

Abstract
The antitumour antibiotic mithramycin A (MTA) is a DNA minor-groove binding ligand. It binds to C/G-rich tracts as a dimer that forms in the presence of divalent cations such as Mg2+. Differential scanning calorimetry, UV thermal denaturation, isothermal titration calorimetry and competition dialysis were used, together with computations of the hydrophobic free energy of binding, to determine the thermodynamic profile of MTA binding to DNA. The results were compared to those obtained in parallel using the structurally related mithramycin SK (MSK). The binding of MTA to salmon testes DNA determined by UV melting studies (Kobs = 1.2 (±0.3) × 105 M−1) is tighter than that of MSK (2.9 (±1.0) × 104 M−1) at 25°C. Competition dialysis studies showed a tighter MTA binding to both salmon testes DNA (42% C + G) and Micrococcus lysodeikticus DNA (72% C + G). The thermodynamic analysis of binding data at 25°C shows that the binding of MTA and MSK to DNA is entropically driven, dominated by the hydrophobic transfer of the antibiotics from solution to the DNA-binding site. Direct molecular recognition between MTA or MSK and DNA through hydrogen bonding and van der Waals contacts may also contribute significantly to complex formation.
INTRODUCTION
Mithramycin A (MTA), also known as plicamycin, is an antitumour antibiotic used clinically in the treatment of Paget's disease and testicular carcinoma (1), and it has gained renewed attention as a therapeutic agent in both cancer- and non-cancer-related pathologies (2,3). MTA (Figure 1) and the structurally related chromomycin A3 and olivomycin are members of the aureolic group of antitumour antibiotics. All contain the same tricyclic core moiety, with a unique dihydroxy-methoxy-oxo-pentyl side chain attached at C-3. They vary only slightly with respect to the residue at C-7, which can be either an H atom or a small alkyl side chain. However, these antibiotics differ in the nature of their saccharide chains, which consist of several 2,6-dideoxysugar residues (4,5). The mithramycin biosynthetic pathways have been almost completely elucidated (6–9). Mithramycin SK (MSK) is a secondary metabolite of MTA, which bears a butyl instead of a pentyl chain, and the functional keto and alcohol groups are in different positions (Figure 1). MSK has been generated by insertional inactivation of gene mtmW in Streptomyces argillaceus (8). It has been tested in vitro against several human cancer cell lines, showing an improved therapeutic index compared to MTA (8), in addition to inhibiting transcription of several genes (10).
Figure 1.

Structural formulae of mithramycin A and mithramycin SK.
The primary cellular target of these molecules is DNA, while bivalent cations such as Mg2+ are an essential requirement for their binding to DNA (11–14), which occurs along the minor groove of C/G-rich tracts (4,12,13,15,16). Mg2+-coordinated MTA dimers bind in the minor groove of DNA, with the chromophores parallel to the sugar-phosphate backbone and the saccharide chains partially wrapping the DNA minor groove (16). Equilibrium and kinetic studies indicate that MTA forms two types of complex with magnesium, which are referred to as Complex I (with 1:1 MTA:Mg2+ stoichiometry) and Complex II (2:1 MTA:Mg2+ stoichiometry) (12,17). In Complex II, the metal ion binds to the two oxygen atoms of each chromophore, while two water molecules are involved as the fifth and sixth ligands (14). Upon binding to DNA, the chromophores form hydrogen bonds with the NH2 of guanines, thus determining the selectivity for C/G-rich sequences (4,14,16). Because of this sequence selectivity, MTA blocks the binding of the Sp1 family of transcription factors to C/G-rich sequences in gene promoters and inhibits gene transcription (10,18,19), which in turn alters the regulation of cell proliferation and differentiation (10,19–21).
Here, we present the first attempt at direct thermodynamic characterization of binding in the minor groove of C/G-regions of DNA without intercalation, using direct calorimetric measurements of the interaction of MTA and MSK with DNA. To date, only the binding in the minor groove of AT-rich DNA has been thermodynamically characterized in detail (22–25), while there are some grounds to consider that binding to DNAs of different nucleotide composition might result in changes in the enthalpy and entropy components of binding (26). Hence, a thermodynamic comparison of the binding of minor-groove ligands to different base-pairs is still required. Furthermore, the binding of MTA and MSK is challenging because of the formation of a drug-dication complex (17), which is required for the interaction with DNA. We report direct measurements of the enthalpy of binding to salmon testes DNA, which allowed us to calculate the entropic and enthalpic contributions to the free energy of binding. For MTA, using isothermal titration calorimetry (ITC), the binding enthalpy and the apparent binding constant can be directly determined without invoking the van't Hoff relationship. We have characterized the hydrophobic component of binding (ΔGhyd) for both mithramycins by using a calculation of the solvent-accessible area in mithramycin–DNA complexes, since it was not feasible to measure any heat capacity (ΔCp) effect experimentally. Finally, competition dialysis, which provides a simple and direct quantitative measure of sequence selectivity (27), was used to characterize the differences in the binding of MTA and MSK to DNAs of various nucleotide compositions. We aimed to extend our understanding of the binding in the minor groove of C/G-rich DNAs, which may ultimately be of pharmacological interest in the pursuit of enhancing the DNA-binding capabilities of synthetic molecules.
MATERIALS AND METHODS
Materials
MTA and its secondary metabolite MSK (Figure 1) were isolated and purified from the producing organisms as described previously (7,8). Stocks of MTA or MSK were prepared as 3 mM solutions in 20 mM Hepes (pH 7.4) supplemented with NaCl to obtain the final Na+ concentrations used in the experiments. Except where otherwise indicated, the buffer used throughout (designated HPS buffer) contained 150 mM NaCl, 20 mM Hepes (pH 7.4). Molar extinction coefficients of 10 000 M−1 cm−1 at 400 nm and of 10 600 M-1 cm-1 – at 420 nm were used to determine the concentration of MTA and MSK, respectively (8,12).
Salmon testes DNA (Sigma) and Micrococcus lysodeikticus DNA (Sigma) were dissolved in 10 mM NaCl, 20 mM Hepes (pH 7.4), sonicated, phenol extracted twice and extensively dialysed against 20 mM Hepes (pH 7.4) containing various NaCl concentrations. Poly[d(G-C)2] (Amersham Biosciences) and poly[d(I-C)2] (Roche) were dissolved in HPS buffer and extensively dialysed against the same buffer.
DNA concentrations, in base pairs, were determined spectrophotometrically by using the following molar extinction coefficient (M−1 cm−1) values (28): ε260(salmon testes DNA) = 12 824, ε260(M. lysodeikticus DNA) = 13 846, ε254(poly[d(G-C)2]) = 16 800 and ε251(poly[d(I-C)2]) = 13 800.
Continuous variation binding analysis
The stoichiometry for the binding of MTA and MSK to salmon testes DNA was obtained in HPS buffer using the method of continuous variation (29). The concentrations of either MTA or MSK and DNA were each varied, while the sum of the concentrations was kept constant at 40 μM. Varying volumes of equally concentrated stock solutions of the antibiotics and DNA were mixed to give a mole fraction of ligands ranging from 0 to 0.3. The MgCl2 concentration was double the antibiotic concentrations in all the experiments to ensure the formation of Mg2+-coordinated drug dimers (2:1 MTA:Mg2+ stoichiometry) (12). The difference in absorbance (ΔA400 nm for MTA or ΔA420 nm for MSK) was plotted against the molar fraction of antibiotic.
