the empirical false call rate derived from the experimental sample analysis (black). We chose for the analysis of the (a) Human-1 and (b) HumanHap300 datasets. Sample 10 was excluded from the analysis as this dataset shown unusually high levels of noise. Errors were derived from bootstrap simulations using the empirical and simulated datasets. There appears to be a good matching between the two boundaries for the Human-1 dataset. The comparison using the HumanHap300 data is less favourable, possibly due to the change in number of probes per SNP in versions of the HumanHap300 used in the experiments (see Supplementary Data, Materials and Methods S1 for details).
Accurate mapping of breakpoints
Using QuantiSNP on the combined datasets (∼400,000 SNPs) all breakpoints were mapped with high-resolution (Supplementary Table S2A). In Figure 6, we compare the performance of BeadStudio LOH+ and QuantiSNP in mapping the breakpoint using the HumanHap300 data to the data collected with other technologies (FISH, sequencing, PCR and MLPA). QuantiSNP accurately mapped 12/15 breakpoints analysed, while BeadStudio LOH+ mapping was accurate only in 6 instances. Some of the deletion/duplication events were detected and mapped in multiple segments. While this never happened (0/11) for QuantiSNP, BeadStudio LOH+ broke the deletion/duplication events in 3/11 cases analysed. Using QuantiSNP on sample No.14 (previously characterized by exon-specific PCR to harbour deletion of exons 46–50 of the DMD gene), we were able to design primers for an amplicon of predicted maximum size 7428 bp. The long-range PCR resulted in a 4627 bp fragment, that was subsequently analysed by restriction enzyme mapping allowing the sequencing across the breakpoint of a smaller PCR amplicon (Figure 7). BeadStudio LOH+ did not detect the deletion on chromosome 3p in sample No.18, while this was detected, and correctly mapped, in the combined dataset QuantiSNP analysis.
Figure 6.

Breakpoint mapping. Comparison of breakpoint mapping using BeadStudio (orange arrows) and QuantiSNP (blue arrows) on HumanHap300 data shown in context with previous data (full data in Supplementary Table S1B and S2C and Table 1, respectively). A star indicates the detection of the event in multiple fragments. The schematic image of the chromosome is not to scale: other technology defined deletion/duplication boundary is indicated in black, the deleted/duplicated area is in grey (see Table 1 for details). (a) Samples characterized by FISH (boundary mapped with a ±1 00 000 bp confidence). (b) Samples characterized by molecular genetics; sample No. 18 breakpoint was successfully identified above significance (log Bayes Factor = 37.5) in the combined data only (light blue arrows).
Figure 7.

DMD deletion mapping in Sample 14. (a) QuantiSNP output for sample 14, the chromosome X deletion is identified; (b) Sequence results across the deletion; (c) Mapping of the sequence to the genome location on chromosome X; (d) Blat results for the sequence (in panel c) and the visualization in the UCSC browser. Orange custom QuantiSNP (QS) log Bayes Factor track and in red (deletions)/green(duplications) QuantiSNP (QS) copy number (0 correspond to the normal state). RefSeq genes and SNPs present in different array platforms (including HumanHap300 labelled as ‘Illumina_300 K’) are also shown in the example.
Analysis of copy number variant coverage
To evaluate the possible use of the current Infinium®-based assays for copy number variation detection and the possible effect of CNV on our own analysis, we mapped the SNPs present in the Human-1 and/or the HumanHap300 to known copy number variants from the database of genomic variants (http://projects.tcag.ca/variation/—12th October 2006 Release). To further evaluate the assays we also mapped the SNP content for the HumanHap550, which is the combination of the HumanHap240S and HumanHap300. As expected, known copy number variation regions are underrepresented in these arrays, this is possibly due to the SNP selection process that is in favour of polymorphisms showing clear Mendelian inheritance and thus will tend to exclude SNP's mapping to CNP regions. Overall 46.47% of unique copy number variation loci (excluding inversions) do not have any SNP mapping to them in the combined data (55.65% Human-1; 47.86% HumanHap300). In the combined data from both arrays, 39.79% of the copy number variants are covered by at least five SNP on the array (25.63% Human-1; 37.34% HumanHap300) and thus it should be possible to detect them using QuantiSNP (details in Supplementary Table 3A). We also performed a detailed analysis mapping the SNPs on the arrays to all events (redundant) in the same database and this shows a similar coverage of every single event (Supplementary Table S3B).
