

Abstract
Transposition-based in vitro insertional mutagenesis strategies provide promising new approaches for functional characterization of any cloned gene or genome region. We have extended the methodology and scope of such analysis to a complete viral genome. To map genome regions both essential and nonessential for Potato virus A propagation, we generated a genomic 15-bp insertion mutant library utilizing the efficient in vitro DNA transposition reaction of phage Mu. We then determined the proficiency of 1125 mutants to propagate in tobacco protoplasts by using a genetic footprinting strategy that simultaneously mapped the genomic insertion sites. Over 300 sites critical for virus propagation were identified, and many of them were located in positions previously not assigned to any viral functions. Many genome regions tolerated insertions indicating less important sites for virus propagation and thus pinpointed potential locations for further genome manipulation. The methodology described is applicable to a detailed functional analysis of any viral nucleic acid cloned as DNA and can be used to address many different processes during viral infection cycles.
Potato virus A (PVA) belongs to Potyviridae (genus Potyvirus), the largest virus family infecting plants (Shukla et al. 1994; Urcuqui-Inchima et al. 2001). The PVA genome is composed of a messenger-polarity ssRNA of 9565 to 9572 nt including a 3′-poly(A) tail and a virus-encoded protein (VPg) covalently attached to the 5′-end (Kekarainen et al. 1999; Oruetxebarria et al. 2001). Each end contains a nontranslated region (NTR). The genome encodes a polyprotein that is cleaved into mature proteins via the action of three viral proteinases: P1, HC-Pro, and NIa-Pro (Dougherty and Semler 1993).
All genomic regions and mature proteins of potyviruses are involved in genome amplification (Haldeman-Cahill et al. 1998; Urcuqui-Inchima et al. 2001). The current information regarding potyviral protein function has accumulated mainly from a number of independent studies employing traditional mutagenesis methods (substitutions, deletions, and insertions) on individual proteins, protein-encoding regions, or NTRs in different potyviruses. These strategies are time-consuming and somewhat limited in potyviruses given the genome's large size and a lack of unique restriction sites. The entire potyviral genome has not yet been systematically investigated, except for a study including 19 mutant cDNA clones of Tobacco vein mottling virus, each containing a 12-nt insertion (Klein et al. 1994). Consequently, large regions of the potyviral genome remain to be studied.
We describe here a rapid functional analysis of a complete PVA genome. Our approach is based on the phage Mu in vitro DNA transposition reaction (Savilahti et al. 1995; Haapa et al. 1999a). Following transposition and processing of the reaction intermediates, the resultant viral mutants are inoculated into protoplasts for propagation. Subsequent PCR-based genetic footprinting analysis is used to map genomic regions that are essential/nonessential for virus propagation (Fig. 1). The mutagenesis strategy resulted in a library of PVA mutants, each containing a single 15-bp insertion. The mutated loci were distributed throughout the genome, and 1125 insertions were detected by autoradiography following denaturing polyacrylamide gel electrophoresis. Analysis of the mutants after selection for virus propagation in protoplasts revealed many previously unidentified regions in the PVA genome that are essential for virus propagation. Additionally, many positions in the viral genome tolerated insertions without a detectable effect on virus propagation. Thus, a highly saturated map of essential and nonessential genomic sites was obtained. Subsequent tests on several individual virus mutants in plants demonstrate that the mutant library will also be useful for future studies on various other stages of viral infection at the whole-plant level.
Figure 1.

Schematic presentation of the steps involved in generation and analysis of the insertion mutant library of PVA resulting in a functional map of the genome. pPVA, the infectious cDNA plasmid of PVA.
RESULTS
Generation and Characterization of a 15-bp Insertion Mutant Library
The full-length PVA genome cloned as DNA in a plasmid vector, pPVA, can be used to initiate viral infection (Puurand et al. 1996). To generate an insertion mutant library we first subjected this plasmid to an in vitro DNA transposition reaction catalysed by the phage Mu transposase. Transposition products were nick-translated with DNA polymerase I, digested with NotI, and ligated to produce the mutant clone library (Fig. 2). The resultant library (pPVA-Mu) included approximately 75,000 independent plasmid clones.
Figure 2.

Generation of 15-bp insertion library. (A) A tetramer of MuA transposase (small circles) assembles a DNA transposition complex with a pair of modified (containing NotI site) Mu genome R end-specific DNA segments (dashed line). The complex next executes the transposition reaction by which the Mu end segments are attached to the target plasmid molecules at variable positions, creating a staggered cut. The linear two-ended transposition reaction products are isolated, and gapped reaction products are then processed with DNA polymerase I, digested with NotI, and self-ligated to produce a pool of plasmids with a 15-bp insertion. During the process, 5 bp of the target site is duplicated. (B) The amino acid sequence encoded by the 15-bp insertion (bold type) varies depending on the insertion reading frame. No wild-type amino acids are mutated or deleted and no stop codon is formed. An example of insertions into the coat protein-coding region of PVA is shown. Genomic positions are indicated both by nucleotides and amino acids (numbers). Duplicated target sites are underlined.
Comparative restriction analysis verified the presence of inserts in the library (Fig. 3). NotI digestion did not linearize the original wild-type clone but completely linearized the mutant pool of plasmids. Furthermore, codigestion of the library with SphI, which alone linearized the wild-type clone, produced a smear indicating that a number of different sized restriction fragments were produced. Thus, NotI sites in the library were distributed throughout the plasmid sequence, and virtually all of the DNA molecules in the insertion library contained the predicted NotI site-containing insertion.
Figure 3.

Restriction analysis of pPVA and pPVA-Mu plasmid pools. (A) Plasmid map of pPVA and predicted map of pPVA-Mu are shown. Arrows indicate inserted NotI sites in different molecules. (B) Plasmids were digested with enzymes and analyzed by electrophoresis on a 1.8% TBE gel parallel to nondigested (nc) plasmids. pPVA and pPVA-Mu are denoted as ‘W’ and ‘I,’ respectively. Migration of circular (C) and linear (L) plasmid forms are indicated.
Fine-Mapping of Insertion Sites at the Nucleotide Level and Validation of the Strategy
Ten base pairs in each of the insertion-containing plasmids were derived from the transposon-specific sequence (Fig. 2). This common sequence allowed us to use PCR-based strategies to localize insertion sites. Overlapping genome segments were first amplified with PVA-specific primers, and each PCR product was subsequently used to map the insertion sites by a combination of linear and exponential DNA amplification strategies (Fig. 4, Methods section).
Figure 4.
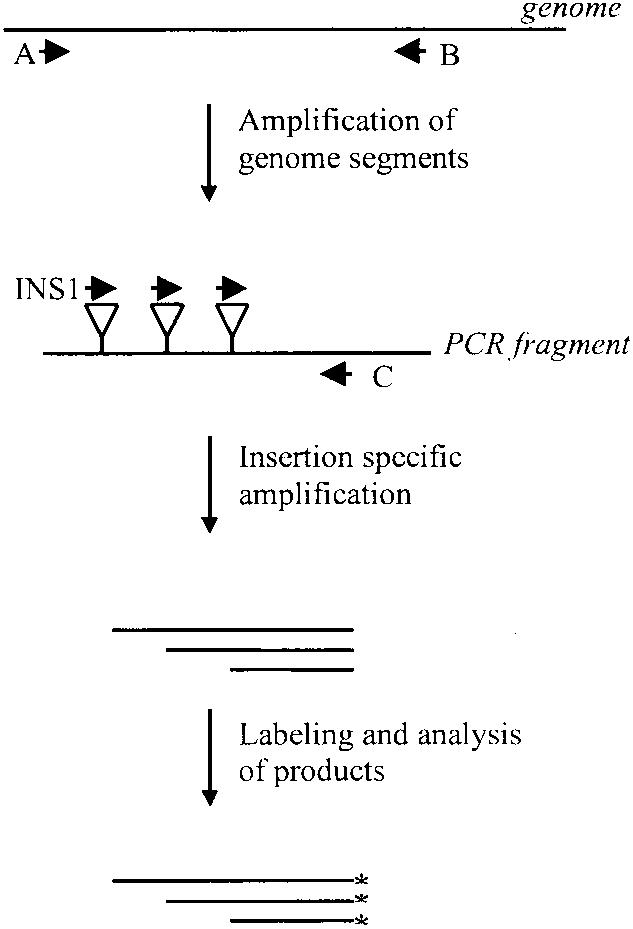
PCR-based footprinting strategy used in fine mapping of insertion sites. Primers are indicated by arrows, insertion sites by triangles, and labeled products by asterisks. Primer INS1: 5′-TATACTCTTCAGATGCGGCCG, with insertion-specific nucleotides underlined. A, B, and C are PVA-specific primers.
