

Abstract
The steroidal alkaloid cyclopamine has both teratogenic and antitumor activities arising from its ability to specifically block cellular responses to vertebrate Hedgehog signaling. We show here, using photoaffinity and fluorescent derivatives, that this inhibitory effect is mediated by direct binding of cyclopamine to the heptahelical bundle of Smoothened (Smo). Cyclopamine also can reverse the retention of partially misfolded Smo in the endoplasmic reticulum, presumably through binding-mediated effects on protein conformation. These observations reveal the mechanism of cyclopamine's teratogenic and antitumor activities and further suggest a role for small molecules in the physiological regulation of Smo.
Keywords: Cyclopamine, Smoothened, Hedgehog signaling, teratogen, development
Plants of the genus Veratrum have a long history of use in the folk remedies of many cultures (Namba 1993; Levetin and McMahon 1996), and the jervine family of alkaloids (Fried and Klingsberg 1953), which constitute a majority of Veratrum secondary metabolites, have been used for the treatment of hypertension and cardiac disease. The association of Veratrum californicum with an epidemic of sheep congenital deformities during the 1950s (Binns et al. 1962) raised the possibility that jervine alkaloids are also potent teratogens. Extensive investigations by the U.S. Department of Agriculture subsequently confirmed that jervine and cyclopamine (11-deoxojervine) given during gestation can directly induce cephalic defects in lambs, including cyclopia in the most severe cases (Keeler and Binns 1965).
It is now known that the teratogenic effects of jervine and cyclopamine are due to their specific inhibition of vertebrate cellular responses to the Hedgehog (Hh) family of secreted growth factors (Cooper et al. 1998; Incardona et al. 1998), as first suggested by similarities between the Vertarum-induced developmental malformations and holoprosencephaly-like abnormalities associated with loss of Sonic hedgehog (Shh) function (Chiang et al. 1996; Roessler et al. 1996). In accordance with this general mechanism, cyclopamine also has shown some promise in the treatment of medulloblastoma tumors caused by inappropriate Hh pathway activation (Berman et al. 2002). How cyclopamine specifically inhibits Hh pathway activation is unclear, but it appears to interfere with the initial events of vertebrate Hh signal reception, which involve the multipass transmembrane (TM) proteins Patched (Ptch) and Smoothened (Smo; Ingham and McMahon 2001). During normal Hh signaling, Hh proteins bind to Ptch (Marigo et al. 1996; Stone et al. 1996; Fuse et al. 1999), thereby alleviating Ptch-mediated suppression of Smo, a distant relative of G-protein-coupled receptors (GCPRs). Smo activation then triggers a series of intracellular events, culminating in the activation of Gli-dependent transcription (Alexandre et al. 1996; Aza-Blanc et al. 1997).
Cyclopamine appears to interfere with these signaling events by influencing Smo function, as it antagonizes Hh pathway activity in a Ptch-independent manner and exhibits attenuated potency toward an oncogenic, constitutively active form of Smo (W539L; SmoA1; Taipale et al. 2000). Although these observations suggest that cyclopamine may regulate Smo activity, they reveal neither the biochemical mechanism of Smo activation nor the molecular basis of cyclopamine action. Studies in Drosophila have shown that Hh stimulation is associated with changes in Smo phosphorylation state, subcellular localization, and perhaps protein conformation (Denef et al. 2000; Ingham et al. 2000). In principle, cyclopamine-mediated inhibition of vertebrate Smo activity could perturb any of these cellular events. How Ptch inhibits Smo function is also unclear, although it appears that Ptch acts catalytically through an indirect mechanism (Taipale et al. 2002).
Here we demonstrate that cyclopamine inhibits Hh pathway activation by binding directly to Smo. This binding interaction is localized to the heptahelical bundle and likely influences the Smo protein conformation. Cyclopamine binding is also sensitive to Ptch function, providing biochemical evidence for an effect of Ptch action on Smo structure. Collectively, these results provide a molecular basis for cyclopamine action and suggest that the regulation of Smo activity by Ptch may involve endogenous small molecules.