Mapping of newly identified events to known copy number variants
As experimental validation of all the CNV events detected by QuantiSNP would be far too time consuming and costly to perform, we attempted to verify these events instead using the database for genomic variants. To perform a reliable comparison and to avoid counting an event multiple times we consolidated the additional unvalidated events, 42 deletions and 56 duplications with at least two SNPs to a non-redundant set of 68 loci (QS copy number summary custom track are available at http://www.well.ox.ac.uk/QuantiSNP) and then compared our findings to the unique loci in the database. Despite the low coverage of the platform(s) (Supplementary Table S3) and the small sample size for this kind of study, many novel events had a partial or complete overlap with events in the database. The different levels of agreement for the boundaries of the CNVs can be due to real sample-related differences and/or the genome coverage of the platform used. In summary, 37/68 loci were mapped to the database and among those 20 were nested within database loci (Supplementary Table S4A). When we analysed the additional loci found with the combined dataset and log Bayes Factor over 30 (Supplementary Table S2A), 11/15 events were found to overlap to database loci and 9 of these were nested within database loci. Several of the CNVs identified were present in more than one sample providing further support for the additional events detected. An interesting example is the duplication event on chromosome 10 found in both samples No. 2 and 8, a mother a and daughter respectively; the same CNV was not present in the father (sample No. 3) (Supplementary Figure S2 shows examples of the browser view of the CNV data). We extended the analysis to the BeadStudio LOH+ detected loci (Supplementary Table S4B and S4C). Using the HumanHap300 data, 53 unique events were identified on autosomes (another 52 events mapped on chromosome X), 48 of these novel loci mapped to the database, but only three were nested in known events. The QuantiSNP loci have a median overlap of 27.5% with database loci, meaning that in general they map to partial events, either nested within or overlapping with the variants in the database. On the contrary, the median overlaps for the Human-1 and HumanHap300 BeadStudio LOH+ analyses are 882 and 1361% respectively, showing that these events are much larger than the database loci. In fact the median size of loci identified with the combined data analysis in QuantiSNP was 294 kb, while the median size for the events identified by BeadStudio LOH+ on HumanHap300 data was 906 kb.
DISCUSSION
The development and validation of novel approaches to accurately and quickly map copy number changes in the human genome is important for the implementation of novel diagnostic strategies. Oligonucleotide array platforms originally developed for SNP genotyping have been successfully used for segmental aneuploidy profiling (4,5,7). Here we present a novel statistical algorithm that uses Objective Bayes inference for a HMM with calibrated prior parameter settings. We validated the technique using Illumina BeadArray™ SNP genotyping technology on well-characterized clinical samples. OB-HMMs resulted in the confident identification with high probability of known copy number alterations, as verified with other molecular cytogenetics and/or molecular biology techniques. Our results show the power of the QuantiSNP approach in accurately mapping breakpoints (12/15 versus only 6 for BeadStudio) (Figure 6) and demonstrate an instance where only the QuantiSNP mapping allowed the direct sequencing and subsequent definition of the breakpoint at the base-pair level (sample No.14) (Figure 7). As for the minimum size in base pairs of copy number changes that can be identified, this is limited only by the resolution and coverage of the SNPs on the arrays. Overall, the SNP array-based approach performed well in the identification of segmental aneuploidy events which makes these platforms a viable and efficient complementary technology to classic karyotyping for molecular characterization of patient samples.