Ultraviolet melting studies and differential scanning calorimetry
Ultraviolet (UV) DNA melting curves were determined using a Jasco V-550 spectrophotometer (Jasco), equipped with an ETC-505T (Jasco) temperature controller. Sonicated salmon testes DNA in 20 mM Hepes (pH 7.4) was used for melting studies, in the presence of various concentrations of NaCl, ranging from 10 to 150 mM. Samples contained 20 μM (bp) DNA, 7 μM MTA or MSK (a concentration enough to saturate the nucleic acid) and 14 μM MgCl2 to obtain Mg2+-coordinated drug dimers (12). The samples were equilibrated at 25°C for 30 min and then heated at a rate of 1°C min−1, while continuously monitoring the absorbance at 260 nm. The Spectra Manager software (Jasco) was used to analyse the data and to calculate the DNA melting temperature (Tm).
Differential scanning calorimetry (DSC) measurements were performed using a MicroCal MC-2 Scanning Calorimeter. Sonicated salmon testes DNA was dissolved in HPS buffer, and extensively dialysed against the same buffer. Samples of MTA or MSK for DSC were prepared by mixing appropriate solutions of either antibiotic and MgCl2 to a 1:2 molar ratio in HPS buffer. In a standard experiment, the antibiotic–DNA complexes consisted of 0.15 mM antibiotic, 0.30 mM MgCl2 (i.e. a pre-formed Mg2+-coordinated antibiotic dimer) and 0.7 mM DNA. After equilibration for 2–3 h in the dark, any undissolved antibiotic was removed by low-speed centrifugation. The amount of drug bound to the DNA was determined spectrophotometrically (12). A heating rate of 0.5°C min−1 was used. Primary data were corrected by subtraction of a buffer–buffer baseline, normalized to the concentration of DNA (in bp), and further baseline-corrected. Data acquisition (excess heat capacity versus temperature) and analysis were performed using the Origin software (MicroCal).
The DSC results and UV melting studies were used to calculate the binding constant of MTA and MSK binding to salmon testes DNA at the DNA melting temperature (KTm), using the equation derived by Crothers (30):
| (1) |
where 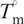 (kelvin) is the UV melting temperature of salmon testes DNA alone, Tm (kelvin) is the melting temperature in the presence of saturating amounts of the drug, ΔHwc is the enthalpy of DNA melting obtained by DSC, R is the gas constant, KTm is the drug binding constant at Tm, a is the free drug activity, which is estimated by one-half of the total drug concentration and n is the size of the drug-binding site determined by continuous variation analysis, as described above.
(kelvin) is the UV melting temperature of salmon testes DNA alone, Tm (kelvin) is the melting temperature in the presence of saturating amounts of the drug, ΔHwc is the enthalpy of DNA melting obtained by DSC, R is the gas constant, KTm is the drug binding constant at Tm, a is the free drug activity, which is estimated by one-half of the total drug concentration and n is the size of the drug-binding site determined by continuous variation analysis, as described above.
The calculated apparent binding constant at Tm can be extrapolated to lower temperatures by use of the van't Hoff equation:
| (2) |
where K is the DNA binding constant of MTA or MSK at temperature T (kelvin) and ΔHb is the enthalpy of binding of the antibiotics to DNA determined by DSC.
The experimental Kobs, calculated at 25°C, and the binding enthalpy for MTA and MSK binding to DNA allowed us to obtain complete thermodynamic profiles. Free energies were calculated from the standard relation ΔG°=−RT ln Kobs and the entropy was evaluated using the standard thermodynamic relationship: ΔS°= (ΔH°− ΔG°)/T.
Salt dependence of the binding constant
Differences between the DNA melting temperatures of DNA alone and in the presence of saturating amounts of MTA or MSK at different NaCl concentrations, in the presence of MgCl2, were used to calculate DNA binding constants for MTA and MSK at the melting temperature. These DNA binding constants were corrected to 25°C by application of Equation (2), assuming that the binding enthalpy is independent of salt concentration. These data can be analysed using the polyelectrolyte theory of Record et al. (31) considering:
| (3) |
The Zψ value is equivalent to the number of counterions released upon binding, which can be used to evaluate the polyelectrolyte contribution to the free energy of binding to salmon testes DNA using the relationship:
| (4) |
and the ΔGpe values were used to calculate ΔGt: the ‘non-polyelectrolyte contribution to binding’ using the following equation:
| (5) |
Isothermal titration calorimetry
Experiments were carried out at 25°C using a MicroCal MCS-ITC calorimeter, and the Origin 2.9 software (MicroCal) was used for data acquisition and analysis. In a typical experiment, 1.34 ml of 0.50 mM (bp) DNA was titrated using a 2 mM antibiotic solution in HPS buffer, containing 4 mM MgCl2 (40 injections of 5 μl each), using a 250 μl syringe rotating at 400 r.p.m. The injection time was 12 s, and the delay between injections was 4 min. The peaks produced during titration were converted to heat output per injection by integration and correction for the cell volume and sample concentration. Heat of dilution corrections were performed for the injections of Mg2+–antibiotic complex into buffer. Moreover, we titrated MgCl2 into DNA, both prepared as solutions in HPS buffer to measure the heat of dilution produced by magnesium, in amounts equivalent to those used in the presence of the antibiotics {i.e. the amount of magnesium needed to obtain dimers of the antibiotics [Complex II, (12)]}. Under these experimental conditions the final Mg2+ concentration was 0.5 mM. We also measured the heat change induced in the formation of Mg2+–antibiotic complexes by the titration of a 1.5 mM antibiotic solution in HPS buffer into a 0.5 mM MgCl2 solution in the same buffer.
Binding enthalpy (ΔH), the stoichiometry (n) and the binding constant (K) were obtained by fitting the corrected data to a model incorporated in the Origin 2.9 software, which considers a single set of identical binding sites.
Calculation of solvent-accessible surface areas
We applied the method described by Haq et al. (32) to estimate the hydrophobic contribution (ΔGhyd) to the drug–DNA-binding free energy from the change in solvent-accessible surface area upon complex formation. To this end, we used the coordinates from PDB entry 146D (16) for the DNA–MTA complex. In this structure, a Mg2+-coordinated MTA dimer is bound to the d(TCGCGA) duplex DNA sequence. For MSK, a model was built manually from the structure of MTA in this complex. Force-field parameters for MTA and MSK were obtained from the PRODRG2 server and adapted to the Amber-99 force field in GROMACS (33). Parameters for the DNA part of the complex were obtained for the same force field directly from GROMACS. An ideal B-DNA model of the same sequence was generated using the 3DNA program (34) to account for the unbound structure. The free structures of MTA and MSK were regarded as their Mg2+-coordinated dimers. Models of complexes and free molecules were subjected to molecular mechanics energy minimization in GROMACS, using the steepest descent method. The resulting coordinates were used for the energy calculations described below.
Accessible surface areas were computed using the analytical method within the NACCESS 2.1.1 program (35). The van der Waals atomic radii used were those defined by Chothia (36) and the probe radius was 1.4 Å. Surfaces of carbon, carbon-bound hydrogen and phosphorus atoms were defined as non-polar, whereas those of all other atoms were considered as polar (32).
