

Abstract
Aging is associated with cardiac hypertrophy and arterial stiffening possibly associated with accumulation of advanced glycation end products (AGEs). We evaluated the effect of aminoguanidine, an inhibitor of AGE production, on end-stage alterations of renal and cardiovascular systems. Normotensive WAG/Rij rats were treated from 24 to 30 mo with aminoguanidine and compared with a control group. Aminoguanidine did not modify body and kidney weights but prevented the age-related cardiac hypertrophy (heart weight: 1276 ± 28 mg and 1896 ± 87 mg in 24- and 30-mo-old control animals and 1267 ± 60 mg in 30-mo-old treated rats, P < 0.01). The increase in mesangial surface in aging rats was reduced by 30% by aminoguanidine. Collagen content of the arterial wall increased between 24 and 30 mo whereas elastin content, media thickness, and smooth muscle cell number remained unchanged. Aminoguanidine did not affect these parameters; however, the age-related increase in aortic impedance (12.4 ± 1.4 and 18.2 ± 1.9 103⋅dyne⋅sec⋅cm−5 in control 24- and 30-mo-old rats, P < 0.01) and the decrease in carotid distensibility (0.79 ± 0.11 and 0.34 ± 0. 07 mm Hg−1 in control 24- and 30-mo-old rats, P < 0.01) were prevented by aminoguanidine. The prevention of arterial stiffening and cardiac hypertrophy in the absence of changes in collagen and elastin content suggests that the effect of aminoguanidine is related to a decrease in the AGE-induced cross-linking of the extracellular matrix.
Aging is characterized by structural and functional changes of the cardiovascular and renal systems. The heart undergoes left ventricular hypertrophy, myocardial fibrosis, and lengthening of action potential (1–4). Arteries exhibit increases in media thickness, wall collagen content, and size of smooth muscle cells, with stiffening of the vessel wall (5–7). Kidney aging is associated with glomerular hypertrophy, increase in intrarenal resistance, proteinuria, thickening of basement membrane, expansion of mesangial matrix, and diffuse focal and segmental glomerulosclerosis (8–10).
Several investigators have proposed that most of these cardiovascular and renal modifications would be related to glycation of proteins and production of advanced glycation end products (AGEs) (11–13). The main experimental arguments for the potential role of AGEs in these processes are derived from diabetic animals studies that have evaluated the effects of aminoguanidine administration, an inhibitor of AGEs accumulation, on the heart, arteries, and kidneys. Aminoguanidine increased carotid and myocardial diastolic compliance, reduced fluid filtration across the carotid wall, and prevented the expansion of the mesangial area of diabetic animals (14–17). More recently, an effect of chronic administration of aminoguanidine on aging of the cardiovascular system also has been reported by Li et al. (18). Fischer 344 and Sprague–Dawley rats treated from 6 to 24 mo had a reduced content of AGEs in plasma, heart, blood vessels, and kidney. The age-related cardiac hypertrophy and the decrease in endothelial dependent vasodilatory response reported in the control aging rats were prevented by aminoguanidine in the older animals. The treatment also reduced proteinuria and glomerular sclerosis without modifications of thickness of glomerular basement membrane.
These effects of long term aminoguanidine treatment suggest that the control of lifelong AGE accumulation may prevent aging of the renal and cardiovascular systems. However, in a recent study on the effects of angiotensin I converting enzyme inhibition on the aging processes, we found that, in addition to the progressive changes occurring from 6 to 24 mo in rats, tremendous alterations had occurred in the last part of life between 24 and 30 mo (17, 10). These modifications were not due to any increase in blood pressure, and it was suggested that modifications of the extracellular matrix could be a key parameter in these aging processes. The hypothesis that AGEs play a role in the end-life structural and functional changes in renal and cardiovascular systems was therefore tested in normotensive rats. Aminoguanidine was administered to 24-mo-old rats for 6 mo compared with a control group receiving placebo. The results indicated that aminoguanidine prevents arterial stiffening and cardiac hypertrophy without significantly affecting arterial blood pressure and cardiac output. The aortic wall histological structure (media thickness, collagen, and elastin content) was not affected by aminoguanidine, suggesting that prevention of cardiac hypertrophy and arterial stiffening resulted from the effect of aminoguanidine on cross-linking of the extracellular matrix proteins.
METHODS
Experiments were performed in normotensive male WAG/Rij rats that were born and maintained in the specific pathogen-free animal facility of the Centre d’Etudes de Saclay (Gif-sur-Yvette, France). The animals were fed ad libitum a commercial diet (DO4, UAR, Epinay sur Orge, France) containing 2% fish protein, 15% vegetable proteins, and a total of 2,900 kcal/kg. They were maintained on a 14:10-h light-dark cycle at 20°C room temperature and 50% humidity. Rats of this strain remain lean even when fed ad libitum, have a constant blood pressure throughout life, and show no sign of hyperglycemia or glycosuria from 6 to 30 mo (7, 8, 10, 19).