Results and Discussion
A photoaffinity derivative of cyclopamine specifically cross-links Smo
To determine whether cyclopamine acts directly on Smo, a photoaffinity reagent (PA-cyclopamine; Fig. 1A) was shown to inhibit Shh signaling in a mouse cultured cell assay (Shh-LIGHT2; Taipale et al. 2000) with an IC50 comparable to that of cyclopamine itself (150 nM versus 300 nM, respectively). Light activation of 125I-labeled PA-cyclopamine in live NIH-3T3 cells did not detectably label endogenous mouse Smo (mSmo, henceforth referred to as Smo). As endogenous Smo in these cells is expressed at low levels (Taipale et al. 2002), we tested whether binding could be detected in COS-1 cells transiently transfected with a construct for high-level expression of Smo C-terminally fused to Myc epitopes. Under these conditions, Smo is observed as two distinctly migrating forms, both of which were readily labeled by 125I-labeled PA-cyclopamine upon photoactivation (Fig. 1B). We observed essentially no cross-linking to presumably nonnative, SDS-resistant Smo aggregates, reflecting the requirement for an intact cyclopamine-binding site. Consistent with the resistance of SmoA1 to cyclopamine, PA-cyclopamine also was unable to efficiently cross-link this oncogenic Smo mutant, which is observed as a single form (Fig. 1B). Thus, the W539L mutation either directly disrupts the cyclopamine-binding site or alters the balance between active and inactive Smo states. To investigate the nature of the differently migrating forms of Smo and SmoA1 we characterized them by digestion with endoglycosidase H (endo H), an enzyme capable of hydrolyzing the simpler glycosyl adducts characteristic of the endoplasmic reticulum (ER), but not the more complex adducts associated with post-ER compartments such as the Golgi or the plasma membrane. One form of Smo is endo H-sensitive and presumably localized to the ER; the second form is endo H-resistant and likely represents post-ER protein (Fig. 1C). All of the SmoA1 protein is completely endo H-sensitive (Fig. 1C), suggesting that SmoA1 is trapped in the ER. This localization is confirmed by colocalization of a constitutively active, fluorescent protein-tagged form of SmoA1 (SmoA1–YFP) with an ER-specific marker (Fig. 1D). Accordingly, SmoA1–YFP does not colocalize with a Golgi-specific marker (Fig. 1D).
Figure 1.

A photoaffinity derivative of cyclopamine cross-links Smo. (A) Chemical structure of PA-cyclopamine and its inhibitory activity on Shh signaling. (B) Upon photoactivation, 125I-labeled PA-cyclopamine cross-links two forms of Smo fused at the C terminus to Myc epitopes (Smo–Myc3) in COS-1 cells, and this labeling is inhibited by 1.5 μM KAAD-cyclopamine (left panel). Nontransfected cells and SmoA1–Myc3-expressing cells do not yield specifically cross-linked products. Western analysis with an anti-Myc antibody demonstrates that Smo–Myc3 and SmoA1–Myc3 expression levels are comparable and are not affected by KAAD-cyclopamine treatment (right panel). (C) The two Smo–Myc3 forms represent different glycosylation states, as one is endo H-sensitive (open arrowhead) and the other endo H-resistant (solid arrowhead). SmoA1–Myc3 is exclusively observed as an endo H-sensitive form. Phosphatase treatment did not alter the mobilities of Smo–Myc3 or SmoA1–Myc3 proteins (data not shown). (D) Endo H-sensitivity is indicative of ER localization, as confirmed by the colocalization of SmoA1–YFP (pseudocolored green, top left panel) and an ER marker (pseudocolored red, top middle panel; merge, top right panel) in C3H/10T1/2 cells. Cells expressing both SmoA1–YFP (pseudocolored green, bottom left panel) and a Golgi marker (pseudocolored red, bottom middle panel) exhibit no colocalization (merge, bottom right panel). (E) KAAD-cyclopamine abrogates Smo–Myc3/PA-cyclopamine cross-linking in a manner that is consistent with its inhibitory activity in the Shh-LIGHT2 assay (left panel) without altering cellular levels of Smo–Myc3 (right panel). Both ER and post-ER forms of Smo–Myc3 are depicted as described above. Cross-linking of an endogenous 160-kD protein (B) was competed by KAAD-cyclopamine only at concentrations significantly higher than those required for pathway inhibition (data not shown).
The specificity of PA-cyclopamine cross-linking of Smo is indicated by its efficient competition by a strongly inhibitory dose of KAAD-cyclopamine, a potent derivative of cyclopamine (IC50 = 20 nM in the Shh-LIGHT2 assay; Taipale et al. 2000; Fig. 1B). Upon titration of this reaction with increasing doses of KAAD-cyclopamine, we found that Smo labeling was competed in a concentration range (Fig. 1E) comparable to that required for inhibition of Shh signaling. The cross-linking competition assay thus appears to faithfully reflect the in vivo properties of cyclopamine derivatives in pathway inhibition.
A fluorescent derivative of cyclopamine specifically binds Smo-expressing cells
The specificity of cyclopamine binding to Smo was further confirmed by assays using BODIPY-cyclopamine (Fig. 2A), a fluorescent derivative that retains potency in Shh signaling inhibition (IC50 = 150 nM). This derivative bound with high capacity to a subpopulation of COS-1 cells transiently transfected for expression of Smo, as determined by fluorescence microscopy and flow cytometry (Fig. 2B,C), but did not bind cells expressing SmoA1, nor to cells expressing the Smo protein from Drosophila (Fig. 2C), in which cyclopamine has no effect on Hh signaling (Taipale et al. 2000). BODIPY-cyclopamine also did not bind cells expressing mouse Frizzled7 protein, the closest structural relative of Smo and a member of the Frizzled family of Wnt receptors, nor to cells expressing mouse Ptch (Fig. 2C). BODIPY-cyclopamine binding to cells expressing Smo was blocked by KAAD-cyclopamine in a dose-dependent manner (Fig. 2B,D), with an apparent dissociation constant for KAAD-cyclopamine (KD = 23 nM) comparable to its biological potency. Similar results were obtained with paraformaldehyde-fixed cells (data not shown), ruling out possible artifacts caused by indirect effects of endocytosis or other trafficking processes. We thus observe in both the covalent PA-cyclopamine cross-linking assay and in the noncovalent BODIPY-cyclopamine-binding assay that cyclopamine interacts specifically with Smo and does so with an affinity that corresponds to its IC50 for pathway inhibition. These results strongly support a direct mechanism of cyclopamine action on Smo.