The change in solvent-accessible surface area (ΔSASA) on binding is the difference between the area of a given complex and the sum of the surface areas of the drug-free native duplex and the ligand molecule separated from each complex. Heat capacity change (ΔCp) values were calculated using the relationship (37):
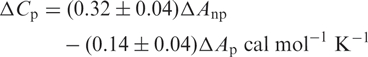 |
where Anp and Ap are the binding-induced accessible surface area changes, in units of Å2, in non-polar and polar surfaces area, respectively. From these values, we estimated the hydrophobic contribution to the antibiotic–DNA-binding free energy using the empirical relationship (38):
Competition dialysis assays
Competition dialysis assays were carried out as described elsewhere (27), with minor modifications. The HPS buffer was used for all experiments. A volume of 0.2 ml (75 μM in bp) of each of the DNA samples, described above, was introduced into separate Slice-A-Lyzer mini dialysis units (Pierce, Rockford, IL). For each competition dialysis assay, all the dialysis units were placed in a beaker containing the dialysate solution, consisting of 200 ml of 1 μM ligand and 4 μM MgCl2 in the same buffer. The dialysis was allowed to equilibrate, with continuous stirring, for 24 h at 25 ± 1°C in the dark. At the end of the equilibration period, DNA samples were removed to microfuge tubes, and were taken to a final concentration of 1% (w/v) sodium dodecyl sulfate (SDS). The total concentration of drug (Ct) within each dialysis unit was then determined spectrophotometrically using wavelengths and extinction coefficients appropriate for MTA and MSK (see above). The free ligand concentration (Cf), which did not vary appreciably from the initial concentration of 1 μM, was determined spectrophotometrically using an aliquot of the dialysate solution. The amount of bound drug was determined by difference: Cb = Ct − Cf.
The apparent binding constant for any DNA can be calculated from the competition dialysis assays (39) by using the following relationship:
where Cb is the drug bound, Cf is the free drug concentration (maintained at 1 μM in the assay) and Ct is the total nucleic acid concentration (75 μM in the assay protocol).
RESULTS
Using the method of continuous variation (Job plots) we determined that the binding of MTA or MSK to salmon testes DNA corresponds approximately to 1:5 (antibiotic:DNA in bp) stoichiometry (Figure S1), in agreement with previous NMR results that showed MTA binding to about 5–6 bp in the minor groove of a double-strand oligonucleotide (4,13).
Determination of the DNA binding constant of MTA and MSK by UV melting studies
UV melting studies and DSC were used to determine the binding constant for the interaction of MTA and MSK with salmon testes DNA. In the absence of either antibiotic, the Tm of salmon testes DNA was 88.4°C. In the presence of a concentration of MTA or MSK sufficient to saturate the DNA lattice, the Tm were 91.0 and 89.5°C, respectively (Figure S2). The enthalpy of DNA melting in the absence of ligand (ΔHm) was determined by DSC to be 8.60 (±0.31) kcal mol−1 (bp) (Figure 2). These values were used to determine the DNA binding constant at the melting temperature by using Equation (1), yielding KTm = 1.5 × 105 M- 1 for MTA and 5.7 × 104 M−1 for MSK. Correction of these values to lower temperature requires knowledge of the binding enthalpy, which we also determined by DSC, as follows.
Figure 2.

Differential scanning calorimetry analysis of MTA and MSK complexes with salmon testes DNA. Melting curves for DNA alone and identical DNA solution in the presence of saturating amounts of MTA or MSK (0.2 mol antibiotic/mol DNA (bp)). The plots show the excess of heat capacity (kcal mol−1 K−1) as a function of temperature. The area under the peaks provides direct estimates of the enthalpy for DNA melting and the MTA–DNA and MSK–DNA complexes.
Figure 2 shows the results of DSC experiments using salmon testes DNA in the presence and absence of saturating amounts of MTA or MSK, in the presence of MgCl2 (a Mg2+–antibiotic complex, see Materials and methods section). By Hess's law, these data may be used to determine the enthalpy of ligand binding to DNA as described elsewhere (40,41). From three independent determinations, we obtained a ΔHb of +1.60 ± 0.32 kcal mol−1for the association of MTA to DNA, while the ΔHb for the association of MSK was +1.02 ± 0.33 kcal mol−1. In both cases, enthalpy values refer to the temperature of their respective Tm.
Once the binding enthalpy was determined, we calculated, by application of Equation (2), the DNA binding constant (Kobs) of MTA and MSK at 25°C to be 1.2 (±0.3) × 105 M−1 and 2.9 (±1.0) × 104 M−1, respectively. Knowledge of the binding constant and binding enthalpy allowed us to construct and compare the complete thermodynamic profiles (ΔG, ΔH and ΔS) for the binding of MTA and MSK to salmon testes DNA (Table 1). The sign of the thermodynamics parameters indicates unambiguously that binding of MTA and MSK to DNA was entropically driven (Table 1).
Table 1.
Comparison of thermodynamic parameters for mithramycin A (MTA) and mithramycin SK (MSK) binding to salmon testes DNAa
| Kobs (M−1) | ΔGobs (kcal mol−1) | ΔGpe (kcal mol−1) | ΔGt (kcal mol−1) | ΔH (kcal mol−1) | TΔS (kcal mol−1) | |
|---|---|---|---|---|---|---|
| MTA | 1.2 (±0.3) × 105 | −6.92 | −0.39 | −6.53 | 1.60 (±0.32) | 8.52 |
| MSK | 2.9 (±1.0) × 104 | −6.07 | −0.42 | −5.65 | 1.02 (±0.33) | 7.09 |
Enthalpy values were determined by DSC (means ± SD, from three independent experiments). aKobs (M−1) is the binding constant for the interaction of the antibiotics with salmon testes DNA in 170 mM Na+ at 25°C. ΔGobs is the binding free energy calculated from ΔGobs = −RT lnK. ΔGpe and ΔGt are the polyelectrolyte and non-polyelectrolyte contributions to the binding free energy, evaluated at 170 mM Na+.
Differences between the DNA melting temperatures of DNA alone and in the presence of saturating amounts of MTA or MSK at different millimolar concentrations of NaCl (Figure S2) were used to calculate DNA binding constants at various ionic strengths, at 25°C, as described above. Figure 3 shows the dependence of the binding constant at 25°C on the concentration of Na+. The binding constants for both antibiotics decreased slightly with increasing salt concentrations. In the presence of 30 mM Na+, the binding constants calculated for MTA and MSK at 25°C were 2.4 (±0.5) × 105 M−1 and 5.7 (±1.3) × 104 M−1 respectively, which were, as expected, higher than those in the presence of 170 mM Na+. These salt-dependent data for minor-groove-binding ligands can be analysed using the polyelectrolyte theory of Record et al. (31). From that theory, the slopes of the lines in Figure 3 are equivalent to the number of Na+ counterions released upon binding of a ligand. For MTA and MSK, we found that 0.37 and 0.40 counterions were liberated respectively from DNA upon binding of each Mg2+-coordinated antibiotic dimer. The polyelectrolyte contributions to the free energy of MTA or MSK binding to DNA were then calculated using these values and Equation (4). At the 170 mM Na+ concentration (HPS buffer) used in our experiments, we calculated that ΔGpe was −0.39 kcal mol−1 for MTA and −0.42 kcal mol−1 for MSK. These are rather modest contributions to the free energy of binding (Table 1). The pKa of MTA is 5.0 (12), so at pH 7.4 it theoretically bears a −0.99 charge. However, these calculations corresponded to free MTA, while a crystallographic analysis of DNA–chromomycin structure revealed that the Mg2+-coordinated antibiotic dimer has no electric charge (chromomycin bears the same tricyclic core moiety as MTA and MSK) (14). For the binding of an uncharged molecule to DNA, we may expect the absence of a polyelectrolyte effect (42). Nevertheless, we observed such an effect experimentally (Figure 3). An explanation to this apparent discrepancy will be presented in the Discussion section.
