


Abstract
The Helicobacter pylori neutrophil-activating protein (HP-NAP), a member of the Dps family, is a fundamental virulence factor involved in H.pylori-associated disease. Dps proteins protect bacterial DNA from oxidizing radicals generated by the Fenton reaction and also from various other damaging agents. DNA protection has a chemical component based on the highly conserved ferroxidase activity of Dps proteins, and a physical one based on the capacity of those Dps proteins that contain a positively charged N-terminus to bind and condense DNA. HP-NAP does not possess a positively charged N-terminus but, unlike the other members of the family, is characterized by a positively charged protein surface. To establish whether this distinctive property could be exploited to bind DNA, gel shift, fluorescence quenching and atomic force microscopy (AFM) experiments were performed over the pH range 6.5–8.5. HP-NAP does not self-aggregate in contrast to Escherichia coli Dps, but is able to bind and even condense DNA at slightly acid pH values. The DNA condensation capacity acts in concert with the ferritin-like activity and could be used to advantage by H.pylori to survive during host-infection and other stress challenges. A model for DNA binding/condensation is proposed that accounts for all the experimental observations.
INTRODUCTION
Helicobacter pylori is a Gram-negative microaerophilic bacillus that unlike other microorganisms colonizes the highly acidic environment of the human gastric mucosa where pH falls to ∼1.5 in the absence of food and to ∼5.0 during the digestive phase (1,2). To accomplish gastric colonization, H.pylori undergoes acid acclimation such that cytoplasmatic pH is 7.0 upon exposure to a pH 6.2 medium and reaches 6.2, that still allows cell growth, upon exposure to pH 4.5 (1,3). The H.pylori infection is associated with chronic gastritis and peptic ulcer. It also elicits an immune response which is not sufficient to either prevent or counteract bacterial colonization (4). This response is mediated by neutrophils and macrophages and culminates in the production of large amounts of reactive oxygen and nitrogen species (ROS and RNS) leading to alteration of the epithelial tight junctions and basal membranes (5). In turn, the inflammatory reaction induced by H.pylori promotes the release of nutrients from the mucosa to support the growth of the bacterium residing within the mucous layer (6).
The H.pylori neutrophil-activating protein (HP-NAP) is one of the toxic factors released by the bacterium which adheres strongly to gastric epithelial cells during infection by means of specialized proteins. HP-NAP crosses the epithelial lining and recruits neutrophils and monocytes to the site of infection. HP-NAP has also other properties: it is a major antigen in the human immune response to H. pylori infection and acts as an adhesin in vitro, a feature attributed to the positively charged protein surface (5,7). In addition, it plays a role in the pathogenesis of H.pylori-associated gastritis (5) and in the ability of the microorganism to survive under oxidative stress conditions (8).
HP-NAP is a highly symmetrical dodecameric molecule of 200 kDa belonging to the Dps (DNA-binding proteins from starved cells) protein family (6). Dps proteins are expressed by most bacteria under a variety of stress conditions in order to protect DNA against oxidative damage and other detrimental factors (9–13). Accordingly, HP-NAP is produced maximally during stationary phase (8). Moreover, the expression is up-regulated upon acid adaptation, e.g. ∼2.4-fold when external pH reaches 4.5 (1).
Dps proteins protect DNA by means of a double mechanism. The first was discovered in Escherichia coli Dps and has given the name to the protein family. It provides a physical shield from damaging agents since it entails binding of DNA and its condensation into large Dps–DNA complexes (13,14). The second mode of protection is due to the ability of Dps proteins to bind and oxidize Fe(II) at the characteristic, highly conserved intersubunit ferroxidase center. Since ferroxidation is carried out most efficiently by hydrogen peroxide, production of the toxic hydroxyl radicals through Fenton chemistry is drastically diminished (15,16). All Dps proteins are endowed with ferroxidase activity, but not all of them are able to bind DNA. Based on the E.coli Dps structure (10), the latter property was attributed to the presence of a positively charged, disordered, freely mobile N-terminus. In accordance with this model, Listeria innocua Dps and Bacillus anthracis Dlp-1 and Dlp-2, that are characterized by an N-terminus of reduced length (17–19), and Agrobacterium tumefaciens Dps, where the N-terminus is immobilized on the protein surface (20), are unable to interact with DNA.
A recent study on E.coli Dps and two N-terminus deletion mutants by Ceci et al. (21) correlated the capacity to bind DNA and form large Dps–DNA complexes, containing many Dps molecules and one or more DNA plasmids, with the number of protonated lysine residues in the N-terminus. Thus, at physiological pH values, native E.coli Dps gives rise to large Dps–DNA aggregates since the intact N-terminus carries three lysine residues that are all protonated. In contrast, the mutant lacking 2 out of 3 N-terminal lysine residues is able to bind DNA, but condensation does not take place, a situation that occurs in native Dps only at pH values above 8.7. In the mutant lacking all three lysine residues even DNA binding is impaired. The study also revealed that under all experimental conditions leading to DNA condensation, E.coli Dps has a strong tendency to self-aggregate and precipitate out of solution in the absence of DNA. This behavior was attributed to the regular disposition of the positive charges on the N-terminus which tend to interact with the negatively charged surface of adjacent protein molecules.
The HP-NAP surface is characterized by the marked prevalence of positive charges (Supplementary Figure 1) at variance with the other Dps proteins known to date (22). Although this characteristic should favor binding of the negatively charged DNA molecule, the experimental data on the occurrence of interaction are contradictory. Thus, agarose gel mobility assays carried out at pH 8.0 did not provide evidence of DNA binding (6), whereas recombinant HP-NAP expressed in E.coli was found to co-localize with the E.coli DNA nucleoid suggesting that such an interaction may take place also in H.pylori (8).