Following the completion of the human genome sequence, the emphasis has shifted towards the characterization of human genetic variation and in the last few years, thanks to novel technologies, more and more structural variation events in the genome have been identified (for review see (41)). The possibility of also using a high-throughput platform for SNP typing to reliably and accurately screen for copy number variants (CNVs) in the genome is appealing. As the resolution of these approaches improves, previously uncharacterized CNV events will become easier to detect and may hinder the optimization of CNV analytical tools, in particular with regard to the control of false positive rates. Several unknown copy number changes were identified in our analysis and 11/15 mapped to CNVs identified in other studies (http://projects.tcag.ca/variation/). Even though the further validation of all novel events was beyond the scope of this study, this overlap with other studies provides circumstantial evidence of a real event which is further strengthened by the frequency with which novel events mapped within (nested) events (9/11) from other studies. Taken together this suggests that our method can accurately identify novel CNVs if SNPs map to the region of interest.
To evaluate the potential of the BeadArray™ platform for CNV mapping, we determined the coverage for different BeadArray™ platforms with regard to CNV events currently present in the public CNV database (Supplementary Table S3). It is clear that the current Infinium®-based array platforms are not offering extensive coverage of known CNVs, both in terms of the number of SNPs that map to each event and the number of events with SNPs. Despite this, a QuantiSNP analysis of the data is robust in the detection of CNV to as few as 5 SNPs per event, thus increasing the utility of these platforms for detection of up to 40% of previously identified events (Supplementary Table S3). Thanks to the potential for customization of the Infinium®-based BeadArrays™, future platforms or custom design arrays could be complemented for CNV detection by interrogating SNPs (or invariant nucleotide positions) mapping in the genomic region of interest. It is conceivable that the same approach could be used to generate BeadArrays™ with a biased distribution of oligonucleotides for CNV discovery, as well as other mapping applications (ChIP on Chip, DNAse protection assays on arrays, global methylation analysis). The great advantage of such custom design is the possibility to detect both potential CNV events and SNPs on the same high-throughput genotyping platform, thus saving both time and biological reagents.
Although we have not applied the multi-sample analysis mode of QuantiSNP to any real datasets, we believe that there is a great scope for use and development of such a technique in population and case-control studies involving large numbers of individuals. Shared copy number variant regions have already been identified in a recent study using the HapMap population (8) and a joint analysis could reveal even more common copy number polymorphisms. As the high-throughput genotyping platform market matures it is now possible to profile larger sized cohorts at ever increasing resolutions. In this environment, analytical tools for the detection of genetic variation need to accommodate increasing volumes of data while moving towards precision that is appropriate for diagnostic and clinical applications. We are also currently working on extending QuantiSNP to integrate information from multiple array platforms (Affymetrix Genechip®, BeadArray™ and oligonucleotides/BAC array CGH) to improve resolution and precision.
In addition, the Bayesian framework of QuantiSNP provides considerable flexibility for extending the model to specific applications. In cancer studies, heterogeneous samples are a common problem in which tumour samples maybe contaminated by the presence of normal gDNA. In such instances, the observed log R ratios and B allele frequencies will be a mixture of the signals due to the two sample components:
 |
15 |
where (x, y) are the intensities due to each allele and μ is the mixing proportion. It is then necessary to deconvolve the mixture by estimating the mixing proportion, which may be assumed to be constant for the whole sample, from the observed data. A strength of our method is that not only is this type of inference possible, via an extension of the observation model for the HMM, it is also possible to generate artificial heterogeneous datasets with pre-specified mixing proportions (such as in (20)) in order to estimate our false positive characteristics for different mixtures. A further feature relevant for cancer studies would be the joint analysis, which should increase the ability to identify common genomic alterations in a set of cancer samples.
We believe our approach is the first application of OB-HMM to high-throughput genomic datasets. In genomic data analysis using HMMs it is often the case that one of the hidden states carries special status as a ‘null’ or normal state. In this scenario, we believe the OB framework provides a powerful approach which allows for calibrated Type I error rates of excursions out of the null state, while affording the benefits of marginal probability calculus that defines the Bayesian approach.