Figure 3.

Salt dependence of MTA and MSK binding constants at 25°C (Record's plot (31)). The linear least-squares fit of the data yields slopes of 0.37 (MTA) and 0.40 (MSK), which are equivalent to the number of ions released upon binding of MTA or MSK to DNA.
Isothermal titration calorimetry analysis of MTA binding to DNA
ITC was used to quantify the binding of an MTA–Mg2+ complex to salmon testes DNA (Figure 4). An MTA–Mg2+ complex was pre-formed in the syringe before titration. Integration with respect to time of the heat produced per serial injection of ligand to the DNA duplex yields the corresponding isotherm (Figure 4A). These data require correction for the heat associated with the addition of ligand to buffer and of buffer to DNA, the latter being negligible. Figure 4B shows that adding Mg2+ ions to a DNA solution was associated with quite a low heat of dilution (about −60 cal mol−1). The titration of Mg2+-coordinated MTA into buffer produced a curve (open squares in Figure 4C) in which the heat signal was dependent on the injected MTA concentration up to 130 μM MTA. This MTA concentration was equivalent to that used to obtain an antibiotic-to-DNA ratio of ∼0.25 in a titration experiment on DNA, while the heat signal remained almost constant afterwards. Although the signal dependence on ligand concentration has been considered a consequence of self-association (32), the titration of MTA on buffer (Figure 4C) can indicate that the heat change reflects a composite effect of the drug dilution after its injection in the large cell together with the re-formation of Mg2+-coordinated MTA dimers. This explanation is consistent with the presence of a plateau in the titration, because this would be the sign once the dimers were re-formed, after dilution in the larger cell volume (compared to that of the syringe), the heat of dilution remained almost constant (Figure 4C). Furthermore, we carried out other titration experiments to monitor the formation of MTA–DNA complexes mediated by Mg2+. For example, Figure S3 shows the results of a representative experiment, in which an MTA solution in HPS buffer was titrated into an MgCl2 solution in the same buffer (see the legend of Figure S3). Several inflections were observed during the ITC titration, which depended on the MTA:Mg2+ molar ratio. The heats per injectant became progressively more positive (Figure S3), changing from −1.6 kcal mol−1 at a magnesium-to-DNA ratio of 1:4, to -1.2 kcal mol−1 at a ratio of 1:2, which corresponds to the formation of Mg+2-coordinated MTA dimers. Nevertheless, we cannot rule out that the heat values obtained in these titrations were also indicative of microaggregation of the antibiotic.
Figure 4.

Isothermal titration curves of MTA binding to salmon testes DNA at 25°C. (A) Titration of a Mg2+–MTA complex in HPS buffer (pre-formed in the syringe of the ITC apparatus) into DNA in the same buffer solution without added Mg2+. ITC raw data are shown in the top panel, while the lower panel shows the uncorrected binding isotherm. (B) Heat of dilution of an MgCl2 solution titrated into a DNA solution in HPS buffer. The distribution of the heats of dilution of Mg2+ corresponds to a ΔH ∼ −60 cal mol−1. (C) Heat change induced in an ITC experiment by adding Mg2+–MTA complexes into buffer in the absence of DNA (open squares). The panel also shows the uncorrected isothermal titration curve of the titration of Mg2+-MTA complex into DNA (black squares) redrawn from (A). (D) Corrected binding isotherm resulting from integration with respect to time, with correction for the measurements presented in (B and C) (see also Figure S3). The heats obtained for each injection in (C) were subtracted from the corresponding raw value (open squares) in the same panel.
To obtain corrected binding isotherms (Figure 4D), we subtracted each value obtained in the experiments shown in Figure 4C from the raw MTA–DNA data (Figure 4A). The fitting of the corrected binding isotherm to a single set of binding sites (see Materials and methods) was used to obtain standard thermodynamic parameters at 25°C: ΔH = +2.62 ± 0.11 kcal mol−1, Kobs = 7.4 (±1.3) × 104 M−1, n = 0.20 ± 0.01 (i.e. a DNA:MTA stoichiometry of ∼5). From these standard parameters, we calculated ΔG = −6.62 kcal mol−1 and TΔS = +9.24 kcal mol−1 at 25°C. The positive enthalpy observed was in keeping with the value determined by DSC (Table 1) and both ITC and DSC results agree with the presence of Mg2+-coordinated MTA dimers (‘Complex II’ (17)) bound to DNA in solution. Our data were also consistent with fluorescence studies showing entropically-driven binding of Mg2+-coordinated MTA dimers to DNA (12).
Evaluation of heat capacity changes involved in the binding of mithramycins to DNA
The experimental characterization of heat capacity changes (ΔCp) that may accompany binding presented an insurmountable difficulty, as we only obtained reproducible data by ITC using MTA in 170 mM Na+ at 25°C (Figure 4). The main problem was the reproducibility of some control (heat of dilution) experiments, which we regard as a consequence of the necessary Mg2+-coordinated antibiotic dimerization coupled to binding along the minor groove of DNA. Therefore, to obtain an estimation of the heat capacity effects in the formation of DNA–MTA and DNA–MSK complexes, we undertook calculations of the non-polar (hydrophobic) and polar (hydrophilic) surfaces of MTA–DNA and MSK–DNA complexes and their component molecules, which allowed us to measure the binding-induced burial of specific molecular surfaces (32).
Inspection of Figure 5 and Table 2 reveals that most of the surface buried from exposure to solvent for the interaction of either mithramycin is hydrophobic rather than polar. Application of an empirical relationship (see Materials and methods) provided a calculated heat capacity change for the binding of MTA and MSK (Table 2). Comparison of surface areas for the native and complexed B-DNA (generated from d(TCGCGA)2 duplexes) showed only a small reduction in exposed DNA surface upon binding of either MTA or MSK, suggesting the absence of large helical rearrangements. From the ΔCp, determined from the surface area buried by MTA or MSK (Table 2), we calculated the hydrophobic component of the free energy of binding, by using the relationship: ΔGhyd = 80 ΔCp (38), to be −9.53 and −9.04 kcal mol−1 for MTA and MSK respectively (Table 1).
Figure 5.

Molecular (water)-accessible surface for the bound MTA molecule in the minor groove of a B-DNA of d(TCGCGA)2 sequence. Structural coordinates were taken from an NMR complex (16), PDB entry 146D. The figure was generated with the Chimera program (43). The right panel shows a detailed view of the region selected on the left panel. It also includes the structurally equivalent MSK atoms. This view highlights the different side-chains bound to the aglycone C-3 atoms in MTA (carbon atoms in blue) and MSK (carbon atoms in yellow). Compared to MTA, the shorter length of the MSK side chain restrains its possibilities of hydrogen bonding to DNA.
Table 2.