In the present work, the ability of HP-NAP to bind DNA was assayed by means of fluorescence quenching, gel mobility and atomic force microscopy (AFM) experiments covering a wide pH range. As mentioned above, in E.coli Dps a linkage exists between the DNA binding/condensation ability and self-aggregation which leads to protein precipitation in the absence of the interacting partner (21). HP-NAP does not self-aggregate, as shown by analytical ultracentrifugation experiments, but is able to bind and even condense DNA at slightly acid pH values. A new interaction mechanism therefore must be involved that differs from that operative in E.coli Dps. A model is proposed that accounts for all the experimental observations. The occurrence of HP-NAP-mediated DNA condensation at low pH provides the bacterium with a fully reversible mechanism that permits protection of DNA from damage in the host environment. In more general terms it points to a possible role of the protein on colonization efficiency.
MATERIALS AND METHODS
HP-NAP cloning
The plasmid containing the hp-nap gene was kindly provided by Dr C. Montecucco. It was introduced into E.coli BL21DE3 and sequenced by dideoxy sequencing to confirm the presence of the correct gene. Escherichia coli strain BL21DE3 was grown at 37°C on LB liquid medium (10 g/l tryptone, 5 g/l yeast extract, 5 g/l NaCl) or on LB plates containing 20 μg/ml chloramphenicol.
Expression purification and characterization of HP-NAP
Escherichia coli BL21DE3 cells harboring the recombinant plasmid were grown at 37°C in 1 l of liquid LB medium containing chloramphenicol (20 μg/ml) to an optical density of 1.6 at 600 nm.
Cells were harvested by centrifugation (15 000 × g for 20 min) and suspended in 10 ml of 50 mM Tris-HCl at pH 7.5, 0.5 mM dithiothreitol, 1 mM EDTA, 500 mM NaCl buffer and disrupted by sonication. The lysate was centrifuged at 15 000 × g for 45 min and the supernatant was precipitated using two ammonium sulfate cuts at 30 and 60% saturation (w/v). HP-NAP precipitates at 60% saturation after centrifugation (15 000 × g for 45 min). The pellet was resuspended and dialyzed overnight against 20 mM Tris-HCl, pH 7.5, and then loaded onto a DEAE cellulose column (DE52) equilibrated with the same buffer. HP-NAP was eluted with 200 mM NaCl. An additional purification step was performed using the Amersharm HiTrap Chelating HP column with 30 mM Tris-HCl, 0.2 M NaCl, pH 8.0 as binding buffer, and 30 mM Tris-HCl, pH 8.0 containing 0.2 M NaCl and 0.2 M imidazole as elution buffer. HP-NAP eluted at 70% of the gradient. It was pooled and stored at −75°C after controlling the purity of the preparation by Coomassie Blue staining of SDS-15% PAGE gels.
Protein concentration was determined spectrophotometrically at 280 nm using a molar extinction coefficient (on a dodecamer basis, 16 933 × 12 = 203 196 Da) of 1.85 × 105 M−1 · cm−1 (ProtParam software, www.expasy.org). Iron loading was performed by addition of ferrous ammonium sulfate at a molar excess of 400 Fe/dodecamer to a 3-μM protein solution in 30 mM MOPS buffer at pH 7.0 (6,17).
The possible protein self-aggregation was assessed by analytical ultracentrifugation experiments on 3 μM protein solutions in 30–50 mM MOPS or Tris-HCl buffer over the pH range 6.5–8.5, in the presence or absence of 3 mM MgCl2. The sedimentation velocity experiments were carried out on a Beckman Instruments Optima XL-I analytical ultracentrifuge at 30 000 r.p.m. Data were analyzed with SEDFIT (23) and the sedimentation coefficient was reduced to s20,w by standard procedures.
Gel retardation assays
The DNA-binding activity of HP-NAP in the apo- or Fe-loaded form was assessed in gel shift assays using 20 nM supercoiled or HindIII linearized pUC9-5S DNA (3115 bp), as a probe. DNA was incubated for 5 min at room temperature with the protein (0.3–16 µM) in 50 mM MOPS or Tris-HCl buffers containing 3 mM MgCl2 at different pH values in the range 6.5–8.5. In the experiments designed to test the effect of salts, 50 mM NaCl or KCl were added. In order to resolve the HP-NAP–DNA complexes, electrophoresis was carried out on 1% agarose gels in 0.04 M Tris-acetate or BisTris-acetate buffers at a constant voltage of 6 V/cm at 20°C. The gels were stained with ethidium bromide and imaged by ImageMaster VDS.
To determine the apparent dissociation constant (KD) of linearized plasmid DNA for HP-NAP at pH 7.5, the protein was titrated onto 20 nM DNA in a total final volume of 15 µl and the gel mobility shift assays were performed as described above. The retardation coefficient R was introduced to quantify mobility shifts (24). In any given lane, R is found by measuring the difference between the migration of free DNA and that of the protein–DNA complex, and normalizing the value thus obtained to the migration of free DNA. Therefore, the fractional saturation (Y) of nucleic acid with protein (i.e. bound DNA/total DNA) in a given lane corresponds to the ratio R/Rmax, where Rmax is the maximum value of R that is attained when the protein concentration is saturating. The apparent KD was calculated by plotting Y against protein concentration and fitting the curve to a hyperbolic function with Origin 7.0 software (OriginLab software Inc., Northampton, MA, USA).
DNA protection against DNAse cleavage
HP-NAP (3 µM) was allowed to interact with 20 nM linearized plasmid DNA for 10 min at room temperature over the pH range 6.5–8.5 in 50 mM MOPS or Tris-HCl buffers containing 3 mM MgCl2. Following incubation, the complex was treated with 0.3 U of DNase I (Deoxyribonuclease I from bovine pancreas, Sigma-Aldrich) for 1 min at 37°C. Reactions were stopped by addition of 3 mM EDTA and incubation at 65°C for 5 min. HP-NAP–DNA complexes at pH 6.5 were disaggregated by raising the pH with an equal volume of 200 mM Tris-HCl pH 8.0. The gels were stained with SYBR Green I and imaged by ImageMaster VDS.