To establish the validity of our approach, we first analyzed a segment spanning the PVA genome region between nt 5152 and nt 6375. This segment was amplified from both pPVA-Mu and pPVA, the latter serving as a negative control. Amplified segments were then subjected to insertion-specific PCR analysis using different primer combinations, and the reaction products were analyzed by denaturing polyacrylamide gel electrophoresis and autoradiography (Fig. 5). The pPVA-Mu generated a distinctive band pattern (lanes 1, 3, 5) that was not produced with pPVA (lanes 2, 4, 6). The same band pattern was generated from both 3′- and 5′-labeled amplified molecules (lanes 1 and 3). Furthermore, PVA-specific primers that hybridize to the PVA genome at 14-nt intervals produced identical patterns with a difference corresponding to 14 nt in their electrophoretic mobility (lanes 3 and 5). These data indicated that insertion clones specifically and reproducibly produced amplification products from the library, and that the methodology could be used to reliably map the insertion sites.
Figure 5.

Specificity of amplification. A genome segment of pPVA (W) and pPVA-Mu (I) spanning the region between nt 5152 and nt 6375 was subjected to insertion-specific amplification reactions with the primer pairs INS1/FO12R (FO12R: 5′-AGCCTGATATCAGTATAGGGACTC) and INS1/FO13R (FO13R: 5′-ATAGGGACTCTCATCTAGTGTGTAC). Products from the former reaction were radioactively labeled with the primer INS2 (INS2: 5′-TATACTCTTCAGATGCGGCCGCA, with insertion-specific nucleotides underlined) or FO12R. Products from the latter reaction were labeled with the primer FO13R. Insertions and primers are illustrated by triangles and arrows, respectively. The radioactive labels are indicated by asterisks. (A) Labeled products were separated by urea-PAGE gel. Numbers to the right refer to the band pattern in lane 5 and indicate the positions of insertions in the PVA genome. Sample numbers are indicated below each lane.
Selection for Propagation and Genetic Footprinting
Functional analysis of genome regions can be obtained by the use of comparative parallel analysis of insertion mutant pools. One such method, genetic footprinting, is based on subjecting a mutant pool to selective conditions and subsequently comparing the selected mutants to the input mutant population. We subjected the PVA insertion mutant pool to selection for virus propagation in plant cells by transfecting the pPVA-Mu plasmid library into tobacco protoplasts. After two days of virus propagation, immunocapture with monoclonal antibodies against the virus coat protein was used to collect a sample for viral cDNA synthesis and subsequent genome segment amplification (the sample included mature virions encapsidating viral RNA). The genome segment from the selected pool was then subjected to insertion-specific PCR and gel analysis (as above). The corresponding amplified fragment from pPVA-Mu prior to selection was used for comparison. Bands missing in the selected pool (compared to the unselected pool) indicated that insertions into those positions were not tolerated during viral propagation. To confirm reproducibility, we produced footprinting patterns from two independent inoculations (Methods section, protoplast pools I and II). Patterns along a 200-nt stretch of interest were essentially identical (data not shown), indicating that the protoplasts were efficiently inoculated and that the identification of the essential sites was accurate and reproducible.
We studied the whole virus genome by first amplifying 11 overlapping fragments (1.1 to 1.3 kb each). Genome analysis was facilitated by the use of 75 PVA-specific labeling primers located at ∼200-bp intervals. The overall strategy yielded overlapping band patterns, and thus about 42% of the genome was analyzed twice. An example of the analysis is shown in Figure 6. Many of the insertion-specific amplification products were generated with both the selected and unselected pools. However, a number of the products obtained from the unselected pool were not generated from the selected pool, clearly indicating positions of deleterious insertions. In rare cases, a product was detected only in the selected pool (e.g., a product generated with primer FO10R and indicating an insertion at the genomic site 6125 in Fig. 6B). These products probably indicated mutants that were represented at low numbers in the pPVA-Mu library but which propagated in protoplasts to detectable levels. A total of 1125 essentially randomly distributed insertion sites were detected in the 9565-nt PVA genome at an average interval of 8 nt. Some insertions were detected at adjacent nucleotide positions, whereas others were more distant. The largest interval was 62 nt. A total of 329 sites were found to be essential for production of mature virions, since these sites were not detected in the selected genome pool.
Figure 6.

Collection and analysis of footprinting data. (A) Insertion-specific amplification from the genome segment spanning the region between nt 5152 and nt 6375 was performed using the primer pairs INS1/FO10R (FO10R: 5′-ATTCAACCGACTCTTTCTTCTGTG) and INS1/FO11R (FO11R: 5′-TTCTTCTGTGGTAGTTCAGCATAGT). The products were labeled as indicated by asterisks. (B) The labeled reaction products were analyzed by urea-PAGE. The patterns generated from pPVA-Mu before and after selection for replication in protoplasts are indicated by ‘I’ and ‘P,’ respectively. The patterns from the wild-type clone (pPVA) are indicated by ‘W.’ Genomic location numbers (as in Fig. 5) refer to products labeled with FO10R. Arrows highlight insertions that are missing after selection. Labeling primers are indicated on the top of the gel, and sample numbers are given below each lane.
Regions of the PVA Genome Essential and Nonessential for Virus Propagation
The insertions that specify genomic locations either essential or nonessential for PVA propagation are summarized in Table 1, and their distribution along genomic regions is illustrated in Figure 7. A specification of known functional characteristics of potyviral proteins and genome segments can be found in recent reviews (Shukla et al. 1994; Revers et al. 1999; Urcuqui-Inchima et al. 2001).
Table 1.
The Genomic Regions of PVA, Their Sizes, and the Numbers of Insertions Detected in Them
| Region | Size (nt)b | No. of insertionsa | ||
|---|---|---|---|---|
| Total | Essential sites | Nonessential sites | ||
| 5′NTR | 161 | 22 | 20 | 2 |
| P1 | 891 | 90 | 44 | 46 |
| HC-Pro | 1374 | 152 | 60 | 92 |
| P3 | 1041 | 135 | 33 | 102 |
| 6K1 | 156 | 13 | 4 | 9 |
| CI | 1905 | 218 | 60 | 158 |
| 6K2 | 159 | 20 | 3 | 17 |
| VPg | 567 | 63 | 20 | 43 |
| NIa-Pro | 729 | 91 | 11 | 80 |
| NIb | 1548 | 188 | 39 | 149 |
| CP | 807 | 102 | 31 | 71 |
| 3′NTR | 227 | 31 | 4 | 27 |
| Total | 9565 | 1125 | 329 | 796 |
5′-NTR and 3′NTR: 5′- and 3′-nontranslated regions, respectively; P1, the first protein; HC-Pro, helper component-proteinase; P3, the third protein; 6K1 and 6K2, 6 kDa proteins 1 and 2; CI, cylindrical inclusion protein; VPg, viral genome-linked protein; NIa-Pro, proteinase; NIb, replicase; CP, coat protein.
Essential sites are those in which insertions prevented virus propagation and/or production of mature virions in protoplasts. Distribution of insertions within each of the genomic regions is shown in Fig. 7.
Size in nucleotides (nt). For positions in the genome, see Fig. 7.
Figure 7.

Functional map of the PVA genome. A typical PVA virion visualized by ISEM and a schematic map of the PVA polyprotein are shown at the top. For functions of the various genomic regions, see Table 1. The genomic sites nonessential (N) and essential (E) for viral genome replication and encapsidation are indicated by the bars protruding upwards and downwards, respectively, from the horizontal lines depicting the various genomic regions of PVA. The positions of the genomic regions are indicated by their first and last nucleotide (nt) or amino acid (aa). The oligonucleotides 61475 and 4141 are complementary to the 5′-proximal nucleotides of the 5′-NTR and the 3′-proximal nucleotides of the 3′-NTR, respectively, of the PVA genome, and insertions within these genomic regions could not be detected.