Determination of the surface area burial for mithramycin A–DNA and mithramycin SK–DNA complexesa
| Mithramycin A | Mithramycin SK | |||||
|---|---|---|---|---|---|---|
| Non-polar surface | Polar surface | Total surface | Non-polar surface | Polar surface | Total surface | |
| DNA–antibiotic complex | 1 588 | 1 657 | 3 245 | 1 559 | 1 657 | 3 216 |
| DNA–unboundb | 920 | 1 608 | 2 528 | 920 | 1 608 | 2 528 |
| Mg+2-coordinated antibiotic dimer | 1 359 | 777 | 2 136 | 1 302 | 759 | 2 061 |
| Induced changes | ΔAnp = −691 | ΔAp = −728 | ΔA = −1419 | ΔAnp = −663 | ΔAp = −710 | ΔA = −1 373 |
| Calculated ΔCpc | −119 (±28) cal mol−1 K−1 | −113 (±27) cal mol−1 K−1 | ||||
| ΔGhydd | −9.53 (±0.28) kcal mol−1 | −9.04 (±0,27) kcal mol−1 | ||||
aArea calculations (Å2) were carried out using NACCESS program (35).
bAn ideal B-DNA structure of d(TCGCGA)2 sequence, generated by the program 3DNA (34).
cHeat capacity change were computed from the induced changes in the surface area burials as described in the Materials and methods section.
dΔGhyd = 0 (±20) ΔCp (38).
If the ΔCp values (Table 2) were used together with the enthalpies determined by DSC (Table 1), the ΔHb values corrected by the heat capacity effects at 25°C would be +9.45 and +8.31 kcal mol−1 for MTA and MSK respectively, which are higher than those presented in Table 1. Furthermore, using these heat-capacity-corrected enthalpy values the apparent binding constants for MTA and MSK would be 7.3 × 105 M−1 and 2.4 × 105 M−1, respectively in 170 mM Na+ at 25°C. These values are moderately higher than those obtained using UV melting data and DSC results, which assumed a zero ΔCp value (Table 1). The negative capacity changes caused ΔH to become more positive at lower temperatures, but the magnitude of such higher ΔH was largely compensated by an increased entropic terms, thus the corrected ΔG values for MTA and MSK were −7.97 and −7.31 kcal mol−1, respectively. There is only a difference of ∼1 kcal mol−1 between these free energy values and those obtained from UV melting experiments, which considered a zero ΔCp (Table 1). Moreover, for MTA these corrected enthalpy and binding constant were also higher than the ΔH and Kb obtained directly by ITC, see above.
Competition dialysis assay of MTA and MSK binding to DNA
A competition dialysis assay (27) was used to study the MTA- and MSK-binding preferences for DNAs of various nucleotide compositions. This experimental approach allowed us to calculate apparent binding constants of mithramycins to C/G-rich DNAs, which cannot be studied using UV thermal denaturation, because their high Tm hinders accurate measurement. An evaluation of the binding to DNAs of diverse nucleotide compositions was used to characterize the differences in the binding of MTA and MSK to different DNA sequences (Figure 6). Both antibiotics bind more tightly to M. lysodeikticus DNA (72% C + G) than to salmon testes DNA (42% C + G), while the binding of MTA to the strictly alternating CG-tracts found in poly[d(G-C)2] is higher than that of MSK (Figure 6). The binding of both ligands to poly[d(I-C)2] was rather weak, as expected, since it does not contain the guanines required for specific binding.
Figure 6.

Competition dialysis experiments using MTA and MSK and various DNAs. Data correspond to experiments performed in triplicate (means ± SD). Values to the right are the apparent binding constants for the displayed DNAs, calculated, as described in the Materials and Methods section, using the bound antibiotic concentrations [Cbound] shown in the figure.
The Kapp for the various DNAs, calculated from the competition dialysis assays, are shown on the right side in Figure 6. These values can be compared with the binding to salmon testes DNA we determined by other methods. The MTA and MSK binding constants are slightly different from those obtained by UV thermal denaturation (cf. Figure 6 and Table 1), reflecting the presence of a small heat capacity effect, which was not considered when the binding data, determined at the Tm, were extrapolated to 25°C by using Equation (6). MTA binding to DNA was entropically driven (Table 1), with enthalpy values ranging from +1.60 kcal mol−1 obtained by DSC at the Tm, to +2.62 kcal mol−1 obtained by ITC (Figure 3). The comparison of these values reveals that any heat capacity effect would be relatively small, since there was a difference of only 1 kcal mol−1 between the ΔH determined at 91.0°C by DSC and the direct measurement at 25°C by ITC.
DISCUSSION
Thermodynamic analysis of drug binding to DNA offers valuable insights into the molecular forces involved in complex formation that cannot be obtained by structural studies alone (23,44). The interpretation of thermodynamic parameters may contribute to the design of new DNA-targeted drugs. We present the first calorimetric study on the binding of natural minor-groove ligands to C/G-rich regions in DNA. So far, the binding in the minor groove of AT-rich sequences has been characterized thermodynamically (24,44), although some features of the binding to C/G-rich DNA are also known (22). Binding to C/G-rich DNA has been addressed for intercalating agents (23,45), yet some of these agents also bind along the minor groove in the vicinity of the intercalating site in DNA tracts of diverse sequences (40,41,46).
Although the ΔH values obtained by DSC or ITC differ from those obtained by spectroscopic techniques (12) they are consistent with the view that positive enthalpy is involved in the binding of Mg2+-coordinated MTA dimers in the minor groove of DNA. Binding of MTA and MSK to DNA is clearly entropically driven (Table 1), and the positive sign of both ΔH and ΔS can be considered hallmarks of a predominantly hydrophobic binding reaction (23,47).
It is interesting to compare the thermodynamic profile of the MTA binding to DNA described here with previous determinations of the enthalpy of binding by spectroscopic titration and fluorescence (12,17). In spectroscopic studies, positive enthalpy was also observed under conditions consistent with the formation of Mg2+-coordinated MTA dimers. In those experiments, the enthalpy of binding (ΔH = +5.1 kcal mol−1) was estimated by application of the van't Hoff relationship to the spectroscopic data (12,17). This value is about 2-fold higher than the enthalpy we obtained directly using ITC at 25°C (ΔH = +2.6 kcal mol−1). These enthalpy values were measured under quite different ionic strength conditions—higher in our experiments—(cf. Table 1, Figures 4 and 6, and Ref. (12)). The differences in the calculated ΔH may be attributed to an ionic dependence of the enthalpy of binding (48). Our observation that the binding enthalpies determined by van't Hoff's analysis were higher than those reported using direct ITC measurements is consistent with previous reports [(48) and references therein]. Besides, the comparison of these ΔH reveals the presence of heat capacity changes (ΔCp), which we determined from the solvent-accessible areas in drug–DNA complexes (Table 2). These changes indicate that ΔH estimates for MTA binding to DNA are temperature dependent, a situation sometimes neglected in van't Hoff's analyses of drug–DNA interactions obtained from spectroscopic data (49). For MTA, correcting the ΔHb obtained by DSC, using the calculated ΔCp, renders an enthalpy, at 25°C, higher than its value determined directly by ITC, which points to intrinsic difficulties in measuring the DNA binding constants for Mg2+-coordinated antibiotics by this technique.