DNA protection against hydroxyl radical formation
Iron-dependent hydroxyl radical formation was assayed as described by Halliwell and Gutteridge (25). Fe(II) salts in solution generate hydroxyl radicals that degrade deoxyribose to a malondialdehyde-like compound that forms a chromogen with thiobarbituric acid. Fluorescence of the chromogen was used to assay for hydroxyl radical formation. The reaction mixture contained the following reagents in a total volume of 1.2 ml at the following final concentration: potassium phosphate buffer (10 mM, pH 7.4), NaCl (63 mM), deoxyribose (1 mM). Variable amounts of HP-NAP were added to the basal reaction mixture. The reaction was initiated by adding ferrous ammonium sulfate (21 µM), and the mixture was incubated at 37°C for 15 min. Thereafter, thiobarbituric acid (1 ml at 1% w/v) plus trichloroacetic acid (1 ml at 2.8% w/v) were added. The whole was heated at 100°C for 10 min, cooled, and the chromogen was measured by fluorescence emission at 553 nm (λex 532 nm) using a FluoroLog-3 spectrofluorimeter (HORIBA Jobin Yvon Inc., NJ, USA).
Atomic force microscopy
Solutions were prepared by incubating HP-NAP (5–30 nM) alone or with DNA (1 nM) at 20°C for 5 min, in 50 mM MOPS or 10 mM Tris-HCl buffers containing 3 mM MgCl2, at different pH values over the range 6.5–8.5. Purified plasmid DNA (pUC9-5S), a 27-bp dsDNA fragment or a 27-nt ssDNA oligonucleotide were used in the experiments. The samples were deposited onto freshly cleaved ruby mica (Mica New York, NY, USA), incubated for ∼1 min, rinsed with 10 drops of water Milli-Q (Millipore) and dried with a weak stream of nitrogen. AFM images were collected in air with a Nanoscope III microscope (Digital Instruments Inc., Santa Barbara, CA, USA) operating in tapping mode. All operations were carried out at room temperature. Commercial diving board silicon cantilevers (MikroMasch Tallinn, Estonia) were used. The microscope was equipped with a type E scanner (12 mm × 12 mm). Images (512 × 512 pixels) were collected with a scan size of 2 or 4 μm at a scan rate varying between two and four scan lines per second.
Fluorimetry
Measurements of intrinsic tryptophan fluorescence emission were made by using a FluoroLog-3 spectrofluorimeter thermostatted at 20°C. A fixed HP-NAP concentration of 0.5 µM and a 1.0 cm pathlength quartz cuvette were used. Linearized plasmid DNA (3115 bp), a 27-bp dsDNA fragment or a 27-nt ssDNA oligonucleotide were titrated onto the fixed amount of HP-NAP Dps in 50 mM MOPS or Tris-HCl buffers containing 3 mM MgCl2 at different pH values in the range 6.5–8.5. Escherichia coli Dps was used as a control. The excitation wavelength was 285 nm (slit width, 3 nm) and emission spectra were recorded from 300 to 450 nm (slit width, 3.5 nm, 1 nm sampling interval). To check for the presence of aggregated particles, 90° light scattering was measured at 20°C with both excitation and emission wavelength set at 480 nm. The apparent dissociation constant KD was calculated by plotting normalized fluorescence against the DNA–protein concentration ratio and curve fitting with Origin 7.0 software.
ABBREVIATIONS
MOPS, 3-(N-morpholino)propanesulfonic acid.
RESULTS AND DISCUSSION
The capacity of HP-NAP to bind DNA is still elusive since previous attempts to demonstrate binding in vitro and in vivo did not provide a definitive answer, possibly because of the different pH conditions used (6,8). This experimental factor influences the binding and condensation processes dramatically given the polyelectrolyte nature of both interacting partners. To establish whether HP-NAP and DNA do interact, gel mobility assays, fluorescence quenching and AFM experiments were performed over the pH range 6.5–8.5 which is compatible with bacterial growth.
HP-NAP state of association at different pH values
Recent work established that in E.coli Dps, the regular arrangement of the positively charged N-termini within the negative protein surface has two effects: it endows the protein with the capacity to bind and condense DNA and with the tendency to self-aggregate in the absence of the interacting partner under conditions of low to moderate ionic strength (21).
On this basis, the possible self-aggregation of HP-NAP (0.3–16 μM) was assessed by means of analytical ultracentrifugation experiments carried out at 20°C over the pH range 6.5–8.5 in buffers of ionic strength 30–50 mM. The effect of 3 mM MgCl2 was also tested because of its use in the AFM experiments to be described below. Under all conditions studied, HP-NAP sediments as a homogeneous peak of 9.9 S (Figure 1, panels A and B). If one assumes a spherical shape and a partial specific volume of 0.736 ml/g (26), the calculated molecular mass is ∼200 kDa corresponding to a dodecamer. Consistently, HP-NAP appears as individual dodecamers in AFM images obtained under the same pH and buffer conditions in the presence of MgCl2 and migrates into the gel upon native agarose gel electrophoresis (Figure 1, panels A and B, and Supplementary Figure 2A).
Figure 1.

State of association of H.pylori HP-NAP at different pH values, monitored by analytical ultracentrifugation, agarose gel electrophoresis and AFM. Panel A: pH 6.5; panel B: pH 8.5. Each panel shows (from left to right): the sedimentation coefficient distribution c(S) as a function of S, agarose gel electrophoresis of 3 μM (lane 1) and 4 μM HP-NAP (lane 2) and AFM images. In these, the white dots represent single HP-NAP dodecamers as indicated by the lateral and vertical dimensions. Buffers: 0.04 M Tris-acetate or BisTris-acetate containing 3 mM MgCl2. Image scan size 2 μm.