At the age of 24 mo, 60 rats were randomized into two groups, control and experimental. Aminoguanidine hemisulfate, (Sigma) was added to the drinking water (1 g/liter) in the experimental group. The water consumption of the animals was checked each week. Control rats drank 15.0 ± 0.3 ml/day, and treated rats drank 16.0 ± 0.5 ml/day. These values were not significantly different and were constant throughout treatment. The mean amount of administered aminoguanidine was 16 mg/day per rat, i.e., 5 mg/day/100 g body weight. An additional group of control 24-mo-old animals was explored in parallel.
Hemodynamic Study—Closed Chest.
General surgical procedures and measurement of hemodynamic variables in anesthetized rats have been described (20). Rats were anesthetized with 50 mg/kg pentobarbital i.p., placed on a heating pad, intubated, and ventilated with a rodent respirator (model 680, Harvard Apparatus). Both left and right common carotid arteries were isolated. Pressure was measured in the right carotid artery with a 2F Millar catheter tip (Houston, TX) pressure transducer. Diameter was measured in the left carotid artery with an echotracking device (NIUS, ASULAB, Switzerland) by using a 10 MHz probe. This device has been evaluated extensively for highly accurate measurements of vascular dimensions in small arteries (21).
Carotid artery compliance was determined by computing the ratio of magnitudes of diameter oscillations (ΔD) and phasic pressure oscillations (ΔP) represented as carotid artery compliance = ΔD:ΔP.
Hemodynamic Study—Open Chest.
A midsternal thoracotomy was performed, and the ascending aorta was dissected free. The pressure transducer was advanced into the ascending aorta. An adapted Doppler probe was positioned around the vessel to measure phasic aortic blood flow. The system was allowed to stabilize for 10 min before aortic blood flow and pressure were recorded and processed by a microcomputer system (Vectra, Hewlett–Packard) with an analog–digital converter. All parameters were calculated on a beat-to-beat basis for 30 sec and then averaged. In steady-state conditions, measurements were obtained of systolic, diastolic, and mean arterial blood pressure, cardiac output, and heart rate. Total peripheral resistance was determined as the quotient of mean arterial blood pressure and cardiac output.
Systemic arterial compliance was calculated from a simple elastic model that discharges during diastole into a single resistance, reflecting the total peripheral resistance. We used the modification of the traditional Windkessel model described by Liu et al. (22), which accounts for a nonlinear exponential pressure–volume relationship by using the area under the diastolic pressure wave form:
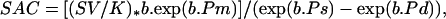 |
where SV is stroke volume; K is an area index (systolic area plus diastolic area/diastolic area) determined from the aortic pressure wave form; b is an experimentally determined coefficient, and Ps, m, and d are systolic, mean, and diastolic aortic pressures, respectively.
Pulsatile pressure and flow signals were subjected to Fourier analysis and impedance modulus, and phase was determined from the harmonic components. Corrections were made for the delay of the pressure transducer and flowmeter that was linear (6.6 Hz) in the frequency range 0–50 Hz. Characteristic impedance was determined from the average of impedance moduli for the second to the sixth harmonic (23).
Artery Morphometry.
A segment of 2–3 cm from the descending thoracic aorta was fixed at operating pressure (corresponding to the mean arterial pressure of each rat) in 10% formaldehyde in saline and embedded in paraffin. Three successive 5-μm sagittal sections were treated by specific staining for the various media structures. Collagen fibers were stained with Sirius red, elastin was stained with orcein, and nuclei were stained with hematoxylin after periodic acid oxidation. Morphometric analysis was performed with a automated image processor (Microvision, Evry, France) as described (24–26). Mean medial thickness was obtained by measuring the distance between the internal and external elastic laminae (70 measurements in each section). The medial elastin and collagen contents were obtained by measuring the relative area/density in 20 contiguous fields in each orcein and Sirius red-stained section. Elastin and collagen densities were defined as the ratio of the area stained by orcein or Sirius red to the area of the studied fields. The total elastin and collagen contents per millimeter of aortic section were calculated as the product of the media surface area per millimeter of longitudinal section and the elastin and collagen densities, respectively. The number of nuclei were obtained within 20 fields of ≈10,000-μm2 area on each section from which was calculated the mean area of each nucleus. Repeated measurements were performed, pooled, and averaged for the three measurements of elastin, collagen, and nuclei in the corresponding stained sections of the aortic wall media of each rat. Morphological analyses were performed twice by two independent observers by using a single-blind protocol.
Glomerular Morphometry.