The Mg2+ requirement for the binding of mithramycin and related antibiotics to DNA has long been known [(15) and references therein] and the presence of a dication is essential for the formation of the dimers that bind to DNA (12). The presence of Mg2+ in the binding buffer merits further consideration not only because of the effects on the ionic strength, but also because of the direct binding (condensation) of the dication to the deoxyribonucleic acids (50). The effect of multivalent cations on the DNA–ligand interactions is greater than expected on the basis of their contribution to the ionic strength (50,51). Our experiments were performed at 170 mM Na+, which may facilitate solubility and reduce possible self-association of the antibiotic, while the concentrations of MgCl2 varied depending on the technique used (see Materials and Methods). In all cases, however, the final concentration was always kept below 1 mM in the presence of DNA. Therefore, Mg2+ effects through direct binding to DNA were minimized, while the relative contribution of MgCl2 to the ionic strength was insignificant. To analyse the effect of magnesium it is impossible to carry out binding experiments with and without dications, because they are necessary for mithramycin binding to DNA. Indeed, the presence of magnesium in the ITC titration of MTA into DNA required us to distinguish between the ‘heat of dilution of the drug’ and the dimerization of MTA when both the antibiotic and the dication became diluted after injecting into the large ITC cell. Addressing this particular issue, we obtained reproducible results for MTA only (Figures 4 and S3).
A small favourable contribution to free energy of binding was found to arise from the polyelectrolyte effect (Figure 3 and Table 1), although Mg2+-coordinated mithramycins are uncharged species. In order to reconcile this apparent discrepancy, we considered that ion release is a consequence of changes in the distance between the phosphates upon binding, due to a widening of the minor groove, which can alter the number of cations associated with each phosphate. We base this interpretation on the analysis of the salt dependence of the binding of uncharged intercalators to DNA (42). This analysis describes the ion release upon binding as a consequence of changes in the distance between the phosphates, which would alter the fraction of cations associated per phosphate. The effect for minor-groove-binding ligands must be lower, as we observed (Figure 3). In line with this explanation, NMR results indicate that binding of Mg2+-coordinated drugs can involve a wider minor groove (13,16). The entropy that accompanies the release of groove-bound ions upon binding of the oligosaccharide moieties into the minor groove also contributes to the free energy of binding (52).
We have determined the hydrophobic transfer free energies (ΔGhyd) from changes of the solvent accessibility areas upon drug binding (Table 2 and Figure 5) because ΔCp could not be measured experimentally. For Mg2+-coordinated MTA or MSK dimers, ΔGhyd is large and ‘favourable’ (negative), as a consequence of various hydration contributions to binding in the minor groove, which include the displacement of the ‘spine of hydration’ (22,24,25,32), and may comprise a dehydration effect produced by Mg2+ cations. These values are larger than the observed free energy of binding (cf. Tables 1 and 2). Therefore, they are enough to counterbalance the unfavourable entropic cost of forming DNA–mithramycin complexes. A negative heat capacity change causes ΔH to become more positive at lower temperatures, which may exaggerate the entropic driving force for binding. When the calculated ΔCp values (Table 2) were used together with thermodynamic parameters derived from UV melting plus DSC analyses (Table 1), the derived ΔH values were larger, see the Results section, yet the free energy of binding (ΔG) changed moderately due to entropy/enthalpy compensation. Besides, it is evident that using either the theoretically derived ΔCp (Table 2) or considering the ∼1 kcal mol−1 difference between the ΔH for MTA, obtained from DSC (at melting temperature) and ITC (at 25°C) experiments (Table 1 and Figure 4), renders small negative heat capacity changes. These negative heat capacity changes are consistent with the hydrophobic interactions, which are the major driving forces for the formation of DNA–mithramycin complexes. NMR studies indicate local changes in the C/G-rich binding site that consist of the presence of A-DNA sugar puckers and glycosidic torsion angles (13,16), which may induce the enhanced reactivity of some bases in chemical footprinting (11). A small local transition to A-DNA conformation upon drug binding might result in some water release since A-DNA can be considered a dehydrated form of DNA (53). Another factor that would involve positive entropy of binding is the release of counterions following this local conformational change and the widening of the minor groove. The oligosaccharide moieties (which are the same in MTA and MSK) participate in the entropic component of binding by disturbing both the water spine in the minor groove around AT base pairs (53) and the other DNA solvation shells, which should be less sensitive to sequence (22). The consequence would be a net favourable contribution to the free energy of binding (54). As mentioned above, our results are in keeping with that a large contribution of the hydrophobic transfer (ΔGhyd) might overwhelm the unfavourable contributions from changes in conformation and the entropic cost of forming a bimolecular complex (23,44), thereby a favourable final ΔG is achieved.
Differences in the free energy of binding for MTA and MSK, other than those provided by the hydrophobic contributions to the free energy of binding and the small polyelectrolyte contribution (Table 1), arise from direct molecular interactions in the C/G-rich binding sites. These include hydrogen bonds, van der Waals interactions and other weak forces (32,40), which would participate in the higher MTA-binding affinity (Table 1). A preferred MTA-binding site in DNA contains at least two contiguous C/G base pairs (4,15). However, there are discrepancies on the exact arrangement of the bases within the preferred binding site and the DNA sequences in their vicinity, since MTA extends its binding in the minor groove about 5–6 bp (Figure S1) and (4,16). We detected differences in the binding preferences of MTA and MSK to DNA of different C + G-content by competition dialysis (Figure 6). Both antibiotics failed to interact significantly with poly[d(I-C) 2], indicating that sequence-selective interaction is achieved via the binding of the chromophore to the amino group of guanines (4,11,13,15,16). However, some sequences containing guanines, for example G/A-rich tracts, do not bind MTA (11,55). Specific recognition in the minor groove of C/G-rich DNA by MTA involves direct hydrogen bonding between the aglycone C8 hydroxyl oxygens of Mg2+-coordinated antibiotic dimers (Figure 1) and the 2-amino of guanines (13,16). MTA and MSK contain the same tricyclic core moiety and oligosaccharide side chains, but they differ in the side chain at C-3. This chain is longer in MTA and bears a higher number, and different arrangement, of potential donors and acceptors of hydrogen bonds (Figure 1). Hence, a higher (more negative) free binding energy (ΔG) for MTA is consistent with the formation of extra hydrogen bonds. The oligosaccharide moieties of the mithramycins are the same. They are involved in the binding within the DNA minor groove, forming equivalent intermolecular contacts with the sugar-phosphate backbone (4,13).
The higher binding constant determined for MTA is consistent with its capacity to displace Sp1 protein from its putative C/G-rich binding sites at concentrations below those at which MSK does this (10). The comparison between MTA and MSK binding to DNAs of diverse C + G content (Figure 6) highlights the fact that small substitutions in naturally occurring DNA-binding molecules have significant effects on intermolecular interactions, which for mithramycin analogues appears to result in different biological properties (20).
SUPPLEMENTARY DATA
Supplementary Data is available at NAR Online.
ACKNOWLEDGEMENTS
We thank J. Cifre at the ‘Serveis Cientifico-Tecnics of the Universitat de les Illes Balears’ for his skilful technical assistance. Supported by a grant from the Spanish Ministry of Education and Science (SAF2005-00551) and the FEDER program of the European Community. This work was carried out within the framework of the Centre de Referencia en Biotecnologia of the Generalitat de Catalunya. Funding to pay the Open Access publication charge was provided by the Ministry of Education and Science and CSIC (Spain).
Conflict of interest statement. None declared.