This characterization therefore shows that the HP-NAP dodecamer is stable and does not aggregate between pH 6.5 and 8.5, whereas E.coli Dps self-aggregates over the same pH range (21).
DNA binding by HP-NAP at different pH values
Atomic force microscopy experiments coupled with fluorimetry and gel retardation assays were used to assess binding of linear and supercoiled dsDNA and of ssDNA over the pH range 6.5–8.5.
At pH 6.5 and 7.0, interaction of HP-NAP with both linearized and supercoiled DNA generates complexes too large to migrate into the agarose gel (left panels in Figure 2A and B, and Supplementary Figure 3). Accordingly, fluorimetry experiments show a significant scattering indicative of the presence of aggregated particles in solutions (Supplementary Figure 4). AFM imaging reveals that the majority of dodecamers are sequestered into large protein–DNA condensates characterized by one or more DNA fragments looping out from a large protein core formed by many HP-NAP molecules (right panels in Figure 2A and B). These pictures contrast with those obtained with HP-NAP alone at pH 6.5 (Figure 1A) where the protein appears as individual, spatially separated, dodecamers.
Figure 2.
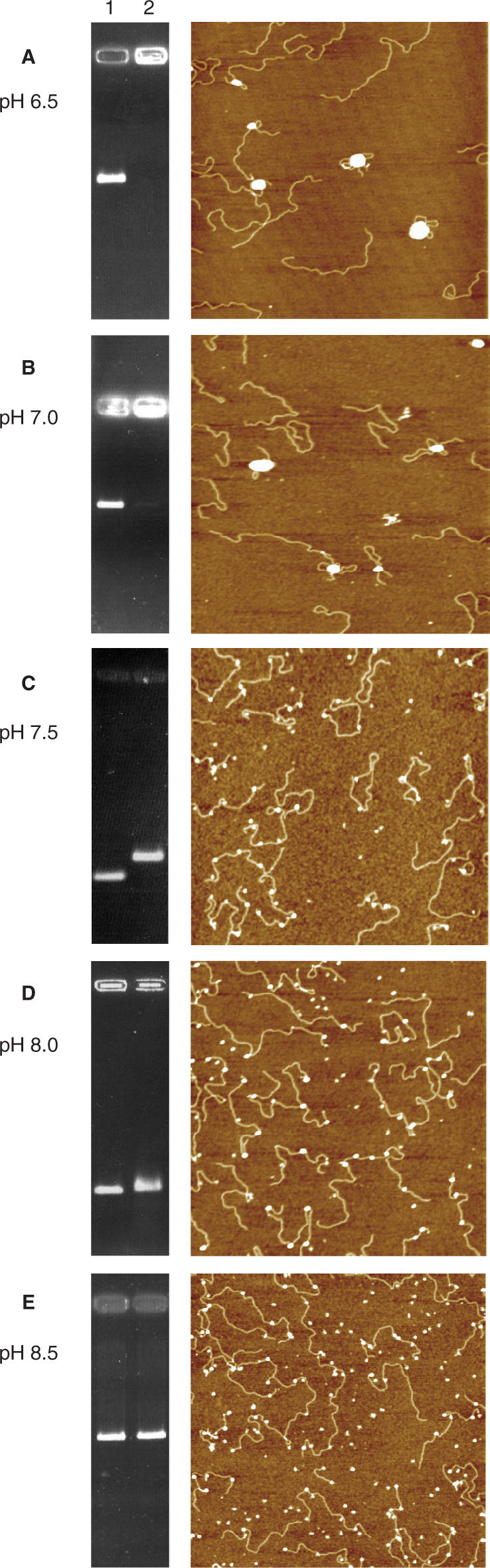
DNA gel retardation assays and AFM images of HP-NAP–DNA complexes at different pH values. (A) pH 6.5; (B) pH 7.0; (C) pH 7.5; (D) pH 8.0; (E) pH 8.5. In all gels (left panels), lane 1 refers to linearized plasmid DNA alone (20 nM) and lane 2 to DNA with HP-NAP at a DNA:protein molar ratio of 1:25. In the AFM images (right panels), the large white globular features in A and B represent HP-NAP–DNA aggregates involving one or few DNA molecules and many HP-NAP dodecamers, those in C, D and E represent single HP-NAP dodecamers. Agarose gels were stained with ethidium bromide. In all AFM images the scan size is 2 μm.
At pH 7.5 and 8.0, HP-NAP is still capable of interacting with DNA as indicated by the change in mobility of the DNA band in the agarose gels (left panels in Figure 2C and D) and by the significant quenching of the protein intrinsic tryptophan fluorescence (Figure 3). In full agreement with the gel and fluorimetry data, AFM imaging shows that at pH 7.5 and 8.0, binding of DNA does not entail formation of the large protein–DNA aggregates observed at lower pH values. Single HP-NAP dodecamers are randomly distributed along the DNA resembling beads-on-a-string (Figure 2C, D and Supplementary Figure 2B). Binding of the protein to supercoiled DNA does not appear to change the super-helical density of the plectonemes significantly, thus suggesting the absence of large DNA deformations (Supplementary Figure 3).
Figure 3.