A highly accurate statistical algorithm, such as QuantiSNP, for the detection of CNV events is vital for the meaningful identification of relevant copy number polymorphisms (CNPs) both in genome-wide and region-specific association studies of complex disease and to fully exploit the potential of whole genome genotyping platforms.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors would like to thank Dan Le for technical assistance and Illumina for providing the genotyping data generated in house (San Diego, USA) for both the Sentrix® Human-1 Genotyping and Sentrix® HumanHap300. Special thanks to Illumina Technical Support for considerable assistance in clarifying technical details regarding their algorithm.
This work was supported by the Wellcome Trust. C.Y. is funded by a UK Engineering and Physical Sciences Research Council Life Sciences Interface Doctoral Training Studentship. C.C.H. is partly supported by the UK Medical Research Council. Funding to pay the Open Access publication charge was provided by Wellcome Trust Grant Ref 0754/Z;Z/04/Z.
Conflict of interest statement. None declared.
REFERENCES
- 1.Speicher MR, Carter NP. The new cytogenetics: blurring the boundaries with molecular biology. Nat. Rev. Genet. 2005;6:782–792. doi: 10.1038/nrg1692. [DOI] [PubMed] [Google Scholar]
- 2.Hochstenbach R, Ploos van Amstel HK, Poot M. Microarray-based genome investigation: molecular karyotyping or segmental aneuploidy profiling? Eur. J. Hum. Genet. 2006;14:262–265. doi: 10.1038/sj.ejhg.5201553. [DOI] [PubMed] [Google Scholar]
- 3.Bignell GR, Huang J, Greshock J, Watt S, Butler A, West S, Grigorova M, Jones KW, Wei W, et al. High-resolution analysis of DNA copy number using oligonucleotide microarrays. Genome Res. 2004;14:287–295. doi: 10.1101/gr.2012304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rauch A, Ruschendorf F, Huang J, Trautmann U, Becker C, Thiel C, Jones KW, Reis A, Nurnberg P. Molecular karyotyping using an SNP array for genomewide genotyping. J. Med. Genet. 2004;41:916–922. doi: 10.1136/jmg.2004.022855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhao X, Li C, Paez JG, Chin K, Janne PA, Chen TH, Girard L, Minna J, Christiani D, et al. An integrated view of copy number and allelic alterations in the cancer genome using single nucleotide polymorphism arrays. Cancer Res. 2004;64:3060–3071. doi: 10.1158/0008-5472.can-03-3308. [DOI] [PubMed] [Google Scholar]
- 6.Herr A, Grutzmann R, Matthaei A, Artelt J, Schrock E, Rump A, Pilarsky C. High-resolution analysis of chromosomal imbalances using the Affymetrix 10K SNP genotyping chip. Genomics. 2005;85:392–400. doi: 10.1016/j.ygeno.2004.07.015. [DOI] [PubMed] [Google Scholar]
- 7.Slater HR, Bailey DK, Ren H, Cao M, Bell K, Nasioulas S, Henke R, Choo KH, Kennedy GC. High-resolution identification of chromosomal abnormalities using oligonucleotide arrays containing 1 16,204 SNPs. Am. J. Hum. Genet. 2005;77:709–726. doi: 10.1086/497343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, et al. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komura D, Shen F, Ishikawa S, Fitch KR, Chen W, Zhang J, Liu G, Ihara S, Nakamura H, et al. Genome-wide detection of human copy number variations using high-density DNA oligonucleotide arrays. Genome Res. 2006;16:1575–1584. doi: 10.1101/gr.5629106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lucito R, Healy J, Alexander J, Reiner A, Esposito D, Chi M, Rodgers L, Brady A, Sebat J, et al. Representational oligonucleotide microarray analysis: a high-resolution method to detect genome copy number variation. Genome