REFERENCES
- 1.Chabner BA, Longo DL. Cancer Chemotherapy and Biotherapy: Principles and Practice, 3rd. Lippincott-Williams & Wilkins; 2001. [Google Scholar]
- 2.Fibach E, Bianchi N, Borgatti M, Prus E, Gambari R. Mithramycin induces fetal hemoglobin production in normal and thalassemic human erythroid precursor cells. Blood. 2003;102:1276–1281. doi: 10.1182/blood-2002-10-3096. [DOI] [PubMed] [Google Scholar]
- 3.Ferrante RJ, Ryu H, Kubilus JK, D'Mello S, Sugars KL, Lee J, Lu P, Smith K, Browne S, et al. Chemotherapy for the brain: the antitumor antibiotic mithramycin prolongs survival in a mouse model of Huntington's disease. J. Neurosci. 2004;24:10335–10342. doi: 10.1523/JNEUROSCI.2599-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Keniry MA, Banville DL, Simmonds PM, Shafer R. Nuclear magnetic resonance comparison of the binding sites of mithramycin and chromomycin on the self-complementary oligonucleotide d(ACCCGGGT)2. Evidence that the saccharide chains have a role in sequence specificity. J. Mol. Biol. 1993;231:753–767. doi: 10.1006/jmbi.1993.1324. [DOI] [PubMed] [Google Scholar]
- 5.Wohlert SE, Kunzel E, Machinek R, Méndez C, Salas JA, Rohr J. The structure of mithramycin reinvestigated. J. Nat. Prod. 1999;62:119–121. doi: 10.1021/np980355k. [DOI] [PubMed] [Google Scholar]
- 6.Rohr J, Méndez C, Salas JA. The biosynthesis of aureolic group antibiotics. Bioorg. Chem. 1999;27:41–54. [Google Scholar]
- 7.Blanco G, Fernández E, Fernández MJ, Braña AF, Weissbach U, Kunzel E, Rohr J, Méndez C, Salas JA. Characterization of two glycosyltransferases involved in early glycosylation steps during biosynthesis of the antitumor polyketide mithramycin by Streptomyces argillaceus. Mol. Gen. Genet. 2000;262:991–1000. doi: 10.1007/pl00008667. [DOI] [PubMed] [Google Scholar]
- 8.Remsing LL, González AM, Nur-e-Alam M, Fernández-Lozano MJ, Braña AF, Rix U, Oliveira MA, Méndez C, Salas JA, et al. Mithramycin SK, a novel antitumor drug with improved therapeutic index, mithramycin SA, and demycarosyl-mithramycin SK: three new products generated in the mithramycin producer Streptomyces argillaceus through combinatorial biosynthesis. J. Am. Chem. Soc. 2003;125:5745–5753. doi: 10.1021/ja034162h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rodríguez D, Quirós LM, Salas JA. MtmMII-mediated C-methylation during biosynthesis of the antitumor drug mithramycin is essential for biological activity and DNA-drug interaction. J. Biol. Chem. 2004;279:8149–8158. doi: 10.1074/jbc.M312351200. [DOI] [PubMed] [Google Scholar]
- 10.Albertini V, Jain A, Vignati S, Napoli S, Rinaldi A, Kwee I, Nur-e-Alam M, Bergant J, Bertoni F, et al. Novel GC-rich DNA-binding compound produced by a genetically engineered mutant of the mithramycin producer Streptomyces argillaceus exhibits improved transcriptional repressor activity: implications for cancer therapy. Nucleic Acids Res. 2006;34:1721–1734. doi: 10.1093/nar/gkl063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cons BM, Fox KR. Footprinting studies of sequence recognition by mithramycin. Anticancer Drug Des. 1990;5:93–97. [PubMed] [Google Scholar]
- 12.Aich P, Dasgupta D. Role of magnesium ion in mithramycin-DNA interaction: binding of mithramycin-Mg2+ complexes with DNA. Biochemistry. 1995;34:1376–1385. doi: 10.1021/bi00004a032. [DOI] [PubMed] [Google Scholar]
- 13.Sastry M, Patel DJ. Solution structure of the mithramycin dimer-DNA complex. Biochemistry. 1993;32:6588–6604. doi: 10.1021/bi00077a012. [DOI] [PubMed] [Google Scholar]
- 14.Hou MH, Robinson H, Gao YG, Wang AH. Crystal structure of the [Mg2+-(chromomycin A3)2]-d(TTGGCCAA)2 complex reveals GGCC binding specificity of the drug dimer chelated by a metal ion. Nucleic Acids Res. 2004;32:2214–2222. doi: 10.1093/nar/gkh549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fox KR, Howarth NR. Investigations into the sequence-selective binding of mithramycin and related ligands to DNA. Nucleic Acids Res. 1985;13:8695–8714. doi: 10.1093/nar/13.24.8695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sastry M, Fiala R, Patel DJ. Solution structure of mithramycin dimers bound to partially overlapping sites on DNA. J. Mol. Biol. 1995;251:674–689. doi: 10.1006/jmbi.1995.0464. [DOI] [PubMed] [Google Scholar]
- 17.Aich P, Dasgupta D. Role of Mg++ in the mithramycin-DNA interaction: evidence for two types of mithramycin-Mg++ complex. Biochem. Biophys. Res. Commun. 1990;173:689–696. doi: 10.1016/s0006-291x(05)80090-7. [DOI] [PubMed] [Google Scholar]
- 18.Snyder RC, Ray R, Blume S, Miller DM. Mithramycin blocks transcriptional initiation of the c-myc P1 and P2 promoters. Biochemistry. 1991;30:4290–4297. doi: 10.1021/bi00231a027. [DOI] [PubMed] [Google Scholar]
- 19.Kwak HJ, Park MJ, Cho H, Park CM, Moon SI, Lee HC, Park IC, Kim MS, Rhee CH, et al. Transforming growth factor-ß1 induces tissue inhibitor of metalloproteinase-1 expression via activation of extracellular signal-regulated kinase and Sp1 in human fibrosarcoma cells. Mol. Cancer Res. 2006;4:209–220. doi: 10.1158/1541-7786.MCR-05-0140. [DOI] [PubMed] [Google Scholar]
- 20.Remsing LL, Bahadori HR, Carbone GM, McGuffie EM, Catapano CV, Rohr J. Inhibition of c-src transcription by mithramycin: structure-activity relationships of biosynthetically produced mithramycin analogues using the c-src promoter as target. Biochemistry. 2003;42:8313–8324. doi: 10.1021/bi034091z. [DOI] [PubMed] [Google Scholar]
- 21.Koutsodontis G, Kardassis D. Inhibition of p53-mediated transcriptional responses by mithramycin A. Oncogene. 2004;23:9190–9200. doi: 10.1038/sj.onc.1208141. [DOI] [PubMed] [Google Scholar]
- 22.Marky LA, Breslauer KJ. Origins of netropsin binding affinity and specificity: correlations of thermodynamic and structural data. Proc. Natl. Acad. Sci. U.S.A. 1987;84:4359–4363. doi: 10.1073/pnas.84.13.4359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaires JB. Energetics of drug-DNA interactions. Biopolymers. 1997;44:201–215. doi: 10.1002/(SICI)1097-0282(1997)44:3<201::AID-BIP2>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 24.Haq I, Ladbury JE, Chowdhry BZ, Jenkins TC, Chaires JB. Specific binding of Hoechst 33258 to the d(CGCAAATTTGCG)2 duplex: calorimetric and spectroscopic studies. J. Mol. Biol. 1997;271:244–257. doi: 10.1006/jmbi.1997.1170. [DOI] [PubMed] [Google Scholar]