Figure 2.

A fluorescent derivative of cyclopamine binds Smo-expressing cells. (A) Chemical structure of BODIPY-cyclopamine and its inhibitory activity on Shh signaling. (B) BODIPY-cyclopamine binds to a subpopulation of COS-1 cells transfected with a Smo expression construct, and KAAD-cyclopamine inhibits this interaction (KAAD-cyclopamine concentrations shown in boldface type). (C) Specific BODIPY-cyclopamine binding to Smo-expressing COS-1 cells can also be detected by flow cytometry (black trace; left and right panels), as this subpopulation exhibits high fluorescence intensity (brackets). In contrast, cells expressing SmoA1 (blue trace; left panel), mouse Ptch (green trace; left panel), mouse Frizzled 7 (red trace; right panel), or Drosophila Smo (blue trace; right panel) fail to bind BODIPY-cyclopamine in a specific manner. (D) Flow cytometric quantitation of specific BODIPY-cyclopamine binding to Smo-expressing cells (bracket; left panel) can be used to determine the affinities of Smo ligands through binding competitions (black trace, 0 nM; orange trace, 80 nM; red trace, 3 μM KAAD-cyclopamine; left panel), yielding an apparent KD of 23 nM for the KAAD-cyclopamine/Smo complex (right panel).
Cyclopamine binding is localized to the Smo heptahelical bundle
Having established Smo as the direct cellular target of cyclopamine, we investigated the structural determinants of Smo required for its binding. We found that BODIPY-cyclopamine can bind cells expressing Smo proteins that lack either the N-terminal, extracellular cysteine-rich domain (SmoΔCRD) or the cytoplasmic C-terminal domain (SmoΔCT; Fig. 3A), and that binding to either protein is sensitive to competition by KAAD-cyclopamine (Fig. 3B). The different levels of BODIPY-cyclopamine binding associated with Smo, SmoΔCRD, and SmoΔCT likely reflect variations in protein expression levels rather than differences in protein–ligand affinities, as KAAD-cyclopamine inhibited the BODIPY-cyclopamine binding to these different proteins with similar potencies. Thus, despite the importance of the cytoplasmic C-terminal domain of Smo for Hh signaling (J. Taipale and P.A. Beachy, unpubl.), and of the homologous CRD of Frizzled receptors for Wnt-binding and receptor activation (Bhanot et al. 1996), cyclopamine binding of Smo appears not to require these domains. Instead, the cyclopamine binding site in Smo is localized to the heptahelical domain of this integral membrane protein.
Figure 3.

Cyclopamine binds to the heptahelical bundle in Smo. (A) COS-1 cells expressing either SmoΔCRD (middle panel) or SmoΔCT (right panel) were treated with BODIPY-cyclopamine and analyzed by flow cytometry. As with Smo-expressing cells (left panel), a subpopulation of these cells exhibit specific BODIPY-cyclopamine binding (see brackets). (B) BODIPY-binding to these cells is inhibited by 150 nM KAAD-cyclopamine to similar extents.
Cyclopamine binding can alter the conformation of SmoA1
The binding of cyclopamine to the Smo heptahelical bundle suggests that Smo inhibition by this natural product involves a protein conformational shift. The structurally related GPCR family uses a conformational change to link the binding of extracellular ligands to the recruitment of intracellular components, in this case G proteins (Christopoulos and Kenakin 2002). Although G proteins have not been implicated in Smo-mediated pathway activation, an effect of cyclopamine binding on Smo structure is supported by the ability of KAAD-cyclopamine to reverse ER retention of SmoA1. We observed that upon treatment with KAAD-cyclopamine, the localization of green fluorescent protein-tagged SmoA1 (SmoA1–GFP) in C3H/10T1/2 cells expanded to include cytoplasmic vesicles and the plasma membrane, thus more closely resembling the subcellular distribution of GFP-tagged wild-type Smo (Smo–GFP; Fig. 4A). In these experiments, higher concentrations of KAAD-cyclopamine than required for inhibition were used to ensure saturation of binding to SmoA1, which has a lower apparent affinity for cyclopamine and its derivatives. This change in localization is confirmed by a corresponding shift in SmoA1 glycosylation state, as evidenced by the partial conversion of SmoA1 to an endo H-resistant form (Fig. 4B). Similar changes in SmoA1 localization were observed with an Hh pathway agonist that also acts directly on Smo (SAG; Chen et al. 2002; FrankKamenetsky et al. 2002; Fig. 4A), ruling out activity state changes as a critical determinant of SmoA1 exit from the ER.
Figure 4.