Res. 2003;13:2291–2305. doi: 10.1101/gr.1349003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.van den Ijssel P, Tijssen M, Chin SF, Eijk P, Carvalho B, Hopmans E, Holstege H, Bangarusamy DK, Jonkers J, et al. Human and mouse oligonucleotide-based array CGH. Nucleic Acids Res. 2005;33:e192. doi: 10.1093/nar/gni191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Urban AE, Korbel JO, Selzer R, Richmond T, Hacker A, Popescu GV, Cubells JF, Green R, Emanuel BS, et al. High-resolution mapping of DNA copy alterations in human chromosome 22 using high-density tiling oligonucleotide arrays. Proc. Natl. Acad. Sci. U.S.A. 2006;103:4534–4539. doi: 10.1073/pnas.0511340103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nannya Y, Sanada M, Nakazaki K, Hosoya N, Wang L, Hangaishi A, Kurokawa M, Chiba S, Bailey DK, et al. A robust algorithm for copy number detection using high-density oligonucleotide single nucleotide polymorphism genotyping arrays. Cancer Res. 2005;65:6071–6079. doi: 10.1158/0008-5472.CAN-05-0465. [DOI] [PubMed] [Google Scholar]
- 14.Huang J, Wei W, Chen J, Zhang J, Liu G, Di X, Mei R, Ishikawa S, Aburatani H, et al. CARAT: a novel method for allelic detection of DNA copy number changes using high density oligonucleotide arrays. BMC Bioinformatics. 2006;7:83. doi: 10.1186/1471-2105-7-83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao X, Weir BA, LaFramboise T, Lin M, Beroukhim R, Garraway L, Beheshti J, Lee JC, Naoki K, et al. Homozygous deletions and chromosome amplifications in human lung carcinomas revealed by single nucleotide polymorphism array analysis. Cancer Res. 2005;65:5561–5570. doi: 10.1158/0008-5472.CAN-04-4603. [DOI] [PubMed] [Google Scholar]
- 16.Beroukhim R, Lin M, Park Y, Hao K, Zhao X, Garraway LA, Fox EA, Hochberg EP, Mellinghoff IK, et al. Inferring loss-of-heterozygosity from unpaired tumors using high-density oligonucleotide SNP arrays. PLoS Comput. Biol. 2006;2:e41. doi: 10.1371/journal.pcbi.0020041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ting JC, Ye Y, Thomas GH, Ruczinski I, Pevsner J. Analysis and visualization of chromosomal abnormalities in SNP data with SNPscan. BMC Bioinformatics. 2006;7:25. doi: 10.1186/1471-2105-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gunderson KL, Steemers FJ, Lee G, Mendoza LG, Chee MS. A genome-wide scalable SNP genotyping assay using microarray technology. Nat. Genet. 2005;37:549–554. doi: 10.1038/ng1547. [DOI] [PubMed] [Google Scholar]
- 19.Steemers FJ, Chang W, Lee G, Barker DL, Shen R, Gunderson KL. Whole-genome genotyping with the single-base extension assay. Nat. Methods. 2006;3:31–33. doi: 10.1038/nmeth842. [DOI] [PubMed] [Google Scholar]
- 20.Peiffer DA, Le JM, Steemers FJ, Chang W, Jenniges T, Garcia F, Haden K, Li J, Shaw CA, et al. High-resolution genomic profiling of chromosomal aberrations using Infinium whole-genome genotyping. Genome Res. 2006;16:1136–1148. doi: 10.1101/gr.5402306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Berger JO. The case for objective Bayesian analysis. Bayesian Analysis. 2004;1:1–17. [Google Scholar]
- 22.Bayarri MJ, Berger JO. The interplay of bayesian and frequentist analysis. Statist. Sci. 2004;19:58–80. [Google Scholar]
- 23.Shah SP, Xuan X, DeLeeuw RJ, Khojasteh M, Lam WL, Ng R, Murphy KP. Integrating copy number polymorphisms into array CGH analysis using a robust HMM. Bioinformatics. 2006;22:431–439. doi: 10.1093/bioinformatics/btl238. [DOI] [PubMed] [Google Scholar]