Analysis of the HP-NAP–DNA interaction at pH 7.5 by means of fluorescence quenching and gel retardation experiments. Panel A: fluorescence data presented as the mean (±SD) of three independent experiments obtained upon titration of 0.5 μM HP-NAP with linearized plasmid DNA (closed circle), a 27 bp dsDNA fragment (closed square) and a 27 nt ssDNA oligonucleotide (open square). A representative experiment is shown in the inset. Panel B: gel retardation data presented as the mean (±SD) of three independent experiments obtained upon titration of 20 nM linearized plasmid DNA with HP-NAP. The inset depicts a typical experiment; the agarose gel was stained with SYBR Green I. At the lowest protein:DNA ratio analyzed (lane 2), there is no free DNA as shown by the AFM image obtained under the same experimental conditions (Figure 2C); consistently, only one DNA band is apparent in the gel which is shifted relative to free DNA. Gel retardation increases with increasing protein concentration because of the increased molecular weight of the nucleo-protein complex. The apparent dissociation constants calculated from the change in mobility are given under ‘Results and discussion section’.
At pH 8.5, HP-NAP does not affect DNA mobility of either linearized or supercoiled plasmids (left panel in Figure 2E and Supplementary Figure 3) under the buffer conditions studied. The absence of binding was confirmed by the lack of quenching in the fluorimetry assays (Supplementary Figure 4) and by AFM imaging which shows most of the dodecamers off the DNA (right panel in Figure 2E).
Importantly, formation of DNA condensates is not affected by the addition of 50 mM NaCl or KCl and is fully reversible when pH is raised from 6.5 to 8.0 or above.
The fluorimetry and gel retardation experiments performed at pH 7.5 were used to calculate an apparent dissociation constant for the interaction, KD. Figure 3A presents the fluorescence quenching data obtained upon titration of HP-NAP with linearized plasmid DNA (3115 bp), a 27 bp dsDNA fragment and a 27 nt ssDNA oligonucleotide. In all cases the binding curve reached a clear saturation point. The apparent KD is 3.30 ± 0.17 µM for the linearized plasmid and 9.69 ± 0.47 and 10.38 ± 0.52 µM for the 27 bp dsDNA and ssDNA, respectively. The changes in mobility of linearized DNA on agarose gels observed at the same pH value were analyzed in terms of fractional saturation of nucleic acid with protein as a function of protein concentration. The KD value thus obtained is 1.15 ± 0.09 µM in fair agreement with the fluorescence data (Figure 3B).
The possible effect of iron loading on the ability to bind DNA was assessed in gel retardation assays carried out at pH 6.5 and 7.5 on HP-NAP incorporated with 400 Fe/dodecamer, i.e. with the maximum amount of iron atoms that can be loaded into the protein cavity. Escherichia coli Dps was analyzed in parallel as a control. Iron loading did not affect the DNA-binding properties of HP-NAP or E.coli Dps. (Supplementary Figure 5). These data contrast with studies on Porphyromonas gingivalis Dps (27) and Helicobacter hepaticus Dps (28) which report that iron loading with a large molar excess of the metal (2000 and 4000 atoms/dodecamer, respectively) confers DNA-binding ability to the apoprotein. The significant amount of oxidized iron remaining in the buffer under these conditions may form insoluble polymers or bind to the protein surface and hence may account for the discrepancy.
A further comparison with literature data is in order. In gel retardation experiments carried out at pH 8.0, ∼8.0 µM HP-NAP is required to saturate 20 nM linear plasmid DNA, i.e. a protein:DNA molar ratio of 400:1 indicative of a marked decrease in affinity relative to pH 7.5 (Supplementary Figure 5). The decrease in affinity is such that no significant change in DNA mobility can be expected at the low protein:DNA molar ratios (≤10:1) used in the agarose gel assays by Tonello et al. (6).
The experiments just presented show that HP-NAP binds DNA non-specifically (not sequence-specifically) in a ‘beads-on-a-string fashion’ at pH 8.0 and condenses it into large aggregates at lower pH values. The transition between the two modes of interaction appears to occur between pH 7.5 and 7.0. The ionizable groups on the protein surface that become protonated over the pH range where condensation takes place are most likely histidine residues since their typical pK value is near neutrality (29). In HP-NAP, His 9, His 63, His 64 and His 87 are solvent exposed. Though assessment of the role played by any individual residue in DNA condensation is beyond the scope of the present study, it is of interest on a comparative basis that only His 9 is conserved in L.innocua Dps (17) and none in E.coli Dps (10). At pH 8.0, where only DNA binding takes place, a participation of lysine residues endowed with an anomalously low pK value cannot be excluded. They ought to be located in a positive patch as in cytochrome c (30), a likely possibility given the overall positive charge that characterizes the HP-NAP surface (Figure 1).
The mechanism underlying DNA condensation by HP-NAP must differ from that operative for E.coli Dps. In the latter protein, DNA condensation is tightly linked to protein self-aggregation since both phenomena are based on the presence of positively charged N-termini positioned regularly within a negative protein surface. This structural feature promotes on the one hand E.coli Dps self-aggregation through interaction between adjacent dodecamers and on the other binding of DNA. In turn, DNA-bound protein aggregates are able to interact with other protein molecules and/or with other DNA molecules and thereby give rise to large DNA condensates. HP-NAP is characterized by a positively charged surface (22), in sharp contrast to E.coli Dps and other members of the family, and does not self-aggregate. This finding, taken together with the observation that aggregates such as those seen in Figure 2 are formed only when HP-NAP is in the presence of DNA, points to a direct involvement of DNA in the condensation process.