The left kidney was excised, weighed, and decapsulated. Slices were fixed for 24 h in Bouin’s solution. They were subsequently embedded in paraffin, and 3-μm thick sections were stained by Marinozzi silver staining. The procedure for morphometric determination of the different glomerular domains was previously described (10). Glomeruli, 30 superficial and 30 deep, were analyzed in each kidney. For every investigated glomeruli, the following measurements were made with an automated image-analyzing system NS 15,000 (Microvision, Evry, France): (i) the total glomerular surface area limited by the internal edge of the Bowman’s capsule; (ii) the glomerular tuft area delineated by the glomerular basement membrane; (iii) the mesangial domain area defined by the glomerular tuft surface area minus the areas of the capillary lumens and the capillary free walls; (iv) the total surface area of the capillary lumen profiles; and (v) the number of capillary lumen profiles. The results are expressed as mean ± SEM. Statistical analysis was performed by means of ANOVA and significance was set at P < 0.05.
RESULTS
Survival.
At the age of 24 mo, 30% of the initial cohort of rats born and raised in our animal facilities had died. The 60 animals used in this study then belonged to the 70% survivors. At 30 mo, the end of the 6-mo treatment period, there were 15 surviving animals in the control group and 14 in the aminoguanidine group. We concluded that end-life aminoguanidine administration did not modify the lifespan of male WAG/Rij rats.
Body, Heart, and Kidney Weights.
Mean body weight of the animals was unchanged from 24 to 30 mo and was not modified by aminoguanidine treatment (Table 1). Heart weight markedly increased in the control group from 24 to 30 mo. This cardiac hypertrophy was prevented by aminoguanidine administration (Fig. 1). Kidney weight also significantly increased with age; aminoguanidine had no effect on age-related renal hypertrophy (Table 1).
Table 1.
Body, heart, and kidney weights of 24- and 30-mo-old control rats and 30-mo-old aminoguanidine rats treated for 6 mo
| Rats
|
|||||
|---|---|---|---|---|---|
| 24-mo-old | 30-mo-old control | 30-mo-old aminoguanidine | ANOVA
|
||
| Age | Treatment | ||||
| Body weight, g | 388 ± 5 | 372 ± 14 | 367 ± 24 | NS | NS |
| Heart weight, mg | 1276 ± 28 | 1896 ± 87 | 1267 ± 60 | P < 0.001 | P < 0.001 |
| Two kidneys weight, mg | 2.09 ± 0.07 | 2.43 ± 0.05 | 2.38 ± 0.08 | P < 0.001 | NS |
n = 12 animals in each groups. NS, not significant.
Figure 1.

Heart-to-body weight ratio in control 24- and 30-mo-old rats and in 30-mo-old animals treated with aminoguanidine from 24 to 30 mo. n = 12 rats per group. ∗, Statistically different from the age-matched control group.
Arterial Wall Morphometry.
Media thickness of the thoracic aorta was not significantly different between 24- and 30-mo-old rats or between 30-mo-old control and treated animals (Table 2). The collagen content of the arterial wall significantly increased by 30% from 24 to 30 mo (P < 0.01). Aminoguanidine did not modify this rise in collagen. The elastin content of the arterial wall was unchanged in senescent and treated animals. As a consequence of increased collagen and constant elastin contents, the ratio of elastin over collagen was decreased with age in both the 30-mo-old control and experimental groups. The number and size of the nuclei in the media were not different in 24- and 30-mo-old control and aminoguanidine-treated rats, indicating that there was no smooth muscle cell hypertrophy or hyperplasia at the end of life in these animals. Aminoguanidine did not affect the number and the size of the smooth muscle cells.
Table 2.
Histomorphometric measurements of thoracic aorta in 24- and 30-mo-old control rats and 30-mo-old aminoguanidine rats treated for 6 mo
| Rats
|
|||||
|---|---|---|---|---|---|
| 24-mo-old | 30-mo-old control | 30-mo-old aminoguanidine | ANOVA
|
||
| Age | Treatment | ||||
| Media thickness, μm | 144 ± 6 | 153 ± 6 | 153 ± 6 | NS | NS |
| Collagen content, μm2/mm of length | 23495 ± 1379 | 30967 ± 2204 | 31150 ± 2797 | P < 0.001 | NS |
| Elastin content, μm2/mm of length | 23693 ± 1559 | 22534 ± 1664 | 25884 ± 2535 | NS | NS |
| Elastin over collagen ratio | 1.03 ± 0.07 | 0.85 ± 0.08 | 0.75 ± 0.07 | P < 0.001 | NS |
| Nucleus content, n/mm of length | 388 ± 28 | 367 ± 20 | 387 ± 43 | NS | NS |
| Nucleus size, μm2 | 8.08 ± 0.46 | 8.80 ± 0.54 | 8.39 ± 0.60 | NS | NS |
n = 12 in each group. NS, not significant.
Hemodynamics.
Mean arterial blood pressure as well as diastolic and systolic blood pressures were unchanged with age in the male WAG/Rij rats (Table 3). Values were slightly lower in the aminoguanidine group, although the difference did not reach statistical significance for any of the three parameters. Heart rate and cardiac output were comparable in the 24- and 30-mo-old control and treated animals. Total peripheral resistance did not change from 24 to 30 mo and was slightly, but significantly, decreased by aminoguanidine administration. The characteristic impedance was increased markedly in the 30-mo-old rats as compared with the 24-mo-old rats (Fig. 2). Aminoguanidine administered from 24 to 30 mo prevented the age-related increase in characteristic impedance.