- 25.Han F, Taulier N, Chalikian TV. Association of the minor groove binding drug Hoechst 33258 with d(CGCGAATTCGCG)2: volumetric, calorimetric, and spectroscopic characterizations. Biochemistry. 2005;44:9785–9794. doi: 10.1021/bi047374f. [DOI] [PubMed] [Google Scholar]
- 26.Breslauer KJ, Remeta DP, Chou WY, Ferrante R, Curry J, Zaunczkowski D, Snyder JG, Marky LA. Enthalpy-entropy compensations in drug-DNA binding studies. Proc. Natl Acad. Sci. U.S.A. 1987;84:8922–8926. doi: 10.1073/pnas.84.24.8922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ren J, Chaires JB. Rapid screening of structurally selective ligand binding to nucleic acids. Methods Enzymol. 2001;340:99–108. doi: 10.1016/s0076-6879(01)40419-8. [DOI] [PubMed] [Google Scholar]
- 28.Wells RD, Larson JE, Grant RC, Shortle BE, Cantor CR. Physicochemical studies on polydeoxyribonucleotides containing defined repeating nucleotide sequences. J. Mol. Biol. 1970;54:465–497. doi: 10.1016/0022-2836(70)90121-x. [DOI] [PubMed] [Google Scholar]
- 29.Jenkins TC. Optical absorbance and fluorescence techniques for measuring DNA-drug interactions. Methods Mol. Biol. 1997;90:195–218. doi: 10.1385/0-89603-447-X:195. [DOI] [PubMed] [Google Scholar]
- 30.Crothers DM. Statistical thermodynamics of nucleic acid melting transitions with coupled binding equilibria. Biopolymers. 1971;10:2147–2160. doi: 10.1002/bip.360101110. [DOI] [PubMed] [Google Scholar]
- 31.Record MT, Anderson CF, Lohman TM. Thermodynamic analysis of ion effects on the binding and conformational equilibria of proteins and nucleic acids: the roles of ion association or release, screening, and ion effects on water activity. Q. Rev. Biophys. 1978;11:103–178. doi: 10.1017/s003358350000202x. [DOI] [PubMed] [Google Scholar]
- 32.Haq I, Jenkins TC, Chowdhry BZ, Ren J, Chaires JB. Parsing free energies of drug-DNA interactions. Methods Enzymol. 2000;323:373–405. doi: 10.1016/s0076-6879(00)23374-0. [DOI] [PubMed] [Google Scholar]
- 33.Van der Spoel D, Lindahl E, Hess B, Groenhof G, Mark AE, Berendsen HJ. GROMACS: fast, flexible, and free. J. Comput. Chem. 2005;26:1701–1718. doi: 10.1002/jcc.20291. [DOI] [PubMed] [Google Scholar]
- 34.Lu XJ, Olson WK. 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 2003;31:5108–5121. doi: 10.1093/nar/gkg680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hubbard SJ, Thornton JM. Department Biochemistry and Molecular Biology. London: University College; 1993. ‘NACCESS’, Computer Program. [Google Scholar]
- 36.Chothia C. The nature of the accessible and buried surfaces in proteins. J. Mol. Biol. 1976;105:1–12. doi: 10.1016/0022-2836(76)90191-1. [DOI] [PubMed] [Google Scholar]
- 37.Spolar RS, Record MT., Jr Coupling of local folding to site-specific binding of proteins to DNA. Science. 1994;263:777–784. doi: 10.1126/science.8303294. [DOI] [PubMed] [Google Scholar]
- 38.Ha JH, Spolar RS, Record MT. Role of the hydrophobic effect in stability of site-specific protein-DNA complexes. J. Mol. Biol. 1989;209:801–816. doi: 10.1016/0022-2836(89)90608-6. [DOI] [PubMed] [Google Scholar]
- 39.Chaires JB, Ren J, Hamelberg D, Kumar A, Pandya V, Boykin DW, Wilson WD. Structural selectivity of aromatic diamidines. J. Med. Chem. 2004;47:5729–5742. doi: 10.1021/jm049491e. [DOI] [PubMed] [Google Scholar]
- 40.Leng F, Chaires JB, Waring MJ. Energetics of echinomycin binding to DNA. Nucleic Acids Res. 2003;31:6191–6197. doi: 10.1093/nar/gkg826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Portugal J, Cashman DJ, Trent JO, Ferrer-Miralles N, Przewloka T, Fokt I, Priebe W, Chaires JB. A new bisintercalating anthracycline with picomolar DNA binding affinity. J. Med. Chem. 2005;48:8209–8219. doi: 10.1021/jm050902g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wilson WD, Tanious FA. Kinetic analysis of drug-nucleic acid binding modes: absolute rates and effects of salt concentration. In: Neidle S, Waring M, editors. Molecular Aspects of Anticancer Drug-DNA Interactions. Vol. 2. London: MacMillan; 1994. pp. 243–269. [Google Scholar]
- 43.Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 44.Haq I. Thermodynamics of drug-DNA interactions. Arch. Biochem. Biophys. 2002;403:1–15. doi: 10.1016/S0003-9861(02)00202-3. [DOI] [PubMed] [Google Scholar]
- 45.Ren J, Jenkins TC, Chaires JB. Energetics of DNA intercalation reactions. Biochemistry. 2000;39:8439–8447. doi: 10.1021/bi000474a. [DOI] [PubMed] [Google Scholar]
- 46.Barceló F, Capó D, Portugal J. Thermodynamic characterization of the multivalent binding of chartreusin to DNA. Nucleic Acids Res. 2002;30:4567–4573. doi: 10.1093/nar/gkf558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chaires JB. A thermodynamic signature for drug-DNA binding mode. Arch. Biochem. Biophys. 2006;453:24–29. doi: 10.1016/j.abb.2006.03.027. [DOI] [PubMed] [Google Scholar]
- 48.Chaires JB. Thermodynamics of the daunomycin-DNA interaction: ionic strength dependence of the enthalpy and entropy. Biopolymers. 1985;24:403–419. doi: 10.1002/bip.360240208. [DOI] [PubMed] [Google Scholar]
- 49.Chaires JB. Possible origin of differences between van't Hoff and calorimetric enthalpy estimates. Biophys. Chem. 1997;64:15–23. doi: 10.1016/s0301-4622(96)02205-3. [DOI] [PubMed] [Google Scholar]
- 50.Lohman TM. Kinetics of protein-nucleic acid interactions: use of salt effects to probe mechanisms of interaction. CRC Crit. Rev. Biochem. 1986;19:191–245. doi: 10.3109/10409238609084656. [DOI] [PubMed] [Google Scholar]
- 51.Tan ZJ, Chen SJ. Nucleic acid helix stability: effects of salt concentration, cation valence and size, and chain length. Biophys. J. 2006;90:1175–1190. doi: 10.1529/biophysj.105.070904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Misra VK, Sharp KA, Friedman RA, Honig B. Salt effects on ligand-DNA binding. Minor groove binding antibiotics. J. Mol. Biol. 1994;238:245–263. doi: 10.1006/jmbi.1994.1285. [DOI] [PubMed] [Google Scholar]
- 53.Dickerson RE. DNA structure from A to Z. Methods Enzymol. 1992;211:67–111. doi: 10.1016/0076-6879(92)11007-6. [DOI] [PubMed] [Google Scholar]
- 54.Cooper A. Heat capacity effects in protein folding and ligand binding: a re-evaluation of the role of water in biomolecular thermodynamics. Biophys. Chem. 2005;115:89–97. doi: 10.1016/j.bpc.2004.12.011. [DOI] [PubMed] [Google Scholar]
- 55.Vaquero A, Portugal J. Small ligands that neither bind to nor alter the structure of d(GA-TC)n sequences in DNA. FEBS Lett. 1997;420:156–160. doi: 10.1016/s0014-5793(97)01509-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.