- 24.Marioni JC, Thorne NP, Tavare S. BioHMM: a heterogeneous hidden Markov model for segmenting array CGH data. Bioinformatics. 2006;22:1144–1146. doi: 10.1093/bioinformatics/btl089. [DOI] [PubMed] [Google Scholar]
- 25.LaFramboise T, Weir BA, Zhao X, Beroukhim R, Li C, Harrington D, Sellers WR, Meyerson M. Allele-specific amplification in cancer revealed by SNP array analysis. PLoS Comput. Biol. 2005;1:e65. doi: 10.1371/journal.pcbi.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Laframboise T, Harrington D, Weir BA. Biostatistics. 2006. PLASQ: a generalized linear model-based procedure to determine allelic dosage in cancer cells from SNP array data. Jun20 [epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 27.Dempster AP, Laird NM, Rubin DB. Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B. 1977;39:1–38. [Google Scholar]
- 28.Rabiner LR. A tutorial on hidden markov models and selected applications in speech recognition. Proc. IEEE. 1989;72:257–286. [Google Scholar]
- 29.Wasserman L. Frequentist Bayes is objective (comment on articles by Berger and by Goldstein) Bayesian Analysis. 2006;1:451–456. (electronic) [Google Scholar]
- 30.Jeffreys H. Theory of Probability. 3rd edn. Oxford, UK: Oxford University Press; 1961. [Google Scholar]
- 31.Kass RE, Raftery AE. Bayes Factors. J. Am. Stat. Assoc. 1995;90:773–795. [Google Scholar]
- 32.Wong KK, deLeeuw RJ, Dosanjh NS, Kimm LR, Cheng Z, Horsman DE, MacAulay C, Ng RT, Brown CJ, et al. A comprehensive analysis of common copy-number variations in the human genome. Am. J. Hum. Genet. 2007;80:91–104. doi: 10.1086/510560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ferguson JD. Variable duration models for speech. Proc. Symp. On the Application of HMMs to Text and Speech; 1980. pp. 143–179. [Google Scholar]
- 34.Levinson SE. Continuously variable duration hidden Markov models for automatic speech recognition. Computer Speech and Technology. 1986;1:29–45. [Google Scholar]
- 35.Davies AF, Imaizumi K, Mirza G, Stephens RS, Kuroki Y, Matsuno M, Ragoussis J. Further evidence for the involvement of human chromosome 6p24 in the aetiology of orofacial clefting. J. Med. Genet. 1998;35:857–861. doi: 10.1136/jmg.35.10.857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Davies AF, Mirza G, Sekhon G, Turnpenny P, Leroy F, Speleman F, Law C, van Regemorter N, Vamos E, et al. Delineation of two distinct 6p deletion syndromes. Hum. Genet. 1999;104:64–72. doi: 10.1007/s004390050911. [DOI] [PubMed] [Google Scholar]
- 37.Davies AF, Stephens RJ, Olavesen MG, Heather L, Dixon MJ, Magee A, Flinter F, Ragoussis J. Evidence of a locus for orofacial clefting on human chromosome 6p24 and STS content map of the region. Hum. Mol. Genet. 1995;4:121–128. doi: 10.1093/hmg/4.1.121. [DOI] [PubMed] [Google Scholar]
- 38.Law CJ, Fisher AM, Temple IK. Distal 6p deletion syndrome: a report of a case with anterior chamber eye anomaly and review of published reports. J. Med. Genet. 1998;35:685–689. doi: 10.1136/jmg.35.8.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mirza G, Williams RR, Mohammed S, Clark R, Newbury-Ecob R, Baldinger S, Flinter F, Ragoussis J. Refined genotype-phenotype correlations in cases of chromosome 6p deletion syndromes. Eur. J. Hum. Genet. 2004;12:718–728. doi: 10.1038/sj.ejhg.5201194. [DOI] [PubMed] [Google Scholar]
- 40.Caluseriu O, Mirza G, Ragoussis J, Chow EW, MacCrimmon D, Bassett AS. Schizophrenia in an adult with 6p25 deletion syndrome. Am. J. Med. Genet. A. 2006;140:1208–1213. doi: 10.1002/ajmg.a.31222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat. Rev. Genet. 2006;7:85–97. doi: 10.1038/nrg1767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}