The proposed mechanism is that the interaction of positively charged dodecamers is mediated by interposed negatively charged DNA filaments which act as a glue. To arrive at this proposal, the experiment shown in Figure 4 is crucial. If condensation were to depend on the ability of DNA to bind many dodecamers thereby favoring self-aggregation of DNA-bound Dps molecules due to the increased local protein concentration, 27 bp fragments should not give rise to aggregates because they are able to bind only a few Dps molecules. Since such short DNA fragments do give rise to aggregates, the phenomenon must be based on the interposition of small (polyanionic) DNA fragments between positively charged dodecamers. In other words, DNA condensation is driven by the polyanionic nature of DNA which bridges positively charged HP-NAP molecules leading to the formation of large aggregates. This model for DNA condensation is depicted in Figure 5. At pH 8.5, essentially no DNA binding is observed. Upon decrease in pH, the progressive increase in positively charged residues on the HP-NAP surface leads to DNA binding at pH 7.5 and to condensation at pH 6.5. The similarity between condensates such as those obtained at pH 7.0 (Figure 5) and E. coli nucleoids (31) is striking.
Figure 4.
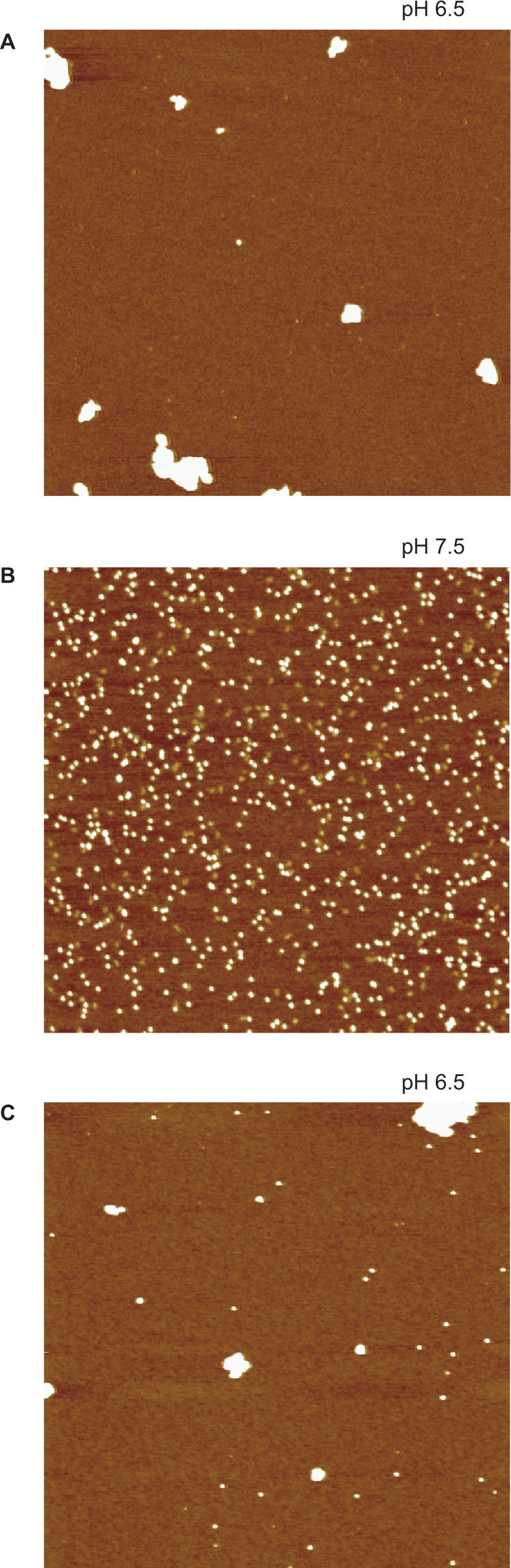
AFM images of H.pylori HP-NAP in the presence of short DNA fragments. (A) HP-NAP incubated with 27 bp dsDNA at pH 6.5. (B) HP-NAP incubated with 27 bp dsDNA at pH 7.5. (C) HP-NAP incubated with 27 nt ssDNA at pH 6.5. The large white globular features in panels A and C represent HP-NAP–DNA aggregates in which DNA is not visible because of its small size. The white dots in panel B represent single HP-NAP dodecamers. The image scan size is 2 μm.
Figure 5.

Proposed model and representative images of DNA binding and condensation by H.pylori HP-NAP. At pH 8.5, HP-NAP dodecamers (blue circles) and DNA (black line) are mainly unbound. A reduction of pH to 7.5 increases the overall positive surface charge of HP-NAP which becomes able to bind DNA non-specifically by interaction with the negatively charged phosphate groups. This primary DNA-binding event generates complexes with a ‘beads-on-a-string’ morphology. Further reduction of pH to 6.5 causes the protonation of surface-exposed amino acid residues (likely histidine residues). Thereby intra- and inter-strand DNA-binding of HP-NAP is rendered possible. This secondary DNA-binding event results in DNA looping and condensation. Thus, HP-NAP–DNA condensation relies on full protonation of amino acid residues which render the overall protein surface positive and capable of multiple binding to the negatively charged DNA polymer. The top and lateral 3D view of selected HP-NAP–DNA condensates at pH 7.0 is given at the bottom. The image scan size is 1 μm.
DNA protection against DNase cleavage and hydroxyl radical formation
To investigate the physiological bearing of the DNA binding/condensation events, DNA protection against DNAse I cleavage and iron-dependent hydroxyl radical formation was assayed.
When DNA is exposed to DNase I following incubation with HP-NAP at different pH values, protection increases progressively with decrease in pH (Figure 6). At pH 6.5, the large HP-NAP–DNA complexes limit nuclease accessibility and DNA is fully protected (Figure 6, lane 3). At pH 7.5, DNA is protected less efficiently due to the occurrence of simple binding (Figure 6, lane 5). At pH 8.5, no protection is observed consistently with the absence of interaction between HP-NAP and DNA (Figure 6, lane 7).
Figure 6.

DNA protection assays against DNase I cleavage. Reaction components and pH conditions are indicated on top of each lane. In each lane linearized plasmid DNA was 20 nM. When present DNase I was 0.3 U and HP-NAP was 2 µM. DNase I treatment was carried out for one minute. All reaction mixtures contained 3 mM MgCl2. Agarose gel was stained with SYBR Green I.