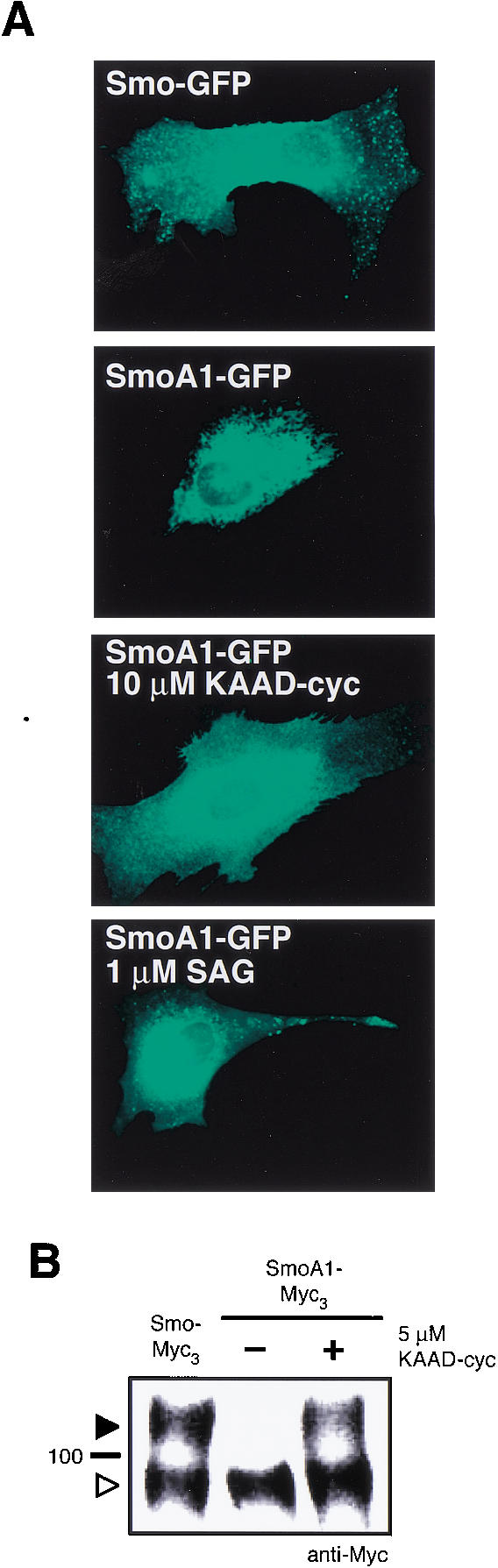
KAAD-cyclopamine binds to SmoA1 and promotes its exit from the ER. (A) Smo–GFP is localized to the plasma membrane and cytoplasmic vesicles of C3H/10T1/2 cells. The ER localization of SmoA1–GFP in C3H/10T1/2 cells is reversed by 10 μM KAAD-cyclopamine or 1 μM SAG, a Hh pathway agonist that directly binds Smo. (B) Glycosylation states of SmoA1–Myc3 upon treatment with 5 μM KAAD-cyclopamine include both endo H-sensitive (open arrowhead) and endo H-resistant (solid arrowhead) forms.
The ER retention of transmembrane proteins, including heptahelical receptors, has been associated with a quality-control mechanism that monitors structurally disordered proteins. For example, the δ opioid receptor is thought to be extensively retained in the ER because of misfolding (Ellgaard et al. 1999), and its export from the ER can be stimulated by the addition of membrane-permeable agonists and antagonists that bind and change receptor structure (Petaja-Repo et al. 2002). Our observations therefore suggest that the W539L mutation produces a partially disordered Smo protein that is retained by the ER quality-control system, and that the binding of small molecules such as cyclopamine or SAG alters SmoA1 structure to resemble a more native state, thus permitting export.
Ptch activity modulates cyclopamine binding to Smo
As both cyclopamine and Ptch negatively regulate Smo activity, we next investigated how Ptch activity influences the ability of Smo to bind cyclopamine. We found that increased levels of mouse Ptch expression in COS-1 cells dramatically enhanced the photoaffinity cross-linking of post-ER Smo by 125I-labeled PA-cyclopamine (Fig. 5A). In contrast, the labeling of ER-localized Smo was not affected, and cellular concentrations of either Smo form were not altered by Ptch expression. Treatment of the Smo- and Ptch-expressing cells with the N-terminal domain of Shh (ShhN) was able to reverse the effect of Ptch expression on PA-cyclopamine/Smo cross-linking, confirming its dependence on Ptch activity (Fig. 5B).
Figure 5.

Ptch activity promotes cyclopamine/Smo complexation. (A) PA-cyclopamine cross-linking of post-ER Smo–Myc3 (solid arrowhead) in COS-1 cells is significantly increased upon Ptch expression in a dose-dependent manner (left panel). The labeling of ER-localized Smo–Myc3 (open arrowhead; left panel) is not affected by Ptch expression, and overall Smo–Myc3 expression levels remain constant (right panel). (B) ShhN reverses the effects of Ptch expression on PA-cyclopamine/Smo cross-linking.