Table 3.
Hemodynamics of 24- and 30-mo-old control rats and 30-mo-old aminoguanidine rats treated for 6 mo
| Rats
|
|||||
|---|---|---|---|---|---|
| 24-mo-old | 30-mo-old control | 30-mo-old aminoguanidine | ANOVA
|
||
| Age | Treatment | ||||
| Mean blood pressure, mm Hg | 122 ± 6 | 126 ± 6 | 111 ± 6 | NS | NS |
| Systolic pressure, mm Hg | 143 ± 6 | 146 ± 8 | 128 ± 8 | NS | NS |
| Diastolic pressure, mm Hg | 113 ± 4 | 109 ± 5 | 96 ± 5 | NS | NS |
| Cardiac output, ml/min | 66 ± 4 | 61 ± 4 | 66 ± 4 | NS | NS |
| Heart rate, beat/min | 355 ± 9 | 326 ± 5 | 320 ± 13 | NS | NS |
| Total peripheral resistance, 103⋅dyne⋅sec⋅cm−5 | 136 ± 8 | 143 ± 15 | 96 ± 6 | NS | P < 0.05 |
n = 12 in each group. NS, not significant.
Figure 2.

Aortic input characteristic impedance (103⋅dyne⋅sec⋅cm−5) measured in control 24- and 30-mo-old rats and in 30-mo-old animals treated with aminoguanidine from 24 to 30 mo. n = 12 rats per group. ∗Statistically different from the age-matched control group.
Carotid Diameter and Compliance.
Carotid diameter measured in situ was significantly enlarged with age (P < 0.05), and this enlargement was prevented by aminoguanidine (P < 0.05): 1043 ± 29 μm in 24-mo-old rats, 1138 ± 16 μm and 1015 ± 41 μm in untreated and treated 30-mo-old animals, respectively. In situ carotid compliance and distensibility were halved between 24 and 30 mo. Aminoguanidine treatment for 6 mo totally prevented the age-related decrease in distensibility (Fig. 3). Arterial distensibility was similar in the 24-mo-old control and 30-mo-old treated rats.
Figure 3.

Carotid distensibility (mm Hg−1) measured in control 24- and 30-mo-old rats and in 30-mo-old animals treated with aminoguanidine from 24 to 30 mo. n = 12 rats per group. ∗Statistically different from the age-matched control group.
Renal Morphometry.
The morphometric parameters of superficial and juxtamedullary glomeruli were not significantly different in the 24- or 30-mo-old animals. Consequently, the values from superficial and deep nephrons were pooled in the subsequent analysis. Glomerular surface area increased with age in both 30-mo-old control and treated rats, as did kidney weight (Table 4). The total capillary surface area and the number of capillary sections in each glomeruli were reduced between 24 and 30 mo. The mesangial area more than doubled from 24 to 30 mo in the control group. Six-month treatment with aminoguanidine did not modify glomerular and capillaries surface but reduced the mean mesangial surface expansion, although the effect was not statistically significant. Examination of individual data, however, indicated that, in half of the 30-mo-old aminoguanidine-treated rats, their mesangial area and capillary surface areas (495 ± 90 μm2 and 2556 ± 91 μm2, respectively) were in the range of the 24-mo-old animals. Mesangial expansion and capillary surface areas were inversely correlated within the 30-mo-old treated group—the larger the mesangial area, the smaller the capillary surface.
Table 4.
Glomerular morphometry in 24-mo-old and 30-mo-old control rats and 30-mo-old aminoguanidine rats treated for 6 mo
| Rats
|
|||||
|---|---|---|---|---|---|
| 24-mo-old | 30-mo-old control | 30-mo-old aminoguanidine | ANOVA
|
||
| Age | Treatment | ||||
| Glomerular surface, μm2 | 10228 ± 308 | 12470 ± 373 | 11525 ± 438 | P < 0.01 | NS |
| Total capillary surface area, μm2 | 2623 ± 125 | 2225 ± 84 | 2127 ± 176 | P < 0.01 | NS |
| Capillary sections/glomerulus, n | 83 ± 2 | 74 ± 1 | 73 ± 2 | P < 0.01 | NS |
| Mesangial surface, μm2 | 610 ± 121 | 1401 ± 160 | 1001 ± 229 | P < 0.05 | NS |
n = 8 rats in each group and 60 glomeruli for each animal. NS, not significant.
DISCUSSION
Administration of aminoguanidine to male rats from 24 to 30 mo prevented arterial stiffening, cardiac hypertrophy, and mesangial expansion without altering neither the collagen and elastin content of the arterial wall, nor the number and size of smooth muscle cells. These effects of aminoguanidine were very similar to those reported in diabetic rats, and it was proposed that common mechanisms may be involved in diabetes and aging. Such a similarity of changes in the cardiovascular and renal systems in diabetic and aging rats would be easily interpreted if the animals became diabetic with age. An increase in plasma Glc has been suggested from age-related insulin resistance that would result in hyperglycemia or in an increase in the integral of Glc concentration over time (27). However, Glc concentration measured in WAG/Rij rats was unchanged from 10 to 30 mo, and no glycosuria was seen in these senescent animals (8, 19). It is therefore unlikely that the reported effect of aminoguanidine on the aging processes was linked to the prevention of diabetes-related diseases appearing at the end of life.
At least two hypotheses have been proposed to explain the effect of aminoguanidine on renal and cardiovascular systems. One attributed a central role to nonenzymatic glycation of proteins, production of AGEs, and collagen cross-linking (12–14, 28). The other was related to vasodilatory dysfunction: AGEs are known to inhibit inducible NO synthase and to quench NO, and prevention of AGEs would favor NO activity (17, 29–31). The two hypotheses may apply to the aging process. Increased production and accumulation of AGEs have been noted in old animals, and treatment with aminoguanidine reduced this accumulation (11, 18, 32). Decreased vasodilatory response and vasodilatory reserve of arteries also have been documented in aging rats, associated with impaired production of NO (18, 33, 34). Depending on the organ, aminoguanidine could act on the aging processes through one of these mechanisms.
Artery.
The most frequently reported morphometric modifications of large arteries with age are lumen enlargement, thickening of the media, rise in collagen content, and drop in elastin-to-collagen ratio (3, 5–7). The media thickening is progressive with age and corresponds to smooth muscle hypertrophy without hyperplasia. The constant, smooth muscle cell population in the arterial wall was confirmed in the present study, whereas thickening was less marked between 24 and 30 mo than it was between 12 and 30 mo. Aminoguanidine did not modify the number of smooth muscle cells, indicating that it did not induce hyperplasia in the last part of life.
The increase in collagen content of the arterial wall is a general characteristic of aging and is common to many tissues, including heart and kidney. AGEs may be implicated in enhanced collagen content by conferring a high resistance to enzymatic proteolysis and a decrease in their rate of degradation. The balance between synthesis and hydrolysis of collagen would thus be shifted in favor of larger amounts of collagen in the arterial wall. In addition to conferring proteolytic resistance, AGEs stimulate the production of extracellular matrix. Receptors of AGEs have been found in smooth muscle cells of the arterial wall as well as in fibroblasts and may contribute to the remodeling of the vessel (12, 13). Although the effect of AGEs on protein synthesis has been documented in vitro and in vivo, their role in the increase in collagen content observed in the arterial wall at the end of life was not supported by the present data. Collagen content of the thoracic aorta increased by 30% from 24 to 30 mo, whether or not the animals were treated with aminoguanidine. The absolute elastin content of the arterial wall was unchanged from 24 to 30 mo, and aminoguanidine did not modify these values. As a result, the ratio of elastin-to-collagen was similarly reduced in control and treated 30-mo-old rats.
Apart from their role in the synthesis and degradation of extracellular matrix, AGEs are implicated in the cross-linking of the extracellular proteins, resulting in a change in mechanical properties of the conjunctive tissue (5, 6, 35). Several authors have proposed that such cross-linking of collagen and other matrix components are responsible for the age- or diabetes-induced stiffening of heart and large arteries. Prevention of AGE production and of protein cross-linking by aminoguanidine was first demonstrated by Brownlee et al. (28) in vitro and in vivo in diabetic rats. Correlatively, carotid compliance was increased and characteristic aortic input impedance was decreased by aminoguanidine in these diabetic rats (15). In the present experiment, distensibility of the thoracic aorta and carotid artery was reduced with age from 24 to 30 mo. This arterial stiffening was prevented by aminoguanidine as indicated by values of carotid compliance and aortic characteristic impedance. Because aminoguanidine did not induce any change in aortic media thickness, elastin and collagen content, or smooth muscle cell hypertrophy and hyperplasia, we hypothesize that the effect of aminoguanidine on the arterial wall mechanical properties would rather correspond to the difference in the molecular arrangement of the collagen and elastin network than to the gross morphological changes of arterial wall.
A role for cross-linking of extracellular matrix in aging arteries also is compatible with the effect of aminoguanidine on their diameter. The arteries were enlarged between 24 and 30 mo, and this enlargement was prevented by aminoguanidine treatment. This would probably not be related to a hemodynamic effect because blood pressure and cardiac output were unchanged in the treated animals.
Heart.
Cardiac hypertrophy in aging male rats is a common feature reported by many investigators (1–3, 7, 18). It is generally accepted that this increase in left ventricular weight is caused by elevation in afterload. Age-related rise in afterload is not necessarily the consequence of an increase in blood pressure, because it occurs even in animals whose mean blood pressure is unchanged throughout life (10). Neither can it be explained by a larger total peripheral resistance, which is also constant in different experimental models of aging. It probably results from the reduced diastolic stiffness of the left ventricle and large arteries characteristic of the aging process.
It is very likely that the prevention of age-related cardiac hypertrophy by aminoguanidine administration toward the end of life corresponds to a drug-induced decrease in left ventricular afterload. Different pharmacological effects of aminoguanidine that can modify vascular hemodynamics and afterload have been described. Aminoguanidine inhibits NO synthase activity and production of the endothelium-derived relaxing factor NO (17, 30, 31). This result was evidenced by increased blood pressure in rats after acute i.v. injection of large doses of aminoguanidine. However, such hypertensive effect of aminoguanidine has not been found during prolonged treatment. As a rule, chronic administration of aminoguanidine did not affect or slightly lower blood pressure in diabetic rats (15–17, 36, 37). In aging rats, increase in blood pressure consecutive to aminoguanidine administration was not detected in the present study. The same results was reported by Li et al. (18) who found that chronic administration of aminoguanidine did not significantly increase but rather tended to decrease arterial blood pressure.
Another mechanism affecting the vasodilation process has been proposed for aminoguanidine. It is based on the ability of AGEs to quench NO, resulting in reduced vasodilation. This is consistent with the reduced vasodilatory capacity of arteries in diabetic and aging rats, with the accumulation of AGEs in the artery and the prevention of vasodilatory dysfunction by aminoguanidine treatment (7, 18, 33, 34). A peripheral vasodilation and reduced blood pressure could thus contribute to reduction in afterload and prevention of cardiac hypertrophy by aminoguanidine. This would be compatible with the decrease in total peripheral resistance noticed in the 30-mo-old treated group, although blood pressure and cardiac output were not significantly changed by aminoguanidine.
The effect of aminoguanidine on cardiac hypertrophy alternatively may be related to reduction of AGEs cross-linking and prevention of the age-related increase in diastolic left ventricular stiffening. This has been well demonstrated in diabetic rats by Norton et al. (16) who found that aminoguanidine treatment prevented heart stiffness and cardiac hypertrophy without any effect on the collagen content of the heart. The hypothesis of a reduced afterload linked to decreased collagen cross-linking in the left ventricle also would be compatible with the parallel low accumulation of AGEs in the heart and prevention of cardiac hypertrophy after administration of aminoguanidine from 6 to 24 mo reported by Li et al. (18).
A change in collagen cross-linking and stiffness of large arteries is another candidate for a reduction of afterload. This has been well documented in diabetic rats whose AGEs accumulation was prevented, and normal arterial compliance was maintained by aminoguanidine (15). In aging normotensive rats, the present data showed that characteristic impedance increased by 47% and carotid distensibility decreased by 56% between 24 and 30 mo and that aminoguanidine maintained these parameters of the 30-mo-old treated rats to a value close to that of the 24-mo-old animals. Such prevention of arterial stiffening by aminoguanidine would contribute to reduce cardiac afterload and would be in part responsible for prevention of age-related cardiac hypertrophy.
Kidney.
Expansion of mesangial matrix is a common feature in aging rats. It occurs even in the absence of glomerulosclerosis and of any decrease in the number of nephrons (8, 9). In WAG/Rij rats, this expansion mainly takes place between 24 and 30 mo and doubles the mean mesangial surface area (5). A role for AGEs in the expansion of mesangial matrix has been evoked from in vivo experiments. Renal accumulation of AGEs has been found in aging men and rats (32). Administration of AGEs induced mesangial expansion,which is prevented by aminoguanidine (13, 37, 38). Reduction of renal accumulation of AGEs and limitation of the mesangial expansion by aminoguanidine administration has been reported in the diabetic rats (36, 39). The underlying mechanisms may involve the AGEs receptors localized on the mesangial cell, stimulation of proteins production, cross-linking of extracellular matrix, or trapping of plasma proteins (37, 40–44). The present morphometric determinations confirmed the previously reported glomerular enlargement, doubling of mesangium, and reduction in the capillary surface in rats between 24 and 30 mo (10). Six-month treatment with aminoguanidine partly reduced mesangial expansion. The reported relation between mesangial expansion and reduction in capillary area in 30-mo-old treated animals would suggest that these two parameters are causally connected. This hypothesis would need additional investigations to be confirmed.
In conclusion, aminoguanidine administered in drinking water to aging rats prevents the end-life increase in arterial stiffness and cardiac hypertrophy. This prevention of cardiovascular dysfunction occurred without any change in collagen and elastin content of the arterial wall and probably results from reduction in the AGE-induced cross-linking of heart and arterial wall extracellular matrix proteins.
Acknowledgments
This study was supported partially by a grant (BVI94023) of AP-HP, Délégation à la Recherche Clinique and by a grant from the Institut de Recherches Internationales Servier (Courbevoie, France). We are especially grateful to Prof. Paul VanHoutte for his permanent support and suggestions.
Footnotes
This paper was submitted directly (Track II) to the Proceedings Office.
Abbreviation: AGE, advanced glycation end product.
References
- 1.Kreher P, Ristori M, Corman B, Verdetti J. J Cardiovasc Pharmacol. 1995;25:75–80. doi: 10.1097/00005344-199501000-00012. [DOI] [PubMed] [Google Scholar]
- 2.Lakatta E G. Basic Res Cardiol. 1993;88:125–133. [PubMed] [Google Scholar]
- 3.Lakatta E G. In: Handbook of Physiology. Masoro E, editor. Oxford: Oxford Univ. Press; 1995. pp. 413–474. [Google Scholar]
- 4.Shreiner D P, Weisfelt M L, Shock N W. Am J Physiol. 1969;217:176–180. doi: 10.1152/ajplegacy.1969.217.1.176. [DOI] [PubMed] [Google Scholar]
- 5.Gerrity R G, Cliff W J. Exp Mol Pathol. 1972;16:382–402. doi: 10.1016/0014-4800(72)90012-3. [DOI] [PubMed] [Google Scholar]
- 6.Guyton G R, Lindsay K L, Dao D T. Am J Pathol. 1983;111:234–246. [PMC free article] [PubMed] [Google Scholar]
- 7.Michel J B, Heudes D, Michel O, Potevin P, Philippe M, Scalbert E, Corman B, Lévy B I. Am J Physiol. 1994;267:R124–R135. doi: 10.1152/ajpregu.1994.267.1.R124. [DOI] [PubMed] [Google Scholar]
- 8.Corman B, Michel J B. Am J Physiol. 1987;253:R555–R560. doi: 10.1152/ajpregu.1987.253.4.R555. [DOI] [PubMed] [Google Scholar]
- 9.François V, Heudes D, Bariety J, Bruneval P, Corman B. Mech Ageing Dev. 1996;91:11–22. doi: 10.1016/0047-6374(96)01773-3. [DOI] [PubMed] [Google Scholar]
- 10.Heudes D, Michel O, Chevalier J, Scalbert E, Ezan E, Bariety J, Zimmerman A, Corman B. Am J Physiol. 1994;266:R1038–R1051. doi: 10.1152/ajpregu.1994.266.3.R1038. [DOI] [PubMed] [Google Scholar]
- 11.Schnider S L, Kohn R R. J Clin Invest. 1980;66:1179–1181. doi: 10.1172/JCI109950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vlassara H, Bucala R, Striker L. Lab Invest. 1994;70:138–151. [PubMed] [Google Scholar]
- 13.Vlassara H, Fuh H, Makita Z, Krungkrai D, Cerami A, Bucala R. Proc Natl Acad Sci USA. 1992;89:12043–12047. doi: 10.1073/pnas.89.24.12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Edelstein D, Brownlee M. Diabetes. 1992;41:26–29. doi: 10.2337/diab.41.1.26. [DOI] [PubMed] [Google Scholar]
- 15.Huijberts M, Wolffenbuttel B, Struijker-Boudier H A, Crijns F, Kruseman A C, Poitevin P, Levy B I. J Clin Invest. 1993;92:1407–1411. doi: 10.1172/JCI116716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Norton G, Candy G, Woodiwiss A. Circulation. 1996;93:1905–1912. doi: 10.1161/01.cir.93.10.1905. [DOI] [PubMed] [Google Scholar]
- 17.Tilton R, Chang K, Hasan K, Smith S, Petrash J, Misko T, Moore W, Currie M, Corbett J, McDaniel M, Williamson J. Diabetes. 1993;42:221–232. doi: 10.2337/diab.42.2.221. [DOI] [PubMed] [Google Scholar]
- 18.Li M L, Steffes M, Donelly T, Liu C, Fuh H, Basgen J, Bucala R, Vlassara H. Proc Natl Acad Sci USA. 1996;93:3902–3907. doi: 10.1073/pnas.93.9.3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bakala H, Verbeke P, Perichon M, Corman B, Shaeverbeke J. Mech Ageing Dev. 1995;78:63–71. doi: 10.1016/0047-6374(94)01527-s. [DOI] [PubMed] [Google Scholar]
- 20.Benessiano J, Levy B I, Michel J B. J Pharmacol Methods. 1985;14:99–110. doi: 10.1016/0160-5402(85)90047-6. [DOI] [PubMed] [Google Scholar]
- 21.Tardy, Y., Hayoz, D., Mignot, J. P., Richard, P., Brunner, H. R. & Meister, J. J. (1992) J. Hypertens. 10, Suppl. 6, S105–S109. [PubMed]
- 22.Liu Z, Brin K P, Yin F. Am J Physiol. 1986;251:H588–H600. doi: 10.1152/ajpheart.1986.251.3.H588. [DOI] [PubMed] [Google Scholar]
- 23.Nichols W, O’Rourke M F. In: McDonald’s Blood Flow in Arteries. Arnold E, editor. Baltimore: William & Wilkins; 1992. [Google Scholar]
- 24.Levy B I, Duriez M, Phillipe M, Poitevin P, Michel J B. Circulation. 1994;90:3024–3033. doi: 10.1161/01.cir.90.6.3024. [DOI] [PubMed] [Google Scholar]
- 25.Salzmann J L, Azizi M, Michel J B, Camilleri J P. Acta Stereol. 1987;6:437–441. [Google Scholar]
- 26.Serra J. Image Analysis and Mathematical Morphology. London: Academic; 1982. pp. 31–52. [Google Scholar]
- 27.Barzilai N, Rosetti L. Am J Physiol. 1996;270:E930–E936. doi: 10.1152/ajpendo.1996.270.6.E930. [DOI] [PubMed] [Google Scholar]
- 28.Brownlee M, Vlassara H, Kooney A, Ulrich P, Cerami A. Science. 1986;232:1629–1632. doi: 10.1126/science.3487117. [DOI] [PubMed] [Google Scholar]
- 29.Bucala R, Tracey K, Cerami A. J Clin Invest. 1991;87:432–438. doi: 10.1172/JCI115014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Corbett J A, Tilton R G, Chang K, Hasan KS, Ido Y, Wang J L, Sweetland M A, Lancaster J R, Williamson J R, McDaniel M L. Diabetes. 1992;41:552–556. doi: 10.2337/diab.41.4.552. [DOI] [PubMed] [Google Scholar]
- 31.Scott J, Machoun M, McCormack D. J Appl Physiol. 1996;80:271–277. doi: 10.1152/jappl.1996.80.1.271. [DOI] [PubMed] [Google Scholar]
- 32.Yamada K, Miyahara Y, Hamaguchi K, Nakayama M, Nakano H, Nozaki O, Miura Y, Suzuki S, Tuchida H, Mimura N, Arataki N, Horiuchi S. Clin Nephrol. 1994;42:354–361. [PubMed] [Google Scholar]
- 33.Baylis C, Fredericks M, Wilson C, Munger K, Collins R. Am J Kidney Dis. 1990;3:244–251. doi: 10.1016/s0272-6386(12)80769-4. [DOI] [PubMed] [Google Scholar]
- 34.Reckelhoff J, Kellum J, Blanchard E, Bacon E, Wesley A, Kruckeberg W. Life Sci. 1994;55:1895–1902. doi: 10.1016/0024-3205(94)00521-4. [DOI] [PubMed] [Google Scholar]
- 35.Milch R A. Monogr Surg Sci. 1965;2:261–341. [PubMed] [Google Scholar]
- 36.Nicholls K, Mandel T. Lab Invest. 1989;60:486–491. [PubMed] [Google Scholar]
- 37.Fuh H, Yang D, Striker L, Vlassara H. Diabetes. 1992;41:9. (abstr.). [Google Scholar]
- 38.Oturai P, Rasch R, Hasselager E, Johansen P, Yokoyama H, Thomsen M, Myrup B, Kofoed-Enevoldsen A, Deckert T. APMIS. 1996;104:259–264. doi: 10.1111/j.1699-0463.1996.tb00715.x. [DOI] [PubMed] [Google Scholar]
- 39.Yang C, Vlassara H, Striker G, Striker L. Kidney Int. 1995;49:S55–S58. [PubMed] [Google Scholar]
- 40.Soulis-Liparota T, Cooper M, Papazoglou D, Clarke B, Jerums G. Diabetes. 1991;40:1328–1334. doi: 10.2337/diab.40.10.1328. [DOI] [PubMed] [Google Scholar]
- 41.Cohen M, Hud E, Van-Yu W, Ziyadeh N. Mol Cell Biochem. 1995;151:61–67. doi: 10.1007/BF01076897. [DOI] [PubMed] [Google Scholar]
- 42.Doi T, Vlassara H, Kirstein M, Yamada Y, Striker G, Striker L. Proc Natl Acad Sci USA. 1992;89:2873–2877. doi: 10.1073/pnas.89.7.2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Skolnik E, Yang Z, Makita Z, Radoff S, Kirstein M, Vlassara H. J Exp Med. 1991;174:931–939. doi: 10.1084/jem.174.4.931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vlassara H, Striker L, Teichberg S, Fuh H, Li Y, Steffes M. Proc Natl Acad Sci USA. 1994;91:11704–11708. doi: 10.1073/pnas.91.24.11704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yang C, Vlassara H, Peten E, He C, Striker G, Striker L. Proc Natl Acad Sci USA. 1994;91:9436–9440. doi: 10.1073/pnas.91.20.9436. [DOI] [PMC free article] [PubMed] [Google Scholar]