Protection against hydroxyl radicals by Dps proteins is unrelated to DNA binding, but is due to their iron oxidation/incorporation capacity that has its structural basis in the ferroxidase center. As expected on the basis of the sequence conservation at the ferroxidase center and of the HP-NAP contribution to H.pylori survival under oxidative stress conditions (6), DNA is protected to a similar extent by HP-NAP and E.coli Dps (Supplementary Table I).
In conclusion, the present work demonstrates unequivocally that the H.pylori neutrophil-activating protein is able to bind and condense DNA by a mechanism different from that described for E.coli Dps (21). Importantly, DNA condensation occurs at pH values that correspond to stress conditions encountered by H.pylori during gastric colonization (1,2). The bacterium therefore exploits the unusual positively charged surface of HP-NAP not only to activate neutrophils, but also to bind and condense DNA. The latter function may explain the high conservation of HP-NAP among geographically distinct H.pylori strains (7).
The correlation that starts emerging is of interest in more general terms: factors reflecting stress conditions also promote and regulate DNA condensation directly (13,21). Further, condensed structures that can be preserved without energy input (14) act not only as physical shields that protect vital components from detrimental factors, but also can be used to advantage for the organism survival.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
ACKNOWLEDGEMENTS
The authors thank ‘Centro Interfacoltà Misure’ at the University of Parma for the AFM facility and Dr V. Consalvi for help in the fluorescence measurements. The work was supported by the Italian Ministry of University and of Scientific and Technological Research grants FIRB-RBNE01KMT9 to C.R., FIRB-RBAU01YMTR, PRIN 2005 and local funds to E.C. Funding to pay the Open Access publication charge was provided by Ministero della Universitàe Ricerca.
Conflict of interest statement. None declared.
REFERENCES
- 1.Wen Y, Marcus EA, Matrubutham U, Gleeson MA, Scott DR, Sachs G. Acid-adaptive genes of Helicobacter pylori. Infect. Immun. 2003;71:5921–5939. doi: 10.1128/IAI.71.10.5921-5939.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sachs G, Weeks DL, Wen Y, Marcus EA, Scott DR, Melchers K. Acid acclimation by Helicobacter pylori. Physiology (Bethesda) 2005;20:429–438. doi: 10.1152/physiol.00032.2005. Review. [DOI] [PubMed] [Google Scholar]
- 3.Scott DR, Weeks D, Hong C, Postius S, Melchers K, Sachs G. The role of internal urease in acid resistance of Helicobacter pylori. Gastroenterology. 1998;114:58–70. doi: 10.1016/s0016-5085(98)70633-x. [DOI] [PubMed] [Google Scholar]
- 4.Dunn BE, Cohen H, Blaser MJ. Helicobacter pylori. Clin. Microbiol. Rev. 1997;10:720–741. doi: 10.1128/cmr.10.4.720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Montecucco C, Rappuoli R. Living dangerously: how Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2001;6:457–466. doi: 10.1038/35073084. [DOI] [PubMed] [Google Scholar]
- 6.Tonello F, Dundon WG, Satin B, Molinari M, Tognon G, Grandi G, Del Giudice G, Rappuoli R, Montecucco C. The Helicobacter pylori neutrophil-activating protein is an iron-binding protein with dodecameric structure. Mol. Microbiol. 1999;34:238–246. doi: 10.1046/j.1365-2958.1999.01584.x. [DOI] [PubMed] [Google Scholar]
- 7.Dundon WG, Nishioka H, Polenghi A, Papinutto E, Zanotti G, Montemurro P, Del Giudice G, Rappuoli R, Montecucco C. The neutrophil-activating protein of Helicobacter pylori. Int. J. Med. Microbiol. 2002;291:545–550. doi: 10.1078/1438-4221-00165. [DOI] [PubMed] [Google Scholar]
- 8.Cooksley C, Jenks PJ, Green A, Cockayne A, Logan RP, Hardie KR. NapA protects Helicobacter pylori from oxidative stress damage, and its production is influenced by the ferric uptake regulator. J. Med. Microbiol. 2003;6:461–469. doi: 10.1099/jmm.0.05070-0. [DOI] [PubMed] [Google Scholar]
- 9.Martinez A, Kolter R. Protection of DNA during oxidative stress by the nonspecific DNA-binding protein Dps. J. Bacteriol. 1997;179:5188–5194. doi: 10.1128/jb.179.16.5188-5194.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Grant RA, Filman DJ, Finkel SE, Kolter R, Hogle JM. The crystal structure of Dps, a ferritin homolog that binds and protects DNA. Nat. Struct. Biol. 1998;5:294–303. doi: 10.1038/nsb0498-294. [DOI] [PubMed] [Google Scholar]
- 11.Nair S, Finkel SE. Dps protects cells against multiple stresses during stationary phase. J. Bacteriol. 2004;186:4192–4198. doi: 10.1128/JB.186.13.4192-4198.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Choi SH, Baumler DJ, Kaspar CW. Contribution of dps to acid stress tolerance and oxidative stress tolerance in Escherichia coli O157:H7. Appl. Environ. Microbiol. 2000;66:3911–3916. doi: 10.1128/aem.66.9.3911-3916.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wolf SG, Frenkiel D, Arad T, Finkel SE, Kolter R, Minsky A. DNA protection by stress-induced biocrystallization. Nature. 1999;400:83–85. doi: 10.1038/21918. [DOI] [PubMed] [Google Scholar]