These results provide some insights into the regulation of Smo by Ptch. First, Ptch appears to act only on post-ER Smo, as the PA-cyclopamine cross-linking of ER-localized Smo is independent of Ptch expression levels. This subcellular compartmentalization of Ptch action is consistent with previous observations that Ptch is primarily localized to endosomal/lysosomal vesicles and the plasma membrane (Capdevila et al. 1994; Fuse et al. 1999; Denef et al. 2000). Second, the ability of Ptch expression to significantly increase post-ER Smo labeling by PA-cyclopamine without influencing overall protein levels suggests that the effect of Ptch activity alters Smo conformation and that Ptch and cyclopamine promote inactive Smo states that may be structurally related.
Endogenous small molecules may regulate Smo activity
How Ptch influences Smo conformation remains enigmatic, despite extensive genetic analyses of the Hh pathway. Although it was initially proposed that Ptch and Smo form a heteromeric receptor (Stone et al. 1996), it is now believed that Smo activity is modulated by Ptch in an indirect, nonstoichiometric manner (Taipale et al. 2002). In the case of the Frizzled family of seven-TM receptors, which are closely related to Smo in structure, receptor activation involves the binding of Wnt ligands to the Frizzled CRD (Bhanot et al. 1996) and recruitment of an LDL receptor-related protein (Pinson et al. 2000; Wehrli et al. 2000). No analogous protein interactions have been associated with Smo activation, and removal of the Smo CRD does not appear to significantly alter Smo function or its suppression by Ptch (Taipale et al. 2002).
These observations coupled with the susceptibility of Smo to cyclopamine suggest that Smo regulation may involve endogenous small molecules rather than direct protein–protein interactions. Consistent with this model, Ptch is structurally related to the resistance-nodulation-cell division family of prokaryotic permeases (Tseng et al. 1999) and to the Niemann-Pick C1 protein (Davies et al. 2000), which are capable of transporting hydrophobic molecules. Ptch action might similarly affect the subcellular and/or intramembrane distribution of endogenous molecules, thus influencing Smo activity by altering the localization of a Smo ligand. Alternatively, this Ptch activity could influence membrane structure and Smo trafficking (Sprong et al. 2001); a shift in Smo localization might then be accompanied by activity-modulating changes in the molecular composition of specific subcellular compartments (Sprong et al. 2001).
Pharmacological modulation of Smo activity may be therapeutically useful
The demonstration of cyclopamine binding to Smo establishes the mechanism of action for this plant-derived teratogen. Our studies show that cyclopamine interacts with the Smo heptahelical bundle, thereby promoting a protein conformation that is structurally similar to that induced by Ptch activity. Equally important, these studies reveal the molecular basis for cyclopamine's antitumor activity (Berman et al. 2002) and validate Smo as a therapeutic target in the treatment of Hh-related diseases. Aberrant Hh pathway activation has been associated with several cancers, such as medulloblastoma and basal cell carcinoma (Taipale and Beachy 2001; Wicking and McGlinn 2001), and many of these tumors involve mutations in Ptch or Smo. As a specific Smo antagonist, cyclopamine may be generally useful in the treatment of such cancers, a therapeutic strategy further supported by the absence of observable toxicity in cyclopamine-treated animals (Keeler and Binns 1968; Berman et al. 2002). Additional Smo antagonists might also be discovered through small molecule screens for specific Hh pathway inhibitors, thus comprising a class of pharmacological agents with possible utility in the treatment of Hh-related oncogenesis.
Materials and methods
Preparation of synthetic compounds
Procedures for the chemical synthesis of KAAD-cyclopamine, PA-cyclopamine, and BODIPY-cyclopamine is described elsewhere (Chen et al. 2002).
Cell-based assays for Hh pathway activation
Assays for Hh pathway activation in Shh-LIGHT2 cells, a clonal NIH-3T3 cell line stably incorporating Gli-dependent firefly luciferase and constitutive Renilla luciferase reporters, were conducted as previously described (Taipale et al. 2000).
Preparation of Smo fusion proteins and deletion mutants
Smo–Myc3 and SmoA1–Myc3 contain three consecutive Myc epitopes at the protein C terminus. SmoΔCRD lacks amino acids 68–182, and SmoΔCT lacks amino acids 556–793. Smo–GFP, SmoA1–YFP, and SmoA1–GFP contain fluorescent proteins at the C terminus. All constructs were generated by PCR and verified by DNA sequencing.
Photoaffinity labeling of Smo proteins
COS-1 cells were cultured in 6-well plates and transfected with Smo–Myc3 or SmoA1–Myc3 expression vectors (1 μg/well). Two days after transfection, each well was incubated with 1 μCi of 125I-labeled PA-cyclopamine (∼0.5 nM final concentration) in phenol red-free DMEM containing 0.5% bovine calf serum and various concentrations of the indicated compounds at 37 °C for 10 min. Solvent vehicle alone (MeOH) was used as a control in these experiments. PA-cyclopamine was then activated by 254-nm light (80,000 μJ/cm2; Stratalinker UV-cross-linker) at room temperature. The cells were chilled on ice, removed from the plates by scraping, and then directly lysed and sonicated in SDS-PAGE loading buffer. Total cell lysates were separated by SDS-PAGE and transferred to nitrocellulose for analysis by autoradiography and Western blotting with an anti-Myc monoclonal antibody (9E10; Santa Cruz Biotechnology).
Endo H digestion of Smo proteins
COS-1 cells were cultured in DMEM containing 10% fetal bovine serum in 6-well plates and transfected with Smo–Myc3 or SmoA1–Myc3 expression vectors (1 μg/well). One day after transfection, 5 μM KAAD-cyclopamine was added to a well of the SmoA1–Myc3-expressing cells. Two days after transfection, each well of cells was washed twice with PBS and lysed with 300 μL of RIPA buffer (50 mM Tris-Cl at pH 7.5, 150 mM NaCl, 1% NP-40, 0.5% sodium deoxycholate, 1 mM EDTA, 1 μg/mL leupeptin, 1 μg/mL aprotinin, 0.2 mM PMSF). Cell lysates were centrifuged at 20,000g at 4°C for 15 min, and the supernatant was then centrifuged at 100,000g for 30 min, and the supernatant of the second centrifugation was used for glycosidase treatments and/or SDS-PAGE. For glycosidase treatments, 45 μL of cell lysate was denatured in 0.5% SDS, 1% β-mercaptoethanol at room temperature for 10 min and then incubated in 50 mM sodium citrate (pH 5.5) and 50 units of endo H at 37°C overnight. The 9E10 antibody was used for Western blotting, following SDS-PAGE separation and protein transfer to nitrocellulose.
Localization studies of Smo and SmoA1 proteins
C3H/10T1/2 cells were cultured in DMEM containing 10% fetal bovine serum, 0.5 μg/mL ZnSO4, and β-mercaptoethanol (3.5 μL/500 mL DMEM) on glass coverslips in 6-cm2 dishes. C3H/10T1/2 cells were transfected with either Smo–GFP, SmoA1–GFP, or SmoA1–YFP expression constructs, all of which yield functionally active proteins (data not shown). To assess SmoA1 subcellular localization, either an ER marker (pECFP-ER; Clontech) or a Golgi marker (pECFP-Golgi; Clontech) was cotransfected with the SmoA1–YFP construct. One day after transfection, SmoA1–GFP-expressing cells were treated with either 10 μM KAAD-cyclopamine or 1 μM SAG for 16–20 h. All cells were imaged 2 d after transfection, at 37°C in a closed observation chamber (FCS2; Bioptechs) with constant laminar flow perfusion of culture medium with or without KAAD-cyclopamine or SAG. Fluorescent protein illumination, detection, and imaging were performed on a Zeiss inverted microscope outfitted with a Xenon light source, single or dual-pass filters, and a cooled CCD camera. Images were acquired with Metamorph software (Universal Imaging).
Fluorescence binding assays
COS-1 cells were transfected in 6-well plates with the described expression vectors (1 μg/well), and after 2 d, incubated in DMEM containing 10% fetal bovine serum, 5 nM BODIPY-cyclopamine, and various concentrations of the indicated competitors at 37°C for 4–6 h. For flow cytometry experiments, the cells were then trypsinized, collected by centrifugation, resuspended in phenol red-free DMEM containing 0.5% bovine calf serum, and analyzed for green fluorescence (FACScan, Beckton Dickinson). A fluorescence intensity range that excludes nontransfected cells was then selected for quantification of specific BODIPY-cyclopamine binding (see brackets in Figs. 2C,D and 3A). Curve-fitting analysis was performed with Kaleidograph (Synergy Software).
Studies of Ptch modulation of Smo photoaffinity labeling
COS-1 cells were cultured in 6-well plates and transfected with Smo–Myc3 (0.5 μg/well) and varying amounts of a mouse Ptch–Myc3 expression construct (0, 1.2, 6, 30, and 150 ng/well). A GFP expression construct was used to normalize total transfected DNA levels. One day after transfection, the COS-1 cells were cross-linked with 125I-labeled PA-cyclopamine and processed as described above. To evaluate the importance of Ptch activity in these assays, COS-1 cells transfected with Smo–Myc3 (0.5 μg/well) and either Ptch–Myc3 or GFP expression constructs (0.1 μg/well) were also treated with either ShhN-conditioned medium or control medium at 37°C for 30 min prior to photoaffinity cross-linking.
Acknowledgments
We thank William Gaffield and Akio Murai for gifts of cyclopamine-containing plant extracts and purified cyclopamine, Tapan Maiti for providing ShhN-expressing cells, Jeff Graham and Alan Kerr for assistance in the synthesis of cyclopamine derivatives, and Brian Gladstone for helpful discussions. We also thank Jeffery Porter for communication of the SAG structure and its functional activity prior to publication. J.K.C. is a recipient of Damon Runyon Cancer Research Foundation and American Cancer Society postdoctoral fellowships. M.K.C. is a recipient of a Career Award in the Biomedical Sciences from the Burroughs Wellcome Fund and an NIH K08 award. This research was supported by an NIH grant. P.A.B. is an investigator of the Howard Hughes Medical Institute.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL pbeachy@jhmi.edu; FAX (410) 955-9124.
Article and publication are at http://www.genesdev.org/cgi/doi/10.1101/gad.1025302.
References
- Alexandre C, Jacinto A, Ingham PW. Transcriptional activation of Hedgehog target genes in Drosophila is mediated directly by the Cubitus interruptus protein, a member of the GLI family of zinc finger DNA-binding proteins. Genes & Dev. 1996;10:2003–2013. doi: 10.1101/gad.10.16.2003. [DOI] [PubMed] [Google Scholar]
- Aza-Blanc P, Ramirez-Weber FA, Laget MP, Schwartz C, Kornberg TB. Proteolysis that is inhibited by Hedgehog targets Cubitus interruptus protein to the nucleus and converts it to a repressor. Cell. 1997;89:1043–1053. doi: 10.1016/s0092-8674(00)80292-5. [DOI] [PubMed] [Google Scholar]
- Berman DM, Karhadkar SS, Hallahan AR, Pritchard JI, Eberhart CG, Watkins N, Chen JK, Cooper MK, Taipale J, Olson JM, et al. Medulloblastoma growth inhibition by Hedgehog pathway blockade. Science. 2002;297:1559–1561. doi: 10.1126/science.1073733. [DOI] [PubMed] [Google Scholar]
- Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J, Nusse R. A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature. 1996;382:225–230. doi: 10.1038/382225a0. [DOI] [PubMed] [Google Scholar]
- Binns W, James LF, Shupe JL, Thacker EJ. Cyclopian-type malformations in lambs. Arch Environ Health. 1962;5:106–108. doi: 10.1080/00039896.1962.10663251. [DOI] [PubMed] [Google Scholar]
- Capdevila J, Pariente F, Sampedro J, Alonso JL, Guerrero I. Subcellular localization of the segment polarity protein patched suggests an interaction with the wingless reception complex in Drosophila embryos. Development. 1994;120:987–998. doi: 10.1242/dev.120.4.987. [DOI] [PubMed] [Google Scholar]
- Chen, J.K., Taipale, J., Young, K.E., Maiti T., and Beachy, P.A. 2002. Small molecule modulation of smoothened activity. Proc. Natl. Acad. Sci. U.S.A. (In press). [DOI] [PMC free article] [PubMed]
- Chiang C, Litingtung Y, Lee E, Young KE, Corden JL, Westphal H, Beachy PA. Cyclopia and defective axial patterning in mice lacking Sonic hedgehog gene function. Nature. 1996;383:407–413. doi: 10.1038/383407a0. [DOI] [PubMed] [Google Scholar]
- Christopoulos A, Kenakin T. G protein-coupled receptor allosterism and complexing. Pharmacol Rev. 2002;54:323–374. doi: 10.1124/pr.54.2.323. [DOI] [PubMed] [Google Scholar]
- Cooper MK, Porter JA, Young KE, Beachy PA. Teratogen-mediated inhibition of target tissue response to Shh signaling. Science. 1998;280:1603–1607. doi: 10.1126/science.280.5369.1603. [DOI] [PubMed] [Google Scholar]
- Davies JP, Chen FW, Ioannou YA. Transmembrane molecular pump activity of Niemann-Pick C1 protein. Science. 2000;290:2295–2298. doi: 10.1126/science.290.5500.2295. [DOI] [PubMed] [Google Scholar]
- Denef N, Neubuser D, Perez L, Cohen SM. Hedgehog induces opposite changes in turnover and subcellular localization of patched and smoothened. Cell. 2000;102:521–531. doi: 10.1016/s0092-8674(00)00056-8. [DOI] [PubMed] [Google Scholar]
- Ellgaard L, Molinari M, Helenius A. Setting the standards: Quality control in the secretory pathway. Science. 1999;286:1882–1888. doi: 10.1126/science.286.5446.1882. [DOI] [PubMed] [Google Scholar]
- Frank-Kamenetsky, M., Zhang, X.M., Bottega, S., Guicherit, O., Wichterle, H., Dudek, H., Bumcrot, D., Wang, F., Jones, S., Shulok, J., et al. 2002. Small molecule modulators of Hedgehog signaling: Indentification and characterization of smoothened agonists and antagonists. J. Biol. (In press). [DOI] [PMC free article] [PubMed]
- Fried J, Klingsberg A. The structure of jervine. III. Degradation to nitrogen-free derivatives. J Am Chem Soc. 1953;75:4929–4938. [Google Scholar]
- Fuse N, Maiti T, Wang B, Porter JA, Hall TM, Leahy DJ, Beachy PA. Sonic hedgehog protein signals not as a hydrolytic enzyme but as an apparent ligand for patched. Proc Natl Acad Sci. 1999;96:10992–10999. doi: 10.1073/pnas.96.20.10992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Incardona JP, Gaffield W, Kapur RP, Roelink H. The teratogenic Veratrum alkaloid cyclopamine inhibits Sonic hedgehog signal transduction. Development. 1998;125:3553–3562. doi: 10.1242/dev.125.18.3553. [DOI] [PubMed] [Google Scholar]
- Ingham PW, McMahon AP. Hedgehog signaling in animal development: Paradigms and principles. Genes & Dev. 2001;15:3059–3087. doi: 10.1101/gad.938601. [DOI] [PubMed] [Google Scholar]
- Ingham PW, Nystedt S, Nakano Y, Brown W, Stark D, van den Heuvel M, Taylor AM. Patched represses the Hedgehog signalling pathway by promoting modification of the Smoothened protein. Curr Biol. 2000;10:1315–1318. doi: 10.1016/s0960-9822(00)00755-7. [DOI] [PubMed] [Google Scholar]
- Keeler RF, Binns W. Teratogenic compounds of Veratrum californicum (Durand). I. Preparation and characterization of fractions and alkaloids for biologic testing. Can J Biochem. 1965;44:819–828. doi: 10.1139/o66-100. [DOI] [PubMed] [Google Scholar]
- ————— Teratogenic compounds of Veratrum californicum (Durand). V. Comparison of cyclopian effects of steroidal alkaloids from the plant and structurally related compounds from other sources. Teratology. 1968;1:5–10. doi: 10.1002/tera.1420010103. [DOI] [PubMed] [Google Scholar]
- Levetin E, McMahon K. Plants and society. Dubuque, IA: Wm. C. Brown; 1996. [Google Scholar]
- Marigo V, Davey RA, Zuo Y, Cunningham JM, Tabin CJ. Biochemical evidence that patched is the Hedgehog receptor. Nature. 1996;384:176–179. doi: 10.1038/384176a0. [DOI] [PubMed] [Google Scholar]
- Namba T. The encyclopedia of Wakan-Yaku (traditional Sino-Japanese medicines) with color pictures. Osaka: Hoikusha; 1993. [Google Scholar]
- Petaja-Repo UE, Hogue M, Bhalla S, Laperriere A, Morello JP, Bouvier M. Ligands act as pharmacological chaperones and increase the efficiency of δ opioid receptor maturation. EMBO J. 2002;21:1628–1637. doi: 10.1093/emboj/21.7.1628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinson KI, Brennan J, Monkley S, Avery BJ, Skarnes WC. An LDL-receptor-related protein mediates Wnt signalling in mice. Nature. 2000;407:535–538. doi: 10.1038/35035124. [DOI] [PubMed] [Google Scholar]
- Roessler E, Belloni E, Gaudenz K, Jay P, Berta P, Scherer SW, Tsui LC, Muenke M. Mutations in the human Sonic hedgehog gene cause holoprosencephaly. Nat Genet. 1996;14:357–360. doi: 10.1038/ng1196-357. [DOI] [PubMed] [Google Scholar]
- Sprong H, van der Sluijs P, van Meer G. How proteins move lipids and lipids move proteins. Nat Rev Mol Cell Biol. 2001;2:504–513. doi: 10.1038/35080071. [DOI] [PubMed] [Google Scholar]
- Stone DM, Hynes M, Armanini M, Swanson TA, Gu Q, Johnson RL, Scott MP, Pennica D, Goddard A, Phillips H, et al. The tumour-suppressor gene patched encodes a candidate receptor for Sonic hedgehog. Nature. 1996;384:129–134. doi: 10.1038/384129a0. [DOI] [PubMed] [Google Scholar]
- Taipale J, Beachy PA. The Hedgehog and Wnt signalling pathways in cancer. Nature. 2001;411:349–354. doi: 10.1038/35077219. [DOI] [PubMed] [Google Scholar]
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, Scott MP, Beachy PA. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature. 2000;406:1005–1009. doi: 10.1038/35023008. [DOI] [PubMed] [Google Scholar]
- Taipale J, Cooper MK, Maiti T, Beachy PA. Patched acts catalytically to suppress the activity of Smoothened. Nature. 2002;418:892–896. doi: 10.1038/nature00989. [DOI] [PubMed] [Google Scholar]
- Tseng TT, Gratwick KS, Kollman J, Park D, Nies DH, Goffeau A, Saier MH., Jr The RND permease superfamily: An ancient, ubiquitous and diverse family that includes human disease and development proteins. J Mol Microbiol Biotechnol. 1999;1:107–125. [PubMed] [Google Scholar]
- Wehrli M, Dougan ST, Caldwell K, O'Keefe L, Schwartz S, Vaizel-Ohayon D, Schejter E, Tomlinson A, DiNardo S. arrow encodes an LDL-receptor-related protein essential for Wingless signalling. Nature. 2000;407:527–530. doi: 10.1038/35035110. [DOI] [PubMed] [Google Scholar]
- Wicking C, McGlinn E. The role of Hedgehog signalling in tumorigenesis. Cancer Lett. 2001;173:1–7. doi: 10.1016/s0304-3835(01)00676-0. [DOI] [PubMed] [Google Scholar]