5′-NTR
Potyviruses and picornaviruses share many similarities in genome organization and expression strategies. The viral polyprotein is translated via a cap-independent mechanism (Niepel and Gallie 1999; Belsham and Sonenberg 2000) that is not well understood in potyviruses. The short 5′-NTR (144–205 nt) of potyviruses lacks predictable stable secondary structures but contains cap-independent regulatory elements that promote internal initiation of translation. Except for the first 31 nt conserved in potyviral 5′-NTRs (Simón-Buela et al. 1997; Kekarainen et al. 1999), rather long sequences from the internal regions of the 5′-NTR can be deleted without loss of infectivity. It was therefore unexpected that only two out of a total of 22 viral mutants with an insertion near the 3′-end of the 5′-NTR were recovered from the protoplasts. Apparently, the 5′-NTR of PVA is highly intolerant of insertions.
P1 Region
Most of the essential sites were located at the N-proximal half of the first protein, P1. Insertions at positions adjacent to the translation initiation codon (−2 and +3) were deleterious. While insertions in the N-proximal region of P1 are not allowed in PVA, propagation proceeds in Tobacco etch potyvirus (TEV) despite the deletion of the corresponding region (Verchot and Carrington 1995). The C-proximal half contains a proteinase domain that cleaves P1 from the polyprotein. It tolerated most insertions. Since no insertions were detected in the pPVA-Mu library at the catalytic triad of the P1 proteinase, this domain was not analyzed. Insertions within the conserved Val-Arg-Gly sequence of potyviruses, known to inhibit P1 proteolysis upon deletion, were detrimental in our analysis. An insertion at the proteinase recognition residue (His297) at the P1/HC-Pro junction was tolerated, reflecting the fact that the Mu transposition system does not substitute or delete any amino acid residues but rather inserts five amino acids into the target sequence. Consequently, His297 was retained in the mutated virus. These data highlight the interesting differences in insertional versus deletion mutations within the P1 region.
HC-Pro Region
Six novel sites essential for virus propagation were located within the putative domain involved in suppression of RNA silencing, an antiviral host defense mechanism (Kasschau and Carrington 2001). This novel finding supports an idea that suppression of RNA silencing may be crucial for infection. All insertions within the region involved in homodimerization of PVA HC-Pro (Guo et al. 1999) were deleterious. This original result indicates that homodimerization may be important for HC-Pro function. PVA tolerated insertions within the invariant sequence Pro606-Thr607-Lys608 that is critical for aphid transmissibility (Atreya and Pirone 1993), agreeing that the PTK motif is important to vector transmissibility rather than to virus propagation. In contrast, an insertion at Arg666 within the putative RNA binding domain prevented propagation of PVA, consistent with the idea that interactions between the propagation proteins and viral RNA are essential for viral RNA amplification. Also, insertions at several other domains conserved in potyviruses, such as the Lys350-Ile351-Thr352-Cys353 motif, were deleterious, consistent with previous studies on Tobacco vein mottling potyvirus (TVMV) (Atreya et al. 1992; Atreya and Pirone 1993).
Twenty-two of the 48 insertions within the C-terminal proteinase domain, which cleaves the polyprotein between HC-Pro and P3, were at essential sites. For example, an insertion at Gly756 flanking the protein cleavage site between HC-Pro and P3 was deleterious; substitution of the corresponding residue is deleterious to TEV (Kasschau and Carrington 1995). However, insertions at the HC-Pro active-site nucleophile (His714) and at Tyr752 and Arg753 at the proteinase cleavage site were tolerated in PVA.
P3 Region
The functions of P3 remain largely obscure (Johansen et al. 2001). Both N- and C-terminal portions of P3 contained many essential sites. PVA did not tolerate insertion at Gly756 at the HC-Pro/P3 cleavage boundary. In addition, two insertions (at Phe1101 and Ala1103) altering the NIa-Pro proteinase recognition site at the P3/6K1 junction were deleterious, but two other insertions located between Phe1101 and Gln1102 and between Gln1102 and Ala1103 were tolerated. An insertion between Ala810 and Leu811 rendered PVA noninfectious, consistent with work on TVMV (Klein et al. 1994). Our comprehensive results identified many previously undefined regions within P3 that are essential for virus propagation, and thus underscored the general importance of P3 in virus propagation.
CI Region
The CI protein is an RNA helicase that contains prototypical superfamily 2 (SF2) helicase domains in the N-proximal region, as shown with tamarillo mosaic virus (Eagles et al. 1994), a strain of PVA (Kekarainen et al. 1999). Many novel essential sites were mapped throughout the CI protein; for example, upstream of helicase motif I, between helicase motifs III and IV, and near the C-terminus. Insertions were tolerated at Gly1244 within helicase motif I (nucleotide binding motif) and at six amino acids within helicase motif IA, but insertions at Pro1267 and Leu1276 within motif IA were detrimental. Some insertions within helicase motif II (Phe1324, Ile1326, and Ile1327) were tolerated, whereas insertion at Pro1360 in the helicase motif III was deleterious. Within helicase motif IV, one deleterious insertion was detected at the conserved Arg1435. Within helicase motif V, insertions at four amino acids were deleterious, whereas those at eight others were tolerated. Two of these deleterious insertions (Thr1455, and between Ile1458 and Glu1459) correspond to conserved residues in which different types of mutations make TVMV and Plum pox potyvirus noninfectious (Klein et al. 1994; Fernández et al. 1997). Insertions within helicase motif VI between Gly1501 and Glu1502, between Val1510 and Gly1511, and at conserved Arg1512 were deleterious. These regions are involved in RNA binding. Taken together, these data indicate that both essential and nonessential sites are distributed throughout the helicase domains of CI, consistent with the three-dimensional structure of the SF2 RNA helicase, in which the different helicase motifs are closely connected in the tertiary structure of the protein and form one large functional domain rather than seven different domains with independent functions (Yao et al. 1997). A significant number of insertions (12 of 32, or 38%) were deleterious within the region between amino acids 1647 and 1722 important for CI RNA unwinding activity. An insertion was tolerated at Ala1785 but not at Gln1787 within the NIa-Pro recognition sequence at the C-terminus.
The 6K1 and 6K2 Regions
The 6K1 and 6K2 regions flank the CI domain. The 6K peptides are quite similar in size, contain hydrophobic stretches, and their positions in the potyviral genome correspond to the picornaviral 2B and 3A peptides involved in viral replication and membrane binding, respectively (Giachetti and Sembler 1991; Johnson and Sarnow 1991). Indeed, a hydrophobic domain within the central part of 6K2 is probably involved in membrane binding of 6K2 and is necessary for infectivity (Schaad et al. 1997). In contrast, no specific functions are known for the 6K1 protein. Deletion of either of the two 6K domains prevents replication of PVA (A. Merits, M.-L. Rajamäki, T. Kekarainen, J.P.T. Valkonen, unpubl.). Four of the 6K1 insertions and three of the 6K2 insertions were deleterious. The essential sites were near the N-terminus in both 6K peptides (Fig. 7), suggesting that these domains may have replicative functions.
VPg Region
The genome-linked protein, VPg, is the N-proximal half of the NIa protein separated from the C-proximal proteinase domain (NIa-Pro) of NIa via a suboptimal proteolytic cleavage site. An insertion at the proteolytic site between 6K2 and VPg (at Gly1848) was deleterious. All insertions within the region corresponding to the domain interacting with the eukaryotic initiation factor eIF(iso)4E in Turnip mosaic potyvirus (Leonard et al. 2000) were tolerated in PVA. The insertion at Met1958 involved in vascular movement of PVA (Hämäläinen et al. 2000) did not prevent propagation in protoplasts. Insertions at the nuclear localization signal (NLS) of VPg were not deleterious, similar to amino acid substitutions that are tolerated in the NLS of TEV (Schaad et al. 1996). An insertion adjacent to Tyr1905 linking VPg to viral RNA had no detectable effect on infectivity. However, novel, essential clusters of amino acids in VPg were mapped between Arg1863 and Ala1873, between the NLS and Tyr1905, between Ser1980 and Asp1986, and between Pro2006 and Ala2024.
NIa-Pro Region
Only 11 of a total of 91 insertions in NIa-Pro were deleterious. The deleterious insertions were located at sites not previously known to be essential for virus infectivity. One deleterious insertion was at the cleavage site between NIa-Pro and NIb (between Gln2274 and at Gly2275). The data are consistent with the fact that the primary function of NIa-Pro is to proteolytically process the viral polyprotein, and the great majority of the insertions did not affect this activity.
NIb Region
NIb, an RNA-dependent RNA polymerase (RdRp), is the most conserved of the potyvirus proteins. The picornaviral polymerase supergroup contains eight conserved motifs within the central region (amino acids 2444–2692 in PVA NIb). One insertion (Arg2450) within conserved motif I was deleterious. Seven insertions were tolerated in the motif II; one (between Val2478 and Asp2479) was not. Within motif III, two (at Trp2505 and Pro2513) of the four insertions were deleterious. All insertions within the polymerase motif V were tolerated. Insertions within motifs VI and VII were tolerated, except at Asn 2626 in motif VI. However, most of the deleterious insertions were upstream of the conserved polymerase motifs, indicating that NIb contains novel domains essential for replication but for which exact functions are unknown.
CP Region
Potyvirus particles are assembled from ∼2000 coat protein (CP) units that encapsidate the viral RNA. The core region of CP forms a structural domain involved in RNA binding, and the N- and C-termini are exposed on the virion surface (Shukla et al. 1994). The essential sites were mapped to the N-proximal half and to a short region close to the C-terminus of CP. Mutants containing insertions in the central region at the conserved amino acid residues within the virion assembly domain (Shukla et al. 1994) were not recovered from protoplasts, except for those containing an insertion at Cys2911 or Pro2918. These results do not necessarily indicate a lack of infectivity; they may simply reflect the inability to form virions (only mutants forming virions in protoplasts were immunocaptured for analysis). A large segment located downstream of the virion assembly domain did tolerate insertions, but insertions at four conserved residues (Leu3027, Glu3035, Arg3040, and Thr3042) within the surface-exposed C-terminus were deleterious.
3′-NTR
Viral positive-strand RNA genomes function as an mRNA as well as a template for minus-strand RNA synthesis in genome replication, and the 3′-NTR interacts with the viral RdRp. Only four insertions (between nt 9363 and nt 9675) of a total of 31 insertions in the 3′-NTR were deleterious. Although the extreme terminus (the last 50 nucleotides) was not analyzed (see Methods), the data nevertheless indicate that most of the 3′-NTR region tolerated the 15-bp insertions without detectable effects on virus propagation. The four insertions not tolerated within a short area of the 3′-NTR may have disrupted important RNA secondary structures; for example, those formed between the 3′-NTR and the 3′-portion of the CP-coding region (Haldeman-Cahill et al. 1998).
Analysis of Individual Insertion Mutants
We selected several independent insertion mutant clones from the pPVA-Mu library to test their individual infectivity in protoplasts and plants. Seven mutants not detected after selection en masse in protoplasts were tested first, and none of them was infectious in protoplasts, as expected (data not shown). Other mutants containing an insertion at different proteins (HC-Pro, P3 (two mutants), CI, NIa-Pro, NIb, or CP) and which were detected after selection en masse, as well as two mutants (insertion in P1 or VPg) that had not been detected in the pPVA-Mu library by footprinting analysis (because low signals were disregarded), were also tested. Following protoplast inoculation, three mutants containing an insertion in P1, HC-Pro, or P3 propagated and accumulated CP antigen to levels detectable by ELISA, in contrast to the other mutants tested. Tobacco plants were systemically infected only with the mutant containing an insertion in P1 and one of the two mutants containing an insertion in P3. Insertions were detected by RT-PCR and subsequent digestion of the PCR product with NotI in the progeny viruses so as to indicate that the mutants had not reverted to wild-type in the systemically infected leaves. Electron microscopy revealed PVA virions of a similar size in the protoplasts infected with the three infectious mutants and the wild-type PVA (data not shown).
These results show that mutants that were actually able to propagate alone in protoplasts were fewer in number than those detected in the genetic footprinting analysis, due most likely to in trans complementation by other mutants during the en masse selection. The mutant containing an insertion in HC-Pro could infect protoplasts but caused no detectable infection in leaves, due presumably to impaired viral genome amplification and/or cell-to-cell movement. Infection of protoplasts by viruses may be less constrained than infection of cells in their natural intercellular context in leaf tissues, because many resistance responses induced by virus infection in leaves and whole plants are not similarly expressed in protoplasts (Chivasa et al. 1997). The insertion in HC-Pro was at a site previously unknown with respect to its functional significance. Taken together, these data demonstrate the potential of the pPVA-Mu library for studies on virus infection at the whole-plant level.
DISCUSSION
We used a Mu in vitro DNA transposition-based strategy to generate a large, genome-wide insertion mutant library for a plant-infecting potyvirus, PVA. Mutants were selected for their ability to propagate in protoplasts and form virus particles, and then screened en masse to map the genomic sites essential for viral propagation. This strategy allowed the efficient examination of genomic regions never before studied for their importance in viral genome amplification.
A total of 1125 essentially randomly distributed insertions were mapped within the PVA genome. Thus, the pPVA-Mu library provided a unique strategy to study functional genomics of the entire PVA genome. Insertions mapped to previously studied genomic regions revealed that a wealth of our pPVA-Mu mutant results are consistent with previous data obtained with fundamentally different methods, thus validating our approach with respect to identifying essential and nonessential sites in the viral genome. A total of 329 PVA mutants were not recovered from protoplasts into which the mutant library was inoculated for propagation, revealing hundreds of essential, previously unknown sites in the potyvirus genome. Further examination of these sites will be the focus of future studies.
An overview of the functional map of the PVA genome constructed in this study (Fig. 7) reveals several interesting features. One of them is the striking difference in the tolerance of short insertions in the 5′- and the 3′-NTRs. Low tolerance of insertions in the 5′-NTR may indicate that insertion mutants possibly contain more stable RNA structures due to the palindromic sequence in the insertion, which may interfere with the initiation of translation. Alternatively, the mutants may be defective in virus assembly (Johansen and Morrow 2000) beginning from near the 5′-terminus of the potyviral genome (Wu and Shaw 1998) and therefore excluded in immunocapture–RT-PCR. Some insertions at the core region of the CP might also have interfered with virus assembly, resulting in exclusion of the mutants from footprinting analyses. The high tolerance of 3′-NTR to short insertions is consistent with the view that only a few predictable, stable secondary structures are important to infectivity in the potyviral 3′-NTR (Haldeman-Cahill et al. 1998).
Another interesting feature is the distribution of the essential sites in the protein-encoding regions of PVA (Fig. 7). A few insertions representing sites essential for propagation were typically located next to each other, followed by tens of codons tolerant of insertions. These data provide confidence that the insertions do, in fact, pinpoint essential sites. Such information will be helpful for future investigation of protein function in large proteins such as P3 that are poorly understood. In our analysis, 33 of the total of 135 insertions in P3 were deleterious. The positions of 22 deleterious insertions matched exactly with, or were one or two nucleotides apart from, the codons for amino acid residues highly conserved among potyviruses (Shukla et al. 1994) and/or identical between PVA and Potato Y potyvirus (data not shown). Taken together, these findings provide validation of the resultant map of essential sites in the PVA genome (Fig. 7).
On the other hand, several protein-encoding regions such as P1, P3, NIa-Pro, and CP contain large regions tolerant of the short insertions. Potyvirus genomes tend to be recalcitrant to insertion of foreign sequences, and identification of suitable insertions sites is particularly cumbersome (German-Retana et al. 1999; Choi et al. 2000). Therefore, data from this study may be helpful for engineering PVA-based gene vectors for expression of foreign peptides or viral proteins with novel epitopes or tags in plants. The tests on tobacco plants performed with two PVA mutants containing an insertion in P1 or P3 showed that they were fully infectious, inducing systemic infection similar to that of wild-type PVA.
In some cases, our results differed from the data obtained using traditional mutagenesis methods in that an insertion within a protein domain known to be important for virus propagation was not deleterious. The most likely explanation for such discrepancies is the fact that the Mu transposition system used does not substitute or delete any amino acid residues, and the original amino acids were retained in the mutated virus. Several insertional mutagenesis studies indicate that insertions do not always perturb protein function (Manoil and Bailey 1997; Nelson et al. 1997; Neuveglise et al. 1998; Laurent et al. 2000). Another explanation may be that in trans complementation (e.g., Li and Carrington 1995) between two viral genomes carrying insertions at different positions and coinfecting protoplasts might mask mutant phenotypes and interfere with detection of certain essential sites. Successful propagation of some insertion mutants during en masse selection and their failure to do so when inoculated to protoplasts one by one (Table 2) is consistent with this possibility. Thus, many of the essential sites mapped in this study by en masse selection probably represent those that cannot be complemented in trans.
Table 2.
Characterization of Nine Insertion Mutants of PVA Isolated from the Mutant cDNA Library
| Position of insertion | Propagation in protoplasts | ||||
|---|---|---|---|---|---|
| Region | nt | En masse selection | Inoculation individually | Systemic infection in tobacco plants (ng/g)a | Plasmid name |
| P1 | 235 | nt | + | 5300 (527) | pM14 |
| HC-Pro | 1831 | + | + | 0 | pM65 |
| P3 | 3249 | + | − | nt | pM9 |
| P3 | 3379 | + | + | 5500 (1233) | pM45 |
| CI | 4820 | + | − | nt | pM60 |
| VPg | 5956 | nt | − | nt | pM25 |
| NIa-Pro | 6595 | + | − | nt | pM56 |
| NIb | 8330 | + | − | nt | pM34 |
| CP | 9149 | + | − | nt | pM49 |
| pPVA | no insertion | ni | + | 6150 (375) | pM52 |
nt, not tested because the mutant was probably represented in low numbers in the pPVA-Mu mutant library and was not detected by footprinting analysis prior or after en masse selection in protoplasts; ni, not included in the en masse selection experiment; +, propagation observed in protoplasts; −, no detectable propagation in protoplasts.
mean amount of PVA coat protein antigen (in nanogram) per gram (g) of infected leaf (SE) tissue as determined by DAS-ELISA using known amounts of PVA virions as a comparison; nt, not tested.
PCR-based footprinting strategies have been used to map insertion sites efficiently in a relatively short genome region (Smith et al. 1995; Singh et al. 1997; Laurent et al. 2000), but the current strategies are not practical for the analysis of long molecules, for example, complete viral genomes. For the analysis of short insertions throughout the whole genome of PVA, we therefore designed a strategy that used a 21-nt primer (INS1) including eight nucleotides complementary to the inserted sequence at the 3′-end and a stretch of 13 nucleotides at the 5′-end which hybridized neither to the viral sequence nor the insert. This primer was used under optimized conditions together with PVA-specific oligonucleotides to map insertions throughout the pPVA-Mu insertion library before and after selection in protoplasts. This footprinting strategy is efficient for the analysis of insertion sites in large plasmids or complete genomes because it is not affected by insertions located outside the region of interest in other molecules present in the mutant pool. Furthermore, a low insertion frequency at any given region does not disturb the analysis.
The number of PVA mutants obtained in this study is remarkable, and the insertion mapping and mutant selection strategies employed yielded information on ∼12 % of all possible sites. However, the Mu integration target site selection is relatively flexible (Haapa et al. 1999a; Haapa-Paananen et al., 2002) and, consequently, the analyzed mutants represent only a fraction of the total number of different mutants present in the library. Indeed, we could isolate individual mutants (pM14 and pM25; Table 2) that were not detected by our footprinting analysis. Although most of the DNA phosphodiester bonds can serve as effective targets for Mu transposition, some of the bonds are preferred to others (Haapa-Paananen et al. 2002). Consequently, the resultant mutant library inevitably contains the representative clones in different numbers, allowing our footprinting assay to identify only those mutant clones that are present in numbers above a certain threshold level. Analysis of additional mutants using the type of assay chosen for our study would require lowering the threshold; for example, by using more cycles in PCR and/or accepting weaker signals.
Besides Mu (Haapa et al. 1999a,b; Laurent et al. 2000), several other in vitro transposition-based methods have been developed for use as tools for molecular biology applications. These methods include bacterial transposons Tn552 (Griffin et al. 1999), Tn7 (Gwinn et al. 1997), and Tn5 (York et al. 1998), the yeast retrotransposon Ty1 (Devine and Boeke 1994), MoMLV retroviral integrase (Singh et al. 1997), and the eukaryotic mariner element Himar 1 (Akerley et al. 1998). Since these diverse systems function similarly, it may be feasible to use them in a manner analogous to our study. The Mu transposon tool is composed of only a few components and utilizes highly efficient, accurate, and essentially random in vitro DNA transposition (Haapa et al. 1999a). The system has enabled us to map many novel sites essential for virus propagation in PVA. Furthermore, this study provides novel data for future studies on protein structure-function relationships in potyviruses. The pPVA-Mu library is a valuable source of specific insertion mutants that can be studied in more detail to characterize the functional domains of different proteins in virus propagation, movement, virion assembly, or for engineering PVA for vector purposes. This is the first study in which an entire virus genome, rather than a genome segment (Laurent et al., 2000), has been mutagenized using an in vitro DNA transposition system and the mutants analyzed using a genetic footprinting technique. Furthermore, the strategy described is applicable to any cloned DNA.
METHODS
Molecular Biology Techniques, Proteins, and Reagents
Standard DNA techniques were used as described (Sambrook et al. 1989). Plasmids, PCR products, and RNA were purified using appropriate purification kits from QIAGEN. The infectious plasmid pPVA (Puurand et al. 1996), containing the PVA-B11 genome (EMBL database accession no. AJ296311) under the control of Cauliflower mosaic virus 35S promoter, was further purified by CsCl gradient centrifugation. Phage Mu transposase (MuA) was purchased from Finnzymes and Taq DNA polymerase from Fermentas. DNA polymerase I (Pol I), T4 DNA polynucleotide kinase (PNK), restriction endonucleases, and the random hexanucleotide mixture (dN)6 were purchased from Promega. T4 DNA ligase and SuperscriptII reverse transcriptase were from Life Technologies, and [γ-33P]-ATP from NEN Life Science Products. All reagents were used under the reaction conditions recommended by the supplier unless otherwise indicated.
In Vitro DNA Transposition Reaction, Processing of Reaction Products, and Electrotransformation
Mu-specific donor DNA segment was produced from the oligonucleotides TK7650 (5′-GTTTTCGCATTTATCGTGAA ACGCTTTCGCGTTTTTCGTGCGCGGCCGCA) and TK7651 (5′-TGCGGCCGCGCACGAAAAACGCGAAAGCGTTTCAC GATAAATGCGAAAAC) by annealing as described (Savilahti et al. 1995). Standard in vitro DNA transposition reactions (50 μL) contained 1320 ng (40 pmol) donor DNA segment, 1670 ng pPVA DNA (0.19 pmol), 8.7 μg MuA (107 pmol), 25 mM Tris-HCl, pH 8.0, 100 μg/mL BSA, 15% (w/v) glycerol, 0.05 % (w/v) Triton X-100, 126 mM NaCl, and 10 mM MgCl2. Reactions were performed at 30°C for 1h 15 min and stopped by freezing in liquid nitrogen. Reaction products were analyzed by electrophoresis on a 1.8% SeaPlaque agarose (BMA) gel in 1× TAE buffer as described (Savilahti et al. 1995). Linear reaction products from a total of 16 transposition reactions were first recovered by electroelution from the gel and then purified by sequential phenol and chloroform extractions, followed by precipitation with ethanol and resuspension in TE-buffer (10 mM Tris-HCl, pH7.5; 0.5 mM EDTA). The product was then subjected to a nick-translation reaction (100 μL) containing ∼2 μg DNA, 5 mM dNTPs, and 9 U Pol I. The reaction was performed at 37°C for 1h 10 min after which the reaction products were purified as above. The DNA was then shortened by digestion with NotI and isolated using the anion exchange column Gen-Pak FAX (Waters) and ethanol precipitation. Plasmid molecules were then recircularized by ligation at a low DNA concentration (∼1 ng/μL) to favor intramolecular ligation. Ligated DNA (600 ng) was purified as above and resuspended in 10 μL of water. Aliquots (1 μL) were electroporated with Genepulser II (BioRad) into 40 μL of E. coli XL1-Blue MRF' electroporation-competent cells (Stratagene). Transformed cells were selected using Luria medium (LB) with ampicillin (100 μg/mL) at 37°C, after which the plasmid DNA (pPVA-Mu) was isolated.
Protoplast Preparation, Electroporation, and Immunocapture of Virions
Protoplasts were prepared from leaves of in vitro-grown plants of Nicotiana tabacum cv. Samsun and electroporated as described (Denecke et al. 1989) using a BioRad Genepulser II. A total of 17 protoplast batches (200 μL, 1 × 106 protoplasts) were electroporated with pPVA-Mu (10 μg) linearized with AgeI. Each batch was then cultured separately in daylight at room temperature for 48 h, after which protoplasts were pelleted by centrifugation (Denecke et al. 1989). Supernatants were removed and protoplasts resuspended in 100 μL of ELISA buffer (Clark and Adams 1977) and disrupted with a syringe by passing them through a needle (gauge 0.9 mm) several times. The absence of intact protoplasts was confirmed by light microscopy. To confirm PVA infection in each protoplast batch, half of the disrupted protoplast sample was analyzed by double antibody sandwich enzyme-linked immunosorbent assay (DAS-ELISA) using a monoclonal antibody (Mab) 58/0 (Adgen) that detects an epitope at the N-terminus of PVA CP (Rajamäki et al. 1998). From the remaining half of the sample, virions were trapped to a microcentrifuge tube with MAb 58/0, followed by reverse transcription (RT) (final vol. 50 μL) of the viral RNA released from virions using Superscript II reverse transcriptase and random hexamers as recommended by the manufacturer. This method, coined as immunocapture-RT-PCR, is described elsewhere (Nolasco et al. 1993). After the RT reactions, the viral cDNA from four and 13 protoplast batches was pooled (pools I and II, respectively), and the pools were used for footprinting analysis.
Genetic Footprinting
The overall strategy used in genetic footprinting is illustrated in Figure 4. Eleven overlapping genomic segments (∼1.1 to 1.3 kb each) were first amplified by PCR using appropriate pairs of PVA-specific oligonucleotides (Table 3). Ten nanograms of plasmid DNA (pPVA or pPVA-Mu) or three microliters of the cDNA obtained from immunocaptured virions was used as template. The reaction (50 μL) was initially heated for 1 min at 95°C, after which 28 cycles of amplification were performed for 1 min at 95°C, 1 min at the estimated optimal annealing temperature of the oligonucleotide pairs (Birren et al. 1997), and 1 min at 72°C. Each PCR product was then purified, and 50 ng of DNA was used as a template for insertion-specific PCR amplification (50 μL). This amplification was initiated using INS1 primer, and after five cycles of linear amplification (1 min at 95°C, 1 min at 45°C, and 1 min at 72°C) a PVA-specific primer was added and 10 more cycles of amplification were performed (1 min at 95°C, 1 min at 58–64°C depending on the oligonucleotide, and 1 min at 72°C). PCR products were purified and equivalent amounts were labeled using the appropriate PVA-specific primer 5′ end-labeled with [γ33-P] ATP and PNK. Ten pmol of labeled oligonucleotide was used in a 25μL labeling-amplification reaction (5 cycles with 25 sec at 95°C, 25 sec at optimal annealing temperature of the labeled oligonucleotide used, and 30 sec at 72°C). The reaction was analyzed using 7 M urea-6% polyacrylamide gel electrophoresis. Visualization/quantitation of the reaction products was performed on a Phosphorimager (Molecular Dynamics) using the ImageQuant program (Molecular Dynamics). Only those band intensity values from the unselected pool that exceeded those of the selected pool by at least a factor of five were considered indicative of deleterious sites. In practice, only clear bands that were missing in the comparison lane were included in the data set. Rarely, control reactions (with pPVA) produced a few nonspecific bands that led to rejection of the corresponding band in the pPVA-Mu footprinting analysis. A few signals were more obvious in the selected sample than in the unselected sample, but they were not studied further.
Table 3.
PVA-Specific Oligonucleotides Used in the Genomic Segment Amplification
| Primer pair (forward-reverse) | Region amplified (first and last nt) | Oligonucleotide sequence | |
|---|---|---|---|
| Forward | Reverse | ||
| 61475-FO15R | 2-1259 | aataaacaactacaaaaca | agtgtcctcatccatgttcaaattc |
| 5960-5961 | 1053-2426 | tattcaacaggggatgttttctggc | tccaaccctgtagtgcttcat |
| 51568-51567 | 2268-3317 | gaattttggttgatcacgatctca | cttacagtactccaattccaggc |
| FO14F-FO14R | 3099-4405 | caggctgtgaatggattgcaag | acttcattataacttgccacgtaca |
| 71832-6695 | 4227-5528 | gtgtccgccacaccaccagg | ctggaactgcactgcctctaagta |
| 71106-71105 | 5152-6375 | ttgagaaattttaaatgtgatgc | tcaagggcccaaaacctaatccaa |
| 72270-6281 | 6256-6983 | caacatctatgtttacaccc | ttgggtgtatactgcctctc |
| FO2F-FO1R | 6799-7570 | ttcccaaaggattcacagaaac | gtttcaattggtgcagctgt |
| FO1F-FO5R | 7180-8260 | gagaggcttacaacacaggatc | tcatgtgatctatcccactcc |
| FO5F-FO7R | 7869-9111 | caccagatggcacagttgtta | ctctcaaggtgcgttgaagacc |
| FO7F-4141 | 8747-9565 | gctatcttacaaaccaaagcaagt | ccctgacagttgaaacataaa |
Genome region coordinates follow the PVA-B11 sequence (AJ296311). Oligonucleotides are shown in the 5′ to 3′ direction.
Insertion Mutants: Isolation, Protoplast Inoculation, Electron Microscopy, and Plant Inoculation
The exact positions of the insertion sites were determined for nine individual plasmid clones by DNA sequence analysis using the appropriate primers, ThermoSequenase kit (Amersham), and ALF DNA Sequencer (Pharmacia). Each plasmid was linearized using AgeI, electroporated into tobacco protoplasts, and the protoplast tested for PVA infection by DAS-ELISA as described above. Virions were studied using immunosorbent electron microscopy (ISEM) using anti-PVA CP MAbs (mixture of MAb 58/0 and MAb 58/6; Rajamäki et al. 1998) as described (Roberts and Harrison 1979). They were visualized with a transmission electron microscope (Philips CM12) at 16–24 × 103-fold magnification at the Wood Ultrastructure Research Centre, SLU.
Linearized plasmids were also used to coat microprojectiles that were subsequently bombarded into the first full-grown true leaves of four-week-old tobacco plants by one or two shots using a Helios Gene Gun (Bio-Rad) as described (Hämäläinen et al. 2000; Kekarainen and Valkonen 2000). The tobacco plants were grown in a growth chamber (photoperiod 18 h, light intensity 250 μEs−1m−2, temperature 19°/17°C day/night, relative humidity 40%). PVA infection was detected in inoculated and upper noninoculated leaves by DAS-ELISA 15 days after bombardment as described (Rajamäki et al. 1998). Total RNA was purified from systemically infected tobacco leaves and cDNA synthesized (reaction volume 20 μL) using Superscript II reverse transcriptase and random hexamers (Life Technologies). One microliter of each cDNA preparation was used in PCR to amplify the regions containing the insertions. PCR products were digested with NotI and analyzed by electrophoresis on a 2% agarose gel in TBE buffer for verification of the insertion in each clone.
Acknowledgments
We thank Saija Haapa, Dr. Igor Oruetxebarria, and Dr. Sabina Vidal for helpful discussions, and Prof. Hans Ronne for critical reading of the manuscript. Financial support from the Academy of Finland (grants 34529, 36256, and 45889), the Technology Development Centre Finland, the Nilsson-Ehle Foundation, and the Swedish Forestry and Agriculture Research Council (SJFR, grant 32.0667/97) is gratefully acknowledged.
The publication costs of this article were defrayed in part by payment of page charges. This article must therefore be hereby marked “advertisement” in accordance with 18 USC section 1734 solely to indicate this fact.
Footnotes
E-MAIL jari.valkonen@vbiol.slu.se; FAX 46 18 67 3392.
Article and publication are at http://www.genome.org/cgi/doi/10.1101/gr.220702.
Article published online before print in March 2002.
REFERENCES
- Akerley BJ, Rubin EJ, Camilli A, Lampe DJ, Robertson HM, Mekalanos JJ. Systematic identification of essential genes by in vitro mariner mutagenesis. Proc Natl Acad Sci. 1998;95:8927–8932. doi: 10.1073/pnas.95.15.8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atreya CD, Atreya PL, Thornbury DW, Pirone TP. Site-directed mutations in the potyvirus HC-Pro gene affect helper component activity, virus accumulation, and symptom expression in infected tobacco plants. Virology. 1992;191:106–111. doi: 10.1016/0042-6822(92)90171-k. [DOI] [PubMed] [Google Scholar]
- Atreya CD, Pirone TP. Mutational analysis of the helper component-proteinase gene of a potyvirus: Effects of amino acid substitutions, deletions, and gene replacement on virulence and aphid transmissibility. Proc Natl Acad Sci. 1993;90:11919–11923. doi: 10.1073/pnas.90.24.11919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belsham GJ, Sonenberg N. Picornavirus RNA translation: Roles for cellular proteins. Trends Microbiol. 2000;8:330–335. doi: 10.1016/s0966-842x(00)01788-1. [DOI] [PubMed] [Google Scholar]
- Birren B, Green ED, Klapholtz S, Myers RM, Roskams J. Genome analysis. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1997. [Google Scholar]
- Chivasa S, Murphy AM, Naylor M, Carr JP. Salicylic acid interferes with tobacco mosaic virus replication via a novel salicylichydroxamic acid-sensitive mechanism. Plant Cell. 1997;9:547–557. doi: 10.1105/tpc.9.4.547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi I-R, Stenger DC, Morris TJ, French R. A plant virus vector for systemic expression of foreign genes in cereals. Plant J. 2000;23:547–555. doi: 10.1046/j.1365-313x.2000.00820.x. [DOI] [PubMed] [Google Scholar]
- Clark MF, Adams AN. Characteristics of the microplate method of enzyme-linked immunosorbent assay (ELISA) for the detection of plant viruses. J Gen Virol. 1977;34:475–483. doi: 10.1099/0022-1317-34-3-475. [DOI] [PubMed] [Google Scholar]
- Denecke J, Gosselé V, Botterman J, Cornelissen M. Quantitative analysis of transiently expressed genes in plant cells. Methods Mol Cell Biol. 1989;1:19–27. [Google Scholar]
- Devine SE, Boeke JD. Efficient integration of artificial transposons into plasmid targets in vitro: A useful tool for DNA mapping, sequencing and genetic analysis. Nucleic Acids Res. 1994;22:3765–3772. doi: 10.1093/nar/22.18.3765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dougherty WG, Semler BL. Expression of virus-encoded proteinases—functional and structural similarities with cellular enzymes. Microbiol Rev. 1993;57:781–822. doi: 10.1128/mr.57.4.781-822.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eagles RM, Balmorimelian E, Beck DL, Gardner RC, Forster RLS. Characterization of NTPase, RNA-binding and RNA-helicase activities of the cytoplasmic inclusion protein of tamarillo mosaic potyvirus. Eur J Biochem. 1994;224:677–684. doi: 10.1111/j.1432-1033.1994.t01-1-00677.x. [DOI] [PubMed] [Google Scholar]
- Fernández A, Guo HS, Sáenz P, Simón-Buela L, Gómez de Cedrón M, García JA. The motif V of plum pox potyvirus CI RNA helicase is involved in NTP hydrolysis and is essential for virus RNA replication. Nucleic Acids Res. 1997;25:4474–4480. doi: 10.1093/nar/25.22.4474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- German-Retana S, Candresse T, Alias E, Delbos R-P, Le Gall O. Effects of green fluorescent protein or β-glucuronidase tagging on the accumulation and pathogenicity of a resistance-breaking Lettuce mosaic virus isolate in susceptible and resistant lettuce cultivars. Mol Plant Microbe Interact. 1999;13:316–324. doi: 10.1094/MPMI.2000.13.3.316. [DOI] [PubMed] [Google Scholar]
- Giachetti C, Sembler BL. Role of the viral membrane polypeptide in strand-specific initiation of poliovirus RNA synthesis. J Virol. 1991;65:2647–2654. doi: 10.1128/jvi.65.5.2647-2654.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin TJ, Parsons L, Leschziner AE, DeVost J, Derbyshire KM, Grindley NDF. In vitro transposition of Tn552: A tool for DNA sequencing and mutagenesis. Nucleic Acids Res. 1999;27:3859–3865. doi: 10.1093/nar/27.19.3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo D, Merits A, Saarma M. Self-association and mapping of interaction domains of helper component-proteinase of potato A potyvirus. J Gen Virol. 1999;80:1127–1131. doi: 10.1099/0022-1317-80-5-1127. [DOI] [PubMed] [Google Scholar]
- Gwinn ML, Stellwagen AE, Craig NL, Tomb JF, Smith HO. In vitro Tn7 mutagenesis of Haemophilus influenzae Rd and characterization of the role of atpA in transformation. J Bacteriol. 1997;179:7315–7320. doi: 10.1128/jb.179.23.7315-7320.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapa S, Suomalainen S, Eerikäinen S, Airaksinen M, Paulin L, Savilahti H. An efficient DNA sequencing strategy based on the bacteriophage Mu in vitro DNA transposition reaction. Genome Res. 1999b;9:308–315. [PMC free article] [PubMed] [Google Scholar]
- Haapa S, Taira S, Heikkinen E, Savilahti H. An efficient and accurate integration of mini-Mu transposons in vitro: A general methodology for functional genetic analysis and molecular biology applications. Nucleic Acids Res. 1999a;27:2777–2784. doi: 10.1093/nar/27.13.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapa-Paananen S, Rita H, Savilahti H. DNA transposition of bacteriophage Mu: A quantitative analysis of target site selection in vitro. J Biol Chem. 2002;277:2843–2851. doi: 10.1074/jbc.M108044200. [DOI] [PubMed] [Google Scholar]
- Haldeman-Cahill R, Daros JA, Carrington JC. Secondary structures in the capsid protein coding sequence and 3′ nontranslated region involved in amplification of the tobacco etch virus genome. J Virol. 1998;72:4072–4079. doi: 10.1128/jvi.72.5.4072-4079.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hämäläinen JH, Kekarainen T, Gebhardt C, Watanabe KN, Valkonen JPT. Recessive and dominant genes interfere with the vascular transport of Potato virus A in diploid potatoes. Mol Plant Microbe Interact. 2000;13:402–412. doi: 10.1094/MPMI.2000.13.4.402. [DOI] [PubMed] [Google Scholar]
- Johansen IE, Søgaard Lund O, Fhulsager CK, Laursen J. Recessive resistance in Pisum sativum and potyvirus pathotype resolved in a gene-for-cistron correspondence between host and virus. J Virol. 2001;75:6609–6614. doi: 10.1128/JVI.75.14.6609-6614.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen LK, Morrow CD. The RNA encompassing the internal ribosome entry site in the poliovirus 5′ nontranslated region enhances the encapsidation of genomic RNA. Virology. 2000;273:391–399. doi: 10.1006/viro.2000.0433. [DOI] [PubMed] [Google Scholar]
- Johnson KL, Sarnow P. Three poliovirus 2B mutants exhibit non-complementable defects in viral RNA amplification and display dosage-dependent dominance over wild-type poliovirus. J Virol. 1991;65:4341–4349. doi: 10.1128/jvi.65.8.4341-4349.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasschau KD, Carrington JC. Requirement for HC-Pro processing during genome amplification of tobacco etch potyvirus. Virology. 1995;209:268–273. doi: 10.1006/viro.1995.1254. [DOI] [PubMed] [Google Scholar]
- Kasschau KD, Carrington JC. Long-distance movement and replication maintenance functions correlate with silencing suppression activity of potyviral HC-Pro. Virology. 2001;285:71–81. doi: 10.1006/viro.2001.0901. [DOI] [PubMed] [Google Scholar]
- Kekarainen T, Valkonen JPT. Inoculation of viral RNA and cDNA to potato and tobacco plants using the Helios Gene Gun. Bio Rad Bull. 2000;2531:4. [Google Scholar]
- Kekarainen T, Merits A, Oruetxebarria I, Rajamäki ML, Valkonen JPT. Comparison of the complete sequences of five different isolates of Potato virus A (PVA), genus Potyvirus. Arch Virol. 1999;144:2355–2366. doi: 10.1007/s007050050649. [DOI] [PubMed] [Google Scholar]
- Klein PG, Klein RR, Rodríguez-Cerezo E, Hunt AG, Shaw JG. Mutational analysis of the Tobacco vein mottling virus genome. Virology. 1994;204:759–769. doi: 10.1006/viro.1994.1591. [DOI] [PubMed] [Google Scholar]
- Laurent LCL, Olsen MN, Crowley RA, Savilahti H, Brown PO. Functional characterization of the Human immunodeficiency virus type 1 genome by genetic footprinting. J Virol. 2000;74:2760–2769. doi: 10.1128/jvi.74.6.2760-2769.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard S, Plante D, Wittmann S, Daigneault N, Fortin MG, Laliberte J-C. Complex formation between potyvirus VPg and translation eukaryotic initiation factor 4E correlates with virus infectivity. J Virol. 2000;74:7730–7737. doi: 10.1128/jvi.74.17.7730-7737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XH, Carrington JC. Complementation of tobacco etch potyvirus mutants by active RNA polymerase expressed in transgenic cells. Proc Natl Acad Sci. 1995;92:457–461. doi: 10.1073/pnas.92.2.457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manoil C, Bailey J. A simple screen for permissive sites in proteins: Analysis of Escherichia coli lac permease. J Mol Biol. 1997;267:250–263. doi: 10.1006/jmbi.1996.0881. [DOI] [PubMed] [Google Scholar]
- Nelson BD, Manoil C, Traxler B. Insertion mutagenesis of the lac repressor and its implications for structure-function analysis. J Bacteriol. 1997;179:3721–3728. doi: 10.1128/jb.179.11.3721-3728.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuveglise C, Nicaud JM, Ross-Macdonald P, Gaillardin C. A shuttle mutagenesis system for tagging genes in the yeast Yarrowia lipolytica. Gene. 1998;213:37–46. doi: 10.1016/s0378-1119(98)00205-4. [DOI] [PubMed] [Google Scholar]
- Niepel M, Gallie DR. Identification and characterization of the functional elements within the Tobacco etch virus 5′ leader required for cap-independent translation. J Virol. 1999;73:9080–9088. doi: 10.1128/jvi.73.11.9080-9088.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nolasco G, Deblas C, Torres V, Ponz F. A method combining immunocapture and PCR amplification in a microtiter plate for the detection of plant-viruses and subviral pathogens. J Virol Methods. 1993;45:201–218. doi: 10.1016/0166-0934(93)90104-y. [DOI] [PubMed] [Google Scholar]
- Oruetxebarria I, Guo DY, Merits A, Mäkinen K, Saarma M, Valkonen JPT. Identification of the genome-linked protein in virions of Potato virus A, with comparison to other members in genus Potyvirus. Virus Res. 2001;73:103–112. doi: 10.1016/s0168-1702(00)00216-1. [DOI] [PubMed] [Google Scholar]
- Puurand Ü, Valkonen JPT, Mäkinen K, Rabenstein F, Saarma M. Infectious in vitro transcripts from cloned cDNA of the potato A potyvirus. Virus Res. 1996;40:135–140. doi: 10.1016/0168-1702(95)01263-x. [DOI] [PubMed] [Google Scholar]
- Rajamäki M, Merits A, Rabenstein F, Andrejeva J, Paulin L, Kekarainen T, Kreuze JF, Forster RLS, Valkonen JPT. Biological, serological, and molecular differences among isolates of potato a potyvirus. Phytopathology. 1998;88:311–321. doi: 10.1094/PHYTO.1998.88.4.311. [DOI] [PubMed] [Google Scholar]
- Revers F, Le Gall O, Candresse T, Maule AJ. New advances in understanding the molecular biology of plant/potyvirus interactions. Mol Plant Microbe Interact. 1999;12:367–376. [Google Scholar]
- Roberts IM, Harrison BD. Detection of potato leafroll and potato mop-top viruses by immunosorbent electron microscopy. Ann Appl Biol. 1979;93:289–297. [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular cloning. A laboratory manual. Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press; 1989. [Google Scholar]
- Savilahti H, Rice PA, Mizuuchi K. The phage Mu transpososome core: DNA requirements for assembly and function. EMBO J. 1995;14:4893–4903. doi: 10.1002/j.1460-2075.1995.tb00170.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaad MC, Haldeman Cahill R, Cronin S, Carrington JC. Analysis of the VPg-proteinase (NIa) encoded by tobacco etch potyvirus: Effects of mutations on subcellular transport, proteolytic processing, and genome amplification. J Virol. 1996;70:7039–7048. doi: 10.1128/jvi.70.10.7039-7048.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaad MC, Jensen PE, Carrington JC. Formation of plant RNA virus replication complexes on membranes: Role of an endoplasmic reticulum-targeted viral protein. EMBO J. 1997;16:4049–4059. doi: 10.1093/emboj/16.13.4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shukla DD, Ward CW, Brunt AA. The Potyviridae. Wallingford, UK: CAB International; 1994. [Google Scholar]
- Simón-Buela L, Guo HS, García JA. Long sequences in the 5′ noncoding region of plum pox virus are not necessary for viral infectivity but contribute to viral competitiveness and pathogenesis. Virology. 1997;233:157–162. doi: 10.1006/viro.1997.8574. [DOI] [PubMed] [Google Scholar]
- Singh IR, Crowley RA, Brown PO. High-resolution functional mapping of a cloned gene by genetic footprinting. Proc Natl Acad Sci. 1997;94:1304–1309. doi: 10.1073/pnas.94.4.1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith V, Botstein D, Brown PO. Genetic footprinting: A genomic strategy for determining a gene's function given its sequence. Proc Natl Acad Sci. 1995;92:6479–6483. doi: 10.1073/pnas.92.14.6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urcuqui-Inchima S, Haenni AL, Bernardi F. Potyvirus proteins: A wealth of functions. Virus Res. 2001;74:157–175. doi: 10.1016/s0168-1702(01)00220-9. [DOI] [PubMed] [Google Scholar]
- Verchot J, Carrington JC. Debilitation of plant potyvirus infectivity by P1 proteinase-inactivating mutations and restoration by 2nd-site modifications. J Virol. 1995;69:1582–1590. doi: 10.1128/jvi.69.3.1582-1590.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu XJ, Shaw JG. Evidence that assembly of a potyvirus begins near the 5 ‘ terminus of the viral RNA. J Gen Virol. 1998;79:1525–1529. doi: 10.1099/0022-1317-79-6-1525. [DOI] [PubMed] [Google Scholar]
- Yao NH, Hesson T, Cable M, Hong Z, Kwong AD, Le HV, Weber PC. Structure of the hepatitis C virus RNA helicase domain. Nat Struct Biol. 1997;4:463–467. doi: 10.1038/nsb0697-463. [DOI] [PubMed] [Google Scholar]
- York D, Welch K, Goryshin IY, Reznikoff WS. Simple and efficient generation in vitro of nested deletions and inversions: Tn5 intramolecular transposition. Nucleic Acids Res. 1998;26:1927–1933. doi: 10.1093/nar/26.8.1927. [DOI] [PMC free article] [PubMed] [Google Scholar]