- 14.Minsky A, Shimoni E, Frenkiel-Krispin D. Stress, order and survival. Nat. Rev. Mol. Cell Biol. 2002;3:50–60. doi: 10.1038/nrm700. [DOI] [PubMed] [Google Scholar]
- 15.Zhao G, Ceci P, Ilari A, Giangiacomo L, Laue TM, Chiancone E, Chasteen ND. Iron and hydrogen peroxide detoxification properties of DNA-binding protein from starved cells. A ferritin-like DNA-binding protein of Escherichia coli. J. Biol. Chem. 2002;277:27689–27696. doi: 10.1074/jbc.M202094200. [DOI] [PubMed] [Google Scholar]
- 16.Chiancone E, Ceci P, Ilari A, Ribacchi F, Stefanini S. Iron and proteins for iron storage and detoxification. Biometals. 2004;17:197–202. doi: 10.1023/b:biom.0000027692.24395.76. [DOI] [PubMed] [Google Scholar]
- 17.Bozzi M, Mignogna G, Stefanini S, Barra D, Longhi C, Valenti P, Chiancone E. A novel non-heme iron-binding ferritin related to the DNA-binding proteins of the Dps family in Listeria innocua. J. Biol. Chem. 1997;272:3259–3265. doi: 10.1074/jbc.272.6.3259. [DOI] [PubMed] [Google Scholar]
- 18.Ilari A, Stefanini S, Chiancone E, Tsernoglou D. The dodecameric ferritin from Listeria innocua contains a novel intersubunit iron-binding site. Nat. Struct. Biol. 2000;7:38–43. doi: 10.1038/71236. [DOI] [PubMed] [Google Scholar]
- 19.Papinutto E, Dundon WG, Pitulis N, Battistutta R, Montecucco C, Zanotti G. Structure of two iron-binding proteins from Bacillus anthracis. J. Biol. Chem. 2002;277:15093–15098. doi: 10.1074/jbc.M112378200. [DOI] [PubMed] [Google Scholar]
- 20.Ceci P, Ilari A, Falvo E, Chiancone E. The Dps protein of Agrobacterium tumefaciens does not bind to DNA but protects it toward oxidative cleavage: x-ray crystal structure, iron binding, and hydroxyl-radical scavenging properties. J. Biol. Chem. 2003;278:20319–20326. doi: 10.1074/jbc.M302114200. [DOI] [PubMed] [Google Scholar]
- 21.Ceci P, Cellai S, Falvo E, Rivetti C, Rossi GL, Chiancone E. DNA condensation and self-aggregation of Escherichia coli Dps are coupled phenomena related to the properties of the N-terminus. Nucleic Acids Res. 2004;32:5935–5944. doi: 10.1093/nar/gkh915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zanotti G, Papinutto E, Dundon WG, Battistutta R, Seveso M, Del Giudice G, Rappuoli R, Montecucco C. Structure of the neutrophil-activating protein from Helicobacter pylori. J. Mol. Biol. 2002;323:125–130. doi: 10.1016/s0022-2836(02)00879-3. [DOI] [PubMed] [Google Scholar]
- 23.Schuck P. Size-distribution analysis of macromolecules by sedimentation velocity ultracentrifugation and Lamm equation modeling. Biophys. J. 2000;78:1606–1619. doi: 10.1016/S0006-3495(00)76713-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lim WA, Sauer RT, Lander AD. Analysis of DNA-protein interactions by affinity coelectrophoresis. Methods Enzymol. 1991;208:196–210. doi: 10.1016/0076-6879(91)08014-9. [DOI] [PubMed] [Google Scholar]
- 25.Halliwell B, Gutteridge JM. Formation of thiobarbituric-acid-reactive substance from deoxyribose in the presence of iron salts: the role of superoxide and hydroxyl radicals. FEBS Lett. 1981;128:347–352. doi: 10.1016/0014-5793(81)80114-7. [DOI] [PubMed] [Google Scholar]
- 26.Laue TM, Shah BD, Ridgeway TM, Pelletier SL. In: Analytical Ultracentrifugation in Biochemistry and Polymer Science. Harding SE, Rowe AJ, Horton JC, editors. Cambridge: The Royal Society of Chemistry; 1992. pp. 90–125. [Google Scholar]
- 27.Ueshima J, Shoji M, Ratnayake DB, Abe K, Yoshida S, Yamamoto K, Nakayama K. Purification, gene cloning, gene expression, and mutants of Dps from the obligate anaerobe. Porphyromonas gingivalis. Infect. Immun. 2003;71:1170–1178. doi: 10.1128/IAI.71.3.1170-1178.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hong Y, Wang G, Maier RJ. Helicobacter hepaticus Dps protein plays an important role in protecting DNA from oxidative damage. Free Radic. Res. 2006;40:597–605. doi: 10.1080/10715760600618882. [DOI] [PubMed] [Google Scholar]
- 29.Huyghues-Despointes BM, Langhorst U, Steyaert J, Pace CN, Scholtz JM. Hydrogen-exchange stabilities of RNase T1 and variants with buried and solvent-exposed Ala → Gly mutations in the helix. Biochemistry. 1999;38:16481–16490. doi: 10.1021/bi9919450. [DOI] [PubMed] [Google Scholar]
- 30.Crnogorac MM, Ullmann GM, Kostic NM. Effects of pH on protein association: modification of the proton-linkage model and experimental verification of the modified model in the case of cytochrome c and plastocyanin. J. Am. Chem. Soc. 2001;123:10789–10798. doi: 10.1021/ja003818t. [DOI] [PubMed] [Google Scholar]
- 31.Ohniwa RL, Morikawa K, Kim J, Ohta T, Ishihama A, Wada C, Takeyasu K. Dynamic state of DNA topology is essential for genome condensation in bacteria. EMBO J. 2006;25:5591–5602. doi: 10.1038/sj.emboj.7601414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.DeLano WL. The case for open-source software in drug discovery. Drug Discov. Today. 2005;10:213–217. doi: 10.1016/S1359-6446(04)03